

Abstract
Cytochrome P450 2B6 (CYP2B6) is primarily responsible for the metabolism of the anti-HIV drug efavirenz (EFV). We set out to explore the molecular basis for CYP2B6 activity toward EFV by examining the metabolism of eight EFV analogues. cDNA-expressed CYP2B6 formed monooxygenated metabolites from EFV analogues containing an intact oxazinone or oxazine ring, but not from analogues with a disrupted ring, suggesting this ring is important for metabolism of EFV by CYP2B6. Subsequent substrate depletion analysis of EFV and EFV analogues found to be CYP2B6 substrates revealed further differences between these CYP2B6 substrates. Compounds that were not found to be CYP2B6 substrates were still able to inhibit CYP2B6 activity toward a known substrate, bupropion, suggesting they do gain access to the CYP2B6 active site. Taken together, these data reveal structural characteristics of EFV, namely, the oxazinone ring, that are critical for CYP2B6 metabolism of compounds with the EFV chemical scaffold.
Keywords: Drug metabolism, CYP2B6, efavirenz, structure−activity relationship
The cytochromes P450 (P450s) are a large family of monooxygenase enzymes that play a major role in the metabolism of xenobiotic and endogenous substrates. One such enzyme, human cytochrome P450 2B6 (CYP2B6), participates in the metabolism of an approximate 4% of the top 200 prescribed drugs in the United States,1 including the atypical antidepressant and smoking cessation aid bupropion2 and the non-nucleoside reverse transcriptase inhibitor efavirenz, or EFV.3,4 CYP2B6 has been detected in many tissues, including brain, intestine, and lung,5 but is primarily expressed in the liver where it is present in concentrations ranging from 0.5 to 100 pmol/mg total microsomal protein.5,6 This marked variability in expression is due in part to the highly polymorphic nature of the CYP2B6 gene. To date, 38 distinct CYP2B6 variants have been described,7 some of which have been associated with altered CYP2B6 expression at the mRNA8 and protein6 level as well as altered activity.1,9,10
EFV is a commonly prescribed non-nucleoside reverse transcriptase inhibitor used to treat HIV-1. CYP2B6 is primarily responsible for the formation of the major EFV metabolite, 8-hydroxyefavirenz.3,11 EFV has been shown to induce CYP2B6 transcription12 as well as inhibit CYP2B6 activity.4,13 Multiple crystal structures of the naturally occurring CYP2B6 variant K262R in complex with various inhibitors have been solved;14−16 however, a structure of CYP2B6 in complex with EFV has yet to be reported.
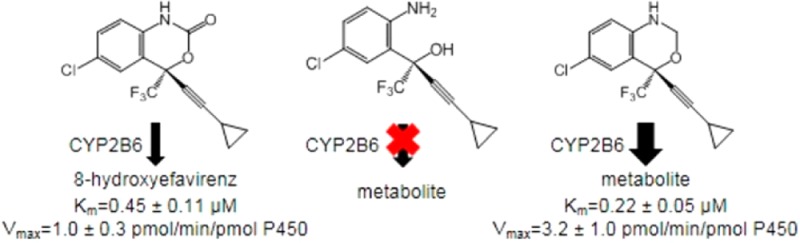
In this study, we set out to determine which regions of the EFV chemical structure contribute to CYP2B6 activity toward EFV. To this end, we analyzed the metabolism of a panel of EFV analogues (Figure 1). Our goals were (1) to determine if any of the analogues are CYP2B6 substrates and (2) to quantitatively compare the kinetics of EFV and EFV analogue metabolism by CYP2B6. In doing so, we observed that CYP2B6 readily metabolized analogues with an intact oxazinone ring (analogues 3, 4, and 8), whereas we did not observe metabolite formation from CYP2B6 incubation with analogues with a disrupted oxazinone ring (analogues 1, 2, 5, 6, and 7). Interestingly, CYP2B6 activity readily produced metabolites of an oxazine EFV analogue (analogue 8), further implicating the integrity of the oxazinone moiety in CYP2B6 metabolism of EFV and not simply the carbonyl oxygen atom.
Figure 1.

Structures of efavirenz (EFV) and EFV analogues 1–8.
Results and Discussion
EFV Analogues Undergo P450-Dependent Metabolism in Pooled Human Liver Microsomes
In order to begin our investigation into the ability of CYP2B6 to metabolize the EFV analogues, we first sought to identify all of the metabolites of these compounds that could be produced by P450 enzymes. To do so, each analogue was incubated with human liver microsomes, which contain a range of P450s, in the presence of NADPH, and product formation was analyzed using ultra high-performance liquid chromatography tandem mass spectrometry (uHPLC–MS/MS). Monooxygenated metabolites of analogues 1, 2, 3, 4, and 8 were detected, while N-dealkylated products were detected from analogues 5 and 6 (Figure 2). No metabolites of analogue 7 were observed. From EFV analogues 1 and 2, which have a disrupted oxazinone ring, monooxygenated metabolites (1-M1 and 2-M1, respectively) were detected, each with a retention time (RT) of 5.8 min (m/z 306, Figure 2). Product ion spectra for all metabolites are shown in Supporting Information Figure S1. The major fragment ions of these metabolites were m/z 268, 247, and 194, indicating a loss of H3Cl, C3H6O, and C3H3OF3. This fragmentation pattern suggests 1-M1 and 2-M1 were formed by oxygen insertion at the 5, 7, or 8 position of the benzene ring. EFV analogue 3, which has a trans-alkene rather than the alkyne of EFV, was metabolized to two different monooxygenated metabolites (m/z 332, 3-M1 and 3-M2) by human liver microsomes (Figure 2). These species corresponded to RTs of 4.98 and 6.43 min, respectively. Metabolite 3-M2, which was the most abundant metabolite observed from EFV analogue 3, displayed fragment ions of m/z 288, 268, 247, and 227, which we propose derived from a loss of CO2, C2H4Cl, C4H5O2, and CF3Cl. These fragment ions suggested the oxygen insertion occurred on the aromatic ring at the 7 or 8 position. The >1 min difference in RT between the two monooxygenated metabolites of EFV analogue 3 suggested that the position of oxygenation for 3-M1 was quite distal compared to 3-M2. Consistent with this notion, the mass spectrum for 3-M1 revealed fragment ions of m/z 254, 171, and 156, which derived from the loss of CO2Cl, C3H2O2F3Cl, and C4H4O2F3Cl. We propose this corresponded to an oxygen insertion at the 14 position.
Figure 2.

Oxidative metabolites of EFV analogues 1–6 and 8 formed by pooled human liver microsomes. Human liver microsomes at a concentration of 2 mg/mL were incubated with 10 μM EFV analogue in the presence of an NADPH regenerating system for 60 min. Metabolites were detected by scanning for the parent m/z plus 16 or 32 using uHPLC–MS/MS in product ion mode. Representative chromatograms are shown for each analogue (N = 3).
In EFV analogue 4, the cyclopropyl ring that is found in EFV is open. Human liver microsome incubations with this analogue resulted in the formation of five monooxygenated metabolites (m/z 332, Figure 2). Metabolite 4-M1 corresponded to a RT of 5.11 min and had characteristic fragment ions of m/z 172, 156, 143, and 128 resulting from the loss of C3O2F3Cl, C3O3F3Cl, C4HO3F3Cl, and C5HO3F3Cl. This fragmentation indicated possible oxygen insertion at the 15 position. Metabolite 4-M2 resulted from oxygen insertion at the 14 position and was characterized by a RT of 5.60 min and fragment ions of m/z 262, 238, and 192, which were the result of a loss of CHF3, C2H3O2Cl, and C4H3O2F3. We detected a third monooxygenated metabolite, 4-M3, at a RT of 5.68 min that represented oxygen insertion on the benzene ring at either the 5 or the 7 position. Metabolite 4-M3 exhibited fragment ions of m/z 268, 244, 218, and 204, which we propose resulted from a loss of C2H5Cl, C4H8O2, C2HO2F3, and C3H4O2F3. Metabolite 4-M4 was observed at a RT of 6.08 min and had fragment ions of m/z 274, 259, 246, and 230. These ions corresponded to a loss of C2H2O2, C3H6O2, C4H6O2, and CF3Cl, suggesting that the oxygen insertion occurred at either the 5 or the 7 position as well. Metabolite 4-M5 was the most abundant metabolite of this analogue and eluted at a RT of 6.98 min. We observed fragment ions of m/z 288, 260, 252, and 228, that corresponded to a loss of CO2, C3H4O2, CO2Cl, and CF3Cl. On the basis of the fragmentation pattern, which closely matches that of 8-hydroxyefavirenz,17 and the hydrophobicity of this analogue, we propose this metabolite resulted from oxygen insertion at the 8 position. The peak eluting immediately after 4-M5 was not denoted as a metabolite because it appeared to be produced within the mass spectrometer since an identical peak was detected when synthetic 8-hydroxyefavirenz alone is injected directly into the mass spectrometer (data not shown).
EFV analogues 5 and 6 differ in stereochemistry about the 4 position, but both lack an intact oxazinone ring and have an additional methoxyphenyl group compared to EFV. No monooxygenated metabolites were observed from these analogues; however, metabolites 5-M1, 6-M1, and 6-M2 (m/z 290), that we propose to be N-dealkylated products were observed. Metabolites 5-M1 and 6-M2 eluted at a RT of 7.31 and 7.25 min, respectively, while 6-M1 was detected at a RT of 6.51 min (Figure 2). All three metabolites exhibited fragment ions of m/z 244, 232, and 178. This fragmentation pattern, representing a loss of C2H6O, C3H6O, and C4H7F3, suggested that these metabolites were the result of an N-dealkylation reaction. Unlike 6-M1, the RT of 5-M1 and 6-M2 were later RTs than analogues 1 and 2 (6.57 min, data not shown), which are structurally identical to the proposed N-dealkylated metabolites. The data suggest that 5-M1 and 6-M2 are less polar than analogues 1 and 2, yet have the same mass spectrum. Analogue 7, which possesses an additional carbonyl compared to analogues 5 and 6, did not exhibit any metabolites from incubation with pooled human liver microsomes.
EFV analogue 8 only lacks the carbonyl oxygen from the oxazinone ring, leaving an intact oxazine ring in its place. Human liver microsome activity produced three monooxygenated metabolites (m/z 318) and one dihydroxylated metabolite (m/z 334) from this analogue (Figure 2). Metabolite 8-M1 had a retention time of 5.00 min and, though it did not fragment readily, produced characteristic ions of m/z 226 and 91. Human liver microsome metabolism assays performed using isotopic water revealed 8-M1 fragments of m/z 228 and 91 (data not shown), indicating that the inserted oxygen was retained in the m/z 226 fragment but not in the m/z 91 fragment. We therefore propose a loss of C3H6OCl and C8H7O2F3Cl for these fragments, suggesting the oxygen was inserted just before the cyclopropyl group at the 14 position. Metabolite 8-M2 was found to have a retention time of 6.43 min with fragment ions of m/z 282, 272, and 237, indicating the loss of H2Cl, C2H6O, and CH2O2Cl. This fragmentation pattern suggested the seven or eight position of the benzene ring as the site of oxygen insertion. Metabolite 8-M3 was the most abundant monooxygenated metabolite detected from metabolism of EFV analogue 8 by human liver microsomes. A retention time of 6.90 min was observed as well as fragment ions of m/z 288, 240, 220, 186, and 141. We propose these fragments corresponded to a loss of CH2O, C4HO, C2H3OCl, C2HOF3, and C3HO2F3. On the basis of this fragmentation pattern as well as hydrophobicity, we propose that metabolite resulted from oxygen insertion on the benzene ring, most likely at the seven or eight position. The fragmentation of dihydroxylated metabolite 8-M4, which corresponds to a retention time of 4.90 min, is nearly identical to that of 8-M1, suggesting that 8-M2 may be formed from 8-M1. Fragment ions of m/z 242 and 91 were observed. Metabolism assays with H218O revealed 8-M4 fragments ions of m/z 246 and 91, suggesting that both oxygens are retained upon formation of m/z 242 species observed with H216O. We propose these fragments resulted from a loss of C3H6OCl and C8H7O2F3Cl. The first oxygen is most likely inserted at the 14 position, as in 8-M1, whereas the second oxygen is more distal, and the insertion may occur at the 5, 7, or 8 position of the benzene ring.
Oxazinone Ring Plays a Critical Role in CYP2B6-Dependent Metabolism of EFV
Following the identification of the metabolites formed by human liver microsomes, we used cDNA-expressed enzymes to determine which of these metabolites could be formed by CYP2B6. The results from these experiments are summarized in Table 1 and Supporting Information Figure S2. Interestingly, incubation of analogues 1, 2, 5, and 6 with CYP2B6 did not result in the formation of detectable metabolites. Each of these analogues possesses a disrupted oxazinone ring. In contrast, from analogues with an intact ring (3, 4, and 8) we did observe metabolites 3-M2, 4-M4, 4-M5, and 8-M2 from incubations with CYP2B6. Since CYP2B6 did not produce all the metabolites we detected using human liver microsomes, we incubated each analogue with a panel of individual P450 enzymes representing the major human drug metabolizing P450s. Metabolites 1-M1 and 2-M1 were detected from incubations with CYP1A2 and CYP2C19. Production of the N-dealkylated metabolites of analogues 5 and 6 were catalyzed by CYP1A2, CYP2C19, and CYP2D6. In addition to CYP2B6, we observed metabolism of analogues 3 and 4 by CYP1A2 and CYP3A5. Analogue 8, however, was only metabolized by CYP2B6 and CYP2C19. Though divergent substrate specificities have been previously observed for CYP2C9 and CYP2C19,18,19 few substrates have been identified that can distinguish the activity of CYP3A4 from CYP3A5, which share 84% amino acid sequence identity. Our group has demonstrated preferential metabolism of maraviroc by CYP3A5,15 while others have shown the metabolism of T-5 to be catalyzed primarily by CYP3A5.20 This panel of EFV analogues may also prove to be useful in distinguishing the activities of these highly homologous P450s.
Table 1. EFV Analogue Metabolites Formed from cDNA-Expressed P450sa.

Blank cells represent no detectable metabolite formation. No metabolites were detected from analogue 7.
A previous study used molecular docking to simulate the presence of EFV in the active site of CYP2B6.21 This study revealed interactions between EFV and residues E301 and T302, which may participate in a hydrogen bonding network involving the EFV oxazinone ring. Indeed, T302 has also been postulated to form polar contacts with amlodipine based on another X-ray crystal structure.22 Since we noted that the absence of an intact oxazinone ring abrogated metabolism by CYP2B6, it is possible that contacts formed by members of this ring are important for catalysis or stabilization of EFV in the active site. Moreover, since analogue 8 was readily metabolized by CYP2B6, the carbonyl moiety of EFV does not appear to be essential for CYP2B6 activity toward EFV. These data suggest that perhaps the increased conformational flexibility afforded to analogues with an open oxazinone ring abrogates the ability of these analogues to be metabolized by CYP2B6.
Kinetic Comparison of CYP2B6 Substrates
Next, we set out to quantitatively compare the metabolism of EFV analogues shown to be CYP2B6 substrates. Since multiple metabolites were detected from CYP2B6 activity with analogue 4 and since CYP2B6 is known to form multiple metabolites from EFV itself,3 substrate depletion was chosen as the method to determine relative kinetic constants (Figure 3).23Km and Vmax values were determined for EFV and analogues 3, 4, and 8 and are reported in Table 2. The observed Km for EFV was 0.45 ± 0.11 μM, while the calculated Vmax was 1.0 ± 0.3 pmol/min/pmol P450 (Table 2). These values are lower than previously reported in studies measuring only 8-hydroxyefavirenz formation.3,4 Since 8-hydroxyefavirenz can be further metabolized by CYP2B6,3 we expect that monitoring EFV disappearance, as we have done in this study, may yield kinetic constants that differ from those obtained in previous reports. The observed 2-fold decrease in Km between EFV and analogue 3 suggests this analogue may have higher affinity for CYP2B6. Conversely, the 2-fold increase in Km observed with analogue 4 suggests this structure with an open cyclopropyl ring is bound less tightly than EFV. Analogue 3 also exhibited a Vmax that was an order of magnitude lower than that observed for EFV, suggesting that, although the Km is lower for this substrate, a catalytic difference may exist between the metabolism of EFV and this analogue with a trans-alkene. The observed improvements in maximal reaction rate as well as substrate affinity for analogue 8 suggest that the absence of the carbonyl oxygen atom does not hinder metabolite formation by CYP2B6 and may in fact enhance it. One possible explanation for this could be that the absence of the carbonyl oxygen atom might relieve steric clashes between EFV and CYP2B6 side chains in the active site.
Figure 3.

Substrate depletion analysis of EFV and EFV analogues 3, 4, and 8. CYP2B6 (10 nM) was incubated with EFV or EFV analogue 3, 4, or 8 at 37 °C in 100 mM potassium phosphate buffer. At 0, 2, 5, 10, 20, and 30 min after the addition of an NADPH regenerating system, 100 μL of the reaction mixture was diluted into equal volume of acetonitrile containing the internal standard fluorinated efavirenz. Analyte and internal standard abundances were measured using uHPLC–MS/MS. Data represent the mean ± SD of three replicate experiments performed in duplicate.
Table 2. Substrate Depletion Kinetic Constants.
| compd | Km (μM) | kdep max (min–1) | Vmax (pmol/min/pmol P450) |
|---|---|---|---|
| EFV | 0.45 ± 0.11 | 0.023 ± 0.001 | 1.0 ± 0.3 |
| analogue 3 | 0.23 ± 0.05 | 0.0057 ± 0.0002 | 0.13 ± 0.03 |
| analogue 4 | 1.10 ± 0.26 | 0.0088 ± 0.0003 | 0.97 ± 0.27 |
| analogue 8 | 0.22 ± 0.05 | 0.14 ± 0.01 | 3.2 ± 1.0 |
Inhibition of CYP2B6 Activity by EFV Analogues
In order to determine if the analogues not found to be CYP2B6 substrates could still interact with the enzyme, we analyzed the metabolism of a known CYP2B6 substrate, bupropion,2 in the presence of each EFV analogue. A concentration of 10 μM EFV or EFV analogue was chosen since this same concentration was used in our metabolism assays. Although spectral binding is commonly employed to investigate substrate interactions with purified P450s, our attempts to detect a spectral shift in the CYP2B6-containing insect microsomes utilized in our studies in response to EFV and EFV analogues were unsuccessful. While the use of this system allowed us to readily examine CYP2B6-dependent activity toward the range of compounds used in the present study, it is possible that the complexity of the insect microsomal system rendered a spectral shift difficult to observe. Coincubations of EFV with bupropion and CYP2B6 resulted in a 93% decrease in hydroxybupropion formation compared to vehicle control (Figure 4). Of EFV analogues found to be CYP2B6 substrates, analogue 4 exhibited the weakest inhibition of bupropion metabolism (27% activity remaining), while analogue 8 showed the greatest inhibition (2% activity remaining). These findings are in congruence with our substrate depletion data since analogue 4 exhibited a higher Km than EFV, while the Km for analogue 8 was lower than EFV. The sizes of analogues 5–7 are greater than the other analogues used in this study, which could provide a clue as to why these analogues were not metabolized by CYP2B6. Analogue 7, which is the largest analogue, was unable to inhibit hydroxybupropion formation, while the slightly smaller analogues 5 and 6 were able to moderately inhibit hydroxybupropion formation (52% and 62% activity remaining, respectively). This suggests that size may influence the interaction of CYP2B6 with compounds possessing the EFV chemical scaffold. Interestingly, although analogues 1 and 2 were not metabolized by CYP2B6, inhibition of CYP2B6 activity toward EFV by these compounds was commensurate with that of EFV as well as analogue 3, which was also a CYP2B6 substrate. These data suggest that analogues 1 and 2 are able to interact with CYP2B6 although this interaction does not result in metabolism of these molecules. This finding lends further evidence to the notion that an intact oxazinone ring is critical to the ability of CYP2B6 to metabolize EFV.
Figure 4.

Inhibition of CYP2B6 bupropion hydroxylase activity by EFV and EFV analogues. CYP2B6 (50 nM) was incubated with bupropion (40 μM) and EFV or EFV analogue (10 μM) in 100 mM potassium phosphate buffer for 10 min at 37 °C. Hydroxybupropion formation was analyzed using uHPLC–MS/MS. Inhibition is reported as a percentage of hydroxybupropion formation in the presence of a vehicle control (no EFV or EFV analogue present). Data reflect the mean ± SD of three replicate experiments performed in duplicate. Symbols for statistical significance represent comparisons to vehicle control (*) or EFV (#). Two symbols, p ≤ 0.01; three symbols, p ≤ 0.001.
In summary, we have demonstrated the importance of the oxazinone ring for metabolism of EFV by CYP2B6. Future studies could test whether compounds that are structurally similar to EFV yet possess other kinds of six-membered rings are also CYP2B6 substrates since our results do not preclude this possibility. No metabolite production was observed from incubations of CYP2B6 with analogues lacking an intact oxazinone ring (analogues 1, 2, 5, 6, and 7), though other P450s were able to catalyze these reactions. Substrate depletion analysis of EFV analogues that were CYP2B6 substrates revealed additional differences between these substrates, namely, that the cyclopropyl ring and alkyne moieties of EFV may play a role in binding affinity and reaction rate, respectively. Interestingly, we determined that an analogue lacking the carbonyl oxygen atom possesses higher affinity for CYP2B6 as well as an increased maximum reaction rate. Thus, the intact oxazinone ring, and not simply the carbonyl functional group, is a determinant of catalytic activity of CYP2B6 toward EFV.
Glossary
ABBREVIATIONS
- CYP2B6
cytochrome P450 2B6
- EFV
efavirenz
- kdep
depletion rate constant
- kdep max
maximum depletion rate constant
- P450
cytochrome P450
- RT
retention time
- uHPLC–MS/MS
ultra high-performance liquid chromatography–tandem mass spectrometry
Supporting Information Available
Complete experimental procedures, MS2 spectra, and metabolism schemes. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
P.M.C. participated in research design, performed experiments, analyzed data, and contributed to writing the manuscript. N.N.B. participated in research design, data analysis, and the writing of the manuscript.
This work was fincially supported by NIH R01GM103853 awarded to N.N.B. and NIH T32GM007445.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zanger U. M.; Turpeinen M.; Klein K.; Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 2008, 392, 1093–108. [DOI] [PubMed] [Google Scholar]
- Hesse L. M.; Venkatakrishnan K.; Court M. H.; von Moltke L. L.; Duan S. X.; Shader R. I.; Greenblatt D. J. CYP2B6 mediates the in vitro hydroxylation of bupropion: potential drug interactions with other antidepressants. Drug Metab. Dispos. 2000, 28, 1176–83. [PubMed] [Google Scholar]
- Ward B. A.; Gorski J. C.; Jones D. R.; Hall S. D.; Flockhart D. A.; Desta Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 2003, 306, 287–300. [DOI] [PubMed] [Google Scholar]
- Bumpus N. N.; Kent U. M.; Hollenberg P. F. Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J. Pharmacol. Exp. Ther. 2006, 318, 345–51. [DOI] [PubMed] [Google Scholar]
- Gervot L.; Rochat B.; Gautier J. C.; Bohnenstengel F.; Kroemer H.; de Berardinis V.; Martin H.; Beaune P.; de Waziers I. Human CYP2B6: expression, inducibility and catalytic activities. Pharmacogenetics 1999, 9, 295–306. [PubMed] [Google Scholar]
- Hofmann M. H.; Blievernicht J. K.; Klein K.; Saussele T.; Schaeffeler E.; Schwab M.; Zanger U. M. Aberrant splicing caused by single nucleotide polymorphism c.516G > T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J. Pharmacol. Exp. Ther. 2008, 325, 284–92. [DOI] [PubMed] [Google Scholar]
- Sim S. C.; Ingelman-Sundberg M. The human cytochrome P450 (CYP) allele nomenclature website: a peer-reviewed database of CYP variants and their associated effects. Hum. Genomics 2010, 4, 278–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukunft J.; Lang T.; Richter T.; Hirsch-Ernst K. I.; Nussler A. K.; Klein K.; Schwab M.; Eichelbaum M.; Zanger U. M. A natural CYP2B6 TATA box polymorphism (−82T → C) leading to enhanced transcription and relocation of the transcriptional start site. Mol. Pharmacol. 2005, 67, 1772–82. [DOI] [PubMed] [Google Scholar]
- Desta Z.; Saussele T.; Ward B.; Blievernicht J.; Li L.; Klein K.; Flockhart D. A.; Zanger U. M. Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics 2007, 8, 547–58. [DOI] [PubMed] [Google Scholar]
- Radloff R.; Gras A.; Zanger U. M.; Masquelier C.; Arumugam K.; Karasi J. C.; Arendt V.; Seguin-Devaux C.; Klein K. Novel CYP2B6 enzyme variants in a Rwandese population: functional characterization and assessment of in silico prediction tools. Hum. Mutat. 2013, 34, 725–34. [DOI] [PubMed] [Google Scholar]
- Ogburn E. T.; Jones D. R.; Masters A. R.; Xu C.; Guo Y.; Desta Z. Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. 2010, 38, 1218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucette S. R.; Zhang T. C.; Moore R.; Sueyoshi T.; Omiecinski C. J.; LeCluyse E. L.; Negishi M.; Wang H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 2007, 320, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse L. M.; von Moltke L. L.; Shader R. I.; Greenblatt D. J. Ritonavir, efavirenz, and nelfinavir inhibit CYP2B6 activity in vitro: potential drug interactions with bupropion. Drug Metab. Dispos. 2001, 29, 100–2. [PubMed] [Google Scholar]
- Gay S. C.; Shah M. B.; Talakad J. C.; Maekawa K.; Roberts A. G.; Wilderman P. R.; Sun L.; Yang J. Y.; Huelga S. C.; Hong W. X.; Zhang Q.; Stout C. D.; Halpert J. R. Crystal structure of a cytochrome P450 2B6 genetic variant in complex with the inhibitor 4-(4-chlorophenyl)imidazole at 2.0-Å resolution. Mol. Pharmacol. 2010, 77, 529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M. B.; Pascual J.; Zhang Q.; Stout C. D.; Halpert J. R. Structures of cytochrome P450 2B6 bound to 4-benzylpyridine and 4-(4-nitrobenzyl)pyridine: insight into inhibitor binding and rearrangement of active site side chains. Mol. Pharmacol. 2011, 80, 1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilderman P. R.; Shah M. B.; Jang H. H.; Stout C. D.; Halpert J. R. Structural and thermodynamic basis of (+)-alpha-pinene binding to human cytochrome P450 2B6. J. Am. Chem. Soc. 2013, 135, 10433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlib A. E.; Chen H.; Nemeth G. A.; Markwalder J. A.; Seitz S. P.; Gan L. S.; Christ D. D. Identification and characterization of efavirenz metabolites by liquid chromatography/mass spectrometry and high field NMR: species differences in the metabolism of efavirenz. Drug Metab. Dispos. 1999, 27, 1319–33. [PubMed] [Google Scholar]
- Lasker J. M.; Wester M. R.; Aramsombatdee E.; Raucy J. L. Characterization of CYP2C19 and CYP2C9 from human liver: respective roles in microsomal tolbutamide, S-mephenytoin, and omeprazole hydroxylations. Arch. Biochem. Biophys. 1998, 353, 16–28. [DOI] [PubMed] [Google Scholar]
- Mancy A.; Antignac M.; Minoletti C.; Dijols S.; Mouries V.; Duong N. T.; Battioni P.; Dansette P. M.; Mansuy D. Diclofenac and its derivatives as tools for studying human cytochromes P450 active sites: particular efficiency and regioselectivity of P450 2Cs. Biochemistry 1999, 38, 14264–70. [DOI] [PubMed] [Google Scholar]
- Li X.; Jeso V.; Heyward S.; Walker G. S.; Sharma R.; Micalizio G. C.; Cameron M. D. Characterization of T-5 N-oxide formation as the first highly selective measure of CYP3A5 activity. Drug Metab. Dispos. 2014, 42, 334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu R. J.; Zheng Q. C.; Zhang J. L.; Zhang H. X. Analysis of clinically relevant substrates of CYP2B6 enzyme by computational methods. J. Mol. Model. 2011, 17, 2839–46. [DOI] [PubMed] [Google Scholar]
- Shah M. B.; Wilderman P. R.; Pascual J.; Zhang Q.; Stout C. D.; Halpert J. R. Conformational adaptation of human cytochrome P450 2B6 and rabbit cytochrome P450 2B4 revealed upon binding multiple amlodipine molecules. Biochemistry 2012, 51, 7225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach R. S.; Reed-Hagen A. E. Measurement of Michaelis constants for cytochrome P450-mediated biotransformation reactions using a substrate depletion approach. Drug Metab. Dispos. 2002, 30, 831–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.