

Abstract
The normal reaction mechanism of cytochrome P450 operates by utilizing two reducing equivalents to reduce atmospheric dioxygen, producing one molecule of water and an oxygenated product in an overall stoichiometry of 2 electrons : 1 dioxygen : 1 product. However, three alternate unproductive pathways exist where the intermediate iron-oxygen states in the catalytic cycle can yield reduced oxygen products without substrate metabolism. The first involves release of superoxide from the oxygenated intermediate while the second occurs after input of the second reducing equivalent. Superoxide rapidly dismutates and hence both processes produce hydrogen peroxide that can be cytotoxic to the organism. In both cases, the formation of hydrogen peroxide involves the same overall stoichiometry as oxygenases catalysis. The key step in the catalytic cycle of cytochrome P450 involves scission of the oxygen-oxygen bond of atmospheric dioxygen to produce a higher valent iron-oxo state termed “Compound I”. This intermediate initiates a radical reaction in the oxygenase pathway but also can uptake two additional reducing equivalents from reduced pyridine nucleotide (NADPH) and the flavoprotein reductase to produce a second molecule of water. This non-productive decay of Compound I thus yields an overall oxygen to NADPH ratio of 1:2 and does not produce hydrocarbon oxidation. This water uncoupling reaction provides one of a limited means to study the reactivity of the critical Compound I intermediate in P450 catalysis. We measured simultaneously the rates of NADPH and oxygen consumption as a function of substrate concentration during the steady-state hydroxylation of testosterone catalyzed by human P450 CYP3A4 reconstituted in Nanodiscs. We discovered that the “oxidase” uncoupling pathway is also operating in the substrate free form of the enzyme with rate of this pathway substantially increasing with the first substrate binding event. Surprisingly, a large fraction of the reducing equivalents used by the P450 system is wasted in this oxidase pathway. In addition, the overall coupling with testosterone and bromocryptine as substrates is significantly higher in the presence of anionic lipids, which is attributed to the changes in the redox potential of CYP3A4 and reductase.
Keywords: Cytochrome P450, CYP3A4, Nanodiscs, P450 mechanism, Uncoupling, Oxidase pathway
Introduction
The catalytic cycle of cytochrome P450 includes the seven reactions shown in the Scheme 1, with an additional three unproductive branches, termed “uncoupling pathways”, which are depicted as dashed arrows. Reducing equivalents are provided to the heme protein by pyridine nucleotide (NADPH) through the action of a di-flavin reductase. The typical hydroxylation and oxygen transfer reactions catalyzed by the cytochromes P450 involve the consumption of one NADPH molecule and one dioxygen molecule, with one oxygen atom inserted into the product and another one forming water molecule. This stoichiometry of NADPH:O2 of 1:1 is summarized in Equation 1. The two uncoupling reactions depicted in Scheme 1 involve release of superoxide from the ferrous dioxygen, [4] → [2], and the release of hydrogen peroxide, [5] → [2], also have an overall NADPH:O2 stoichiometry of 1:1 as is illustrated in Equation 2. Following the generation of a heme peroxo state, the next step in the cyclic catalytic cycle involves cleavage of the O-O bound to produce a higher valent iron-oxo intermediate termed “Compound I” (Cpd I, [6]) following the historical peroxidase nomenclature. Cpd I is a ferryl-oxo porphyrin cation radical and is thought to initiate substrate hydroxylation through the Groves “oxygen rebound” process [1]. However, there is an additional reactivity of Cpd I. Inasmuch as this intermediate is thought to have an extremely positive redox potential [2] it can accept two additional electrons from NADPH and the flavoprotein reductase to reduce the electron deficient heme bound oxygen atom to water. This [6] → [2] pathway shown in Scheme 1 is thus analogous to the reaction of cytochrome oxidase with an NADPH:O2 ratio of 2:1 This oxidase uncoupling is described by Equation 3.
Scheme 1.
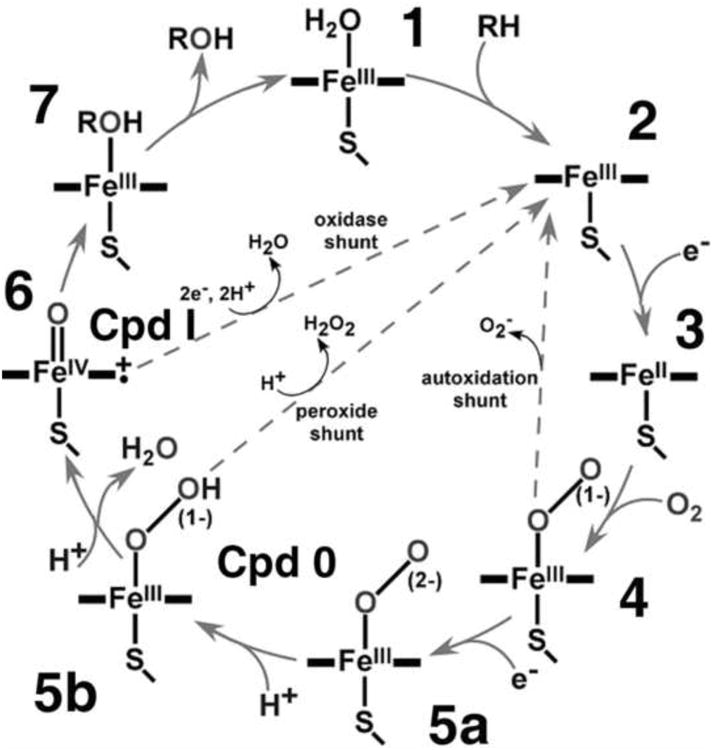
The catalytic cycle of cytochrome P450. Three uncoupling reactions are shown as dashed arrows.
| (1) |
| (2) |
| (3) |
While this reactivity of the cytochrome P450s has been recognized for some time, it has often been thought to represent a small fraction of the overall pyridine nucleotide reducing equivalents used by the system. Previous investigations have been hampered by problems related to the study of membrane proteins outside a bilayer environment. Systems using aggregated P450s are known to have many artifacts, including the lack of simple first order autoxidation behavior [3]. We have overcome these difficulties by using the Nanodisc system to form a homogeneous and monodisperse sample of human P450 CYP3A4 where detailed mechanistic investigations are possible. CYP3A4 is the major player in human drug metabolism in the liver, metabolizing almost 50% of the drugs currently on the market [4]. Due to the need for the enzyme to recognize a wide variety of substrate structures, the protein is thought to have a highly “plastic” active site. A downside of this flexibility is the poor utilization of pyridine nucleotide reducing equivalents. Although a deleterious process, the oxidase reactivity of Cpd I provides an important opportunity to study the fleeting iron-oxo intermediate.
The distinct NADPH:O2 stoichiometry of Equation 3 relative to the other processes allows quantitation of the flux through the oxidase pathway by measuring the difference between the rates of NADPH and oxygen consumption, or between NADPH consumption and the sum of the rates of the product formation and H2O2 production. These measurements directly probe the branch point between hydrogen abstraction and subsequent oxygenase catalysis and the facile reduction of this intermediate by two additional electrons. An NADPH:O2 ratio greater than 1:1 indicates excess water production and provides a measure of the efficiency of Cpd I driven catalytic transformation of a given substrate with a given P450. Thus, more “extra” water is formed in the oxidase uncoupling pathway during metabolism of substrates that offer higher barriers for oxygenation, such as hydroxylation versus epoxidation and as reflected in the presence of deuterated analogs [5].
The oxidase uncoupling pathway has been documented for several mammalian cytochromes P450 in reconstituted systems and in microsomes [6-15]. However, earlier approaches suffered from the lack of a clean monomeric heme protein and were not used to gain information about the reactivity and stability of Cpd I through the direct comparison of the rates of productive and unproductive (oxidase) decompositions. We have systematically studied the steady-state rates of water production and product formation for the metabolism of testosterone (TST) and bromocriptine (BC), as catalyzed by the most abundant human cytochrome P450, CYP3A4, with resultant new information about the properties of the Cpd I intermediate state.
Materials and Methods
Protein expression and purification
Expression and purification of membrane scaffold protein (MSP), cytochrome P450 CYP3A4 and rat P450 reductase (CPR), as well as preparation of CYP3A4 in POPC Nanodiscs (ND) was as described previously [16-18]. Incorporation of CPR into preformed and purified CYP3A4-Nanodiscs was made by direct addition of oligomeric CPR at 1:4 CYP3A4/CPR molar ratio, as described [19]. All experiments have been performed at 37 °C using POPC Nanodisc system similar to our earlier detailed mechanistic studies [11-13] in order to allow direct comparison of results. This reconstitution system provides a stable well characterized and monodisperse preparation of CYP3A4 incorporated into the model lipid bilayer effectively mimicking the native membrane.
Catalytic Activities
The simultaneous measurements of NADPH oxidation and oxygen consumption rates were performed using a temperature-controlled cylindrical plastic cell, stirrer and Clark-type electrode, in conjunction with Cary 3 spectrophotometer (Varian). The signal from the electrode measuring oxygen concentration in the reaction mixture, and the absorption at 340 nm measuring the NADPH concentration, were both recorded as a function of time and analyzed in a parametric plot where the slope corresponds directly to the NADPH/O2 ratio. The reaction buffer (0.1 M HEPES buffer pH 7.4 containing 7.5 mM MgCl2), with or without the substrate, was vigorously stirred for at least 60 min at 37 °C prior to the experiment to ensure the equilibrium oxygen concentration is reached. The buffer then was kept at 37 °C. One ml of the prepared buffer was placed in the cell with stirring and the oxygen electrode signal was monitored until stable (typically 10 minutes). CYP3A4-ND and CRP were added and the sample capped and incubated for 5 min before the reaction is initiated by the addition of 12 μl of 20 mM NADPH. The absorption at 340 nm and oxygen concentration were monitored for 5 min, then the reaction was quenched by addition of 12 μl of 9 M sulfuric acid to bring pH below 4.0. The sample was removed from the cuvette, frozen by immersion in liquid nitrogen, and stored at -80 °C for product analysis. The initial rate of NADPH oxidation was determined from the slope of absorption at 340 nm during the first two minutes using absorption coefficient 6.22 cm-1mM-1. The effective path length of the cylindrical cell (0.94 cm) was determined in a separate experiment. The initial concentration of oxygen was obtained from oxygen solubility tables and varied from 211 to 216 μM depending on the atmospheric pressure. Oxygen solubility in buffer was estimated according to [20] to be 97% of that in water.
Analysis of TST metabolites was performed as described elsewhere [21, 22] with minor modifications. Briefly, 1.5 ml of CH2Cl2 and 25 μl of 90 μM cortexolone solution in methanol were added as an internal standard to 0.5 ml of the sample solution and thoroughly mixed. After separation, the organic phase was isolated and the solvent was removed under stream of nitrogen. The dried sample was dissolved in 70 μl of methanol and 40 μl was injected onto C18 HPLC column, 2.1×150 mm, 4 μm (Waters Nova-Pak). The mobile phase was 30% acetonitrile and 10 mM ammonium acetate in water with the flow rate 0.4 ml/min. Products of testosterone hydroxylation were separated in linear gradient of acetonitrile rising from 30% to 70% over 25 min. The peak integration was performed with GRAMS 32 software (Thermo Fisher Scientific)
Hydrogen peroxide assay
Hydrogen peroxide concentration was determined using the lipid-compatible PeroXOquant essay (Pierce). Standard curves were generated with known amounts of hydrogen peroxide added to the control mixture containing all the reaction components except NADPH. However, significant loss of the H2O2 occurred when it was incubated with the control mixture for 10 minutes. Therefore, since experimental measurements would result in severe underestimation, the rate of hydrogen peroxide production was calculated by the difference in oxygen consumption rate and the sum of the “extra” water production rate and product formation rates.
Results and discussion
Figure 1 shows the absolute rates of NADPH and oxygen consumption, and their difference, which corresponds to the water uncoupling rate (see Scheme 1). Most interestingly, the unproductive water channel exists even in the absence of substrate. This means that Cpd I is formed with a rate of ∼ 8 min-1, or 20 % of the total oxygen consumption rate (∼ 42 min-1) even in the absence of substrate. The formation of Cpd I requires facile protonation of the hydroperoxo-ferric intermediate [5B] (Scheme 1) with a rate faster than dissociative decomposition of [5B]. The substantial fraction of oxidase uncoupling pathway experimentally observed in the substrate-free CYP3A4 indicates that this protonation of [5B] can proceed quickly, as expected in the large and highly hydrated substrate binding pocket of this cytochrome P450. The rates observed when there is no product formation channel provide a rough estimate of the lower limit of the oxidase reaction of Cpd I under these conditions. The rate of water formation is similar to the rates of other uncoupling channels as these channels utilize comparable fractions of the total NADPH consumption. However, all of these processes occur at rates that are likely much faster than the experimentally observed steady-state rate, which is at least partially limited by the rate of the first electron transfer [23].
Figure 1.
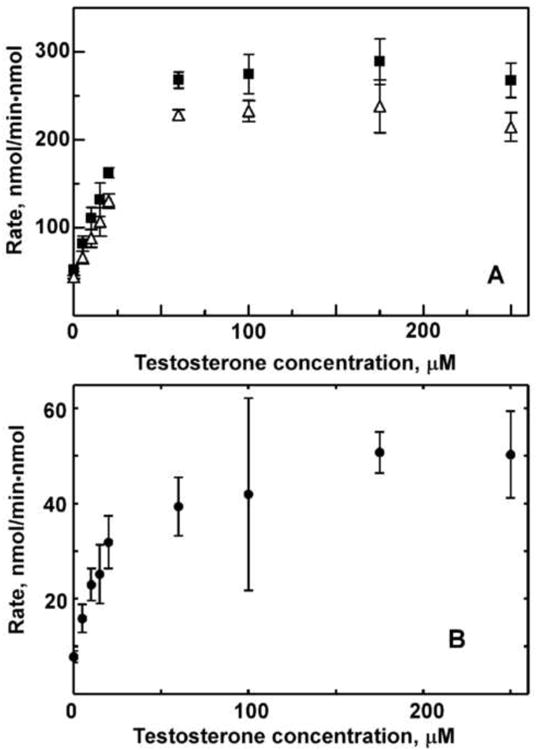
Steady-state rates of NADPH oxidation and oxygen consumption (A) and water production (B) at different testosterone concentrations.
When CYP3A4 is saturated with TST, the rate of water production increases to 52 min-1 (Fig. 1B), while the total oxygen consumption rises to ∼215 min-1 and the product formation rate to 23 min-1. Thus, a moderate increase in the fraction of the oxidase uncoupling channel in substrate saturated CYP3A4 (∼25%) is observed in the TST saturated enzyme at the expense of a moderate decrease of the fraction of peroxide uncoupling; from 80% to 65% of the total oxygen consumption. The absolute rate of the steady-state peroxide production, however, increases in the presence of 250 μM TST up to 140 min-1, i.e. approximately 4-fold from the rate 34 min-1 observed in the absence of substrate.
Figure 2A shows the rate of TST hydroxylation as a function of substrate concentration. The results are identical to those earlier reported for CYP3A4 co-incorporated with CPR in Nanodiscs [24]. For comparison, in Fig. 2B we document the fraction of Cpd I which is involved in product formation. The efficiency of Cpd I utilization for TST hydroxylation increases as a function of substrate concentration and reaches saturation at a maximum of 33% at 100 μM TST. This effect is due to the lack of product formation by CYP3A4 with only one TST molecule bound [18]. As the fraction of CYP3A4 with two and three TST molecules increases, the overall percentage of product formation by Cpd I also increases. However, even at highest TST concentrations the oxidase uncoupling channel consumes oxygen two times faster than product is formed. Clearly, CYP3A4 is not an efficient enzyme with respect to NADPH / O2 consumption, even with relatively good substrates, such as TST.
Figure 2.
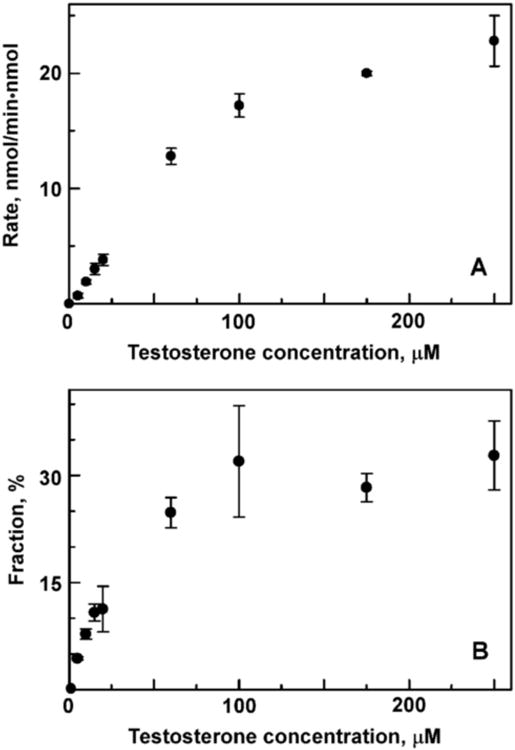
Rate of testosterone hydroxylation (A) and fraction of Cpd I utilized via the productive pathway (B) measured at different testosterone concentrations.
Comparison of Fig. 1B and Fig. 2A reveals that the water production rate increases much faster than the product formation rate as a function of TST concentration. In order to better understand how the rate of water production depends on the number of bound substrate molecules, we calculated the fractional contributions to oxidase uncoupling from each of four binding intermediates. The concentrations of CYP3A4 molecules with zero, one, two or three TST molecules were calculated at each experimentally used TST concentration (Figures 1 and 2) using the determined stepwise dissociation constants K1 = 19 μM, K2 = 37 μM, and K3 = 56 μM [24]. Using these populations of binding intermediates, the fractional contributions to the overall water production are calculated as 7, 39, 50 and 52 min-1 for CYP3A4 with zero, one, two and three TST molecules bound. This result indicates substantial increase in the absolute rate of Cpd I formation with the first substrate binding event in CYP3A4, despite the fact that this binding occurs at a remote site and does not result in product formation [24-26]. Previously we documented the similar increase of the geminate CO recombination amplitude and substantial stabilization of Fe-O2 intermediate in CYP3A4 caused by the first TST binding event [18] and tentatively attributed it to the significant restriction of the diatomic ligand escape caused by substrate positioning at a remote high-affinity site. Stabilization of the oxy-ferrous intermediate [4] in Scheme 1 changes the partitioning of reaction fluxes at the first uncoupling point in favor of the second electron transfer [4] → [5] and formation of peroxo-ferric intermediate and subsequent formation of Cpd I. In the absence of substrate, in the immediate vicinity of the heme-oxygen catalytic moiety, Cpd I decays exclusively via an oxidase pathway, using one more NADPH molecule to generate the additional water. As can be seen from Fig. 2B, addition of the second and third substrate molecules increases the fraction of productive utilization of Cpd I, up to 33% at high TST concentrations.
In order to test the general conclusions based on oxidase uncoupling measurements, we performed the same experiments with two additional substrates, bromocriptine (BC) and tamoxifene, at saturating concentrations. The results shown in Fig. 3 reveal the same pattern as observed with TST. Despite the difference in absolute rates of steady-state NADPH / O2 consumption and product formation, the fractions of oxygen utilized on the productive pathway is low in all three cases, with major uncoupling happening at the peroxide branch point and less, though still substantial, oxidase uncoupling. Interestingly, in the presence of 30% POPS the rate of product formation and overall coupling significantly improve for TST and BCT. Better coupling is due to the inhibition of peroxide uncoupling channel and improved formation of Cpd I, which can be measured as the sum of product formation and oxidase uncoupling rates (Fig. 3). These effects may be tentatively attributed to the faster electron transfer from CPR to CYP3A4 when incorporated into the negatively charged lipid bilayer, based on the observed changes of redox potentials of these proteins [27, 28]. However, the main uncoupling channel in all cases is through peroxide formation (Fig. 3). This fact is commonly accepted as one of the most important sources of the general toxic effect of non-specific drug interactions with cytochromes P450 [2, 4, 5, 7, 10, 13].
Figure 3.
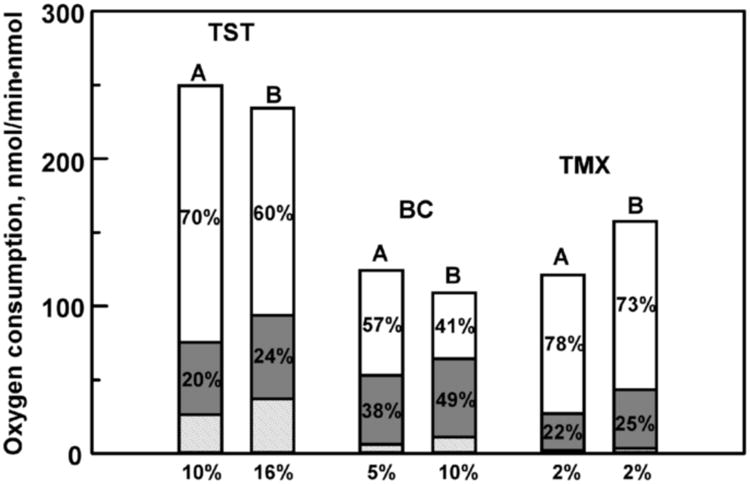
Uncoupling pathways in CYP3A4 catalyzed metabolism of testosterone (250 μM), bromocriptine (10 μM) and tamoxiphene (80 μM) shown as fractional contributions of oxygen consumption. CYP3A4 is incorporated in Nanodiscs assembled with POPC (A) or with 30% POPS + 70% POPC (B). The fraction of peroxide uncoupling is shown on the top bar (white), the oxidase uncoupling fraction as the middle shaded gray bar and the product forming pathway in the dashed bar at the bottom.
Substantial oxidase uncoupling is measured for substrate free CYP3A4. Surprisingly, this is a direct indication of formation of the Cpd I even in the absence of substrate. The Cpd I formed quickly disappears to form water. This step is either direct, involving oxidation of NADPH by Cpd I, or indirect, via oxidation of nearby tyrosine or tryptophan residues and subsequent reduction of these amino acid radicals reaction with the second NADPH molecule. Moreover, it seems that the main channel of Cpd I decay is water production even in the presence of testosterone and bromocriptine. For TST saturated CYP3A4 the formation rates are 50 waters and 20 - 25 hydroxylated TST per minute. With BC the water rate is similar, but the overall turnover is slower.
Theoretically, the absolute rate of water production (or at least a lower limit for this rate) can be estimated from the partitioning ratio product/water and from comparison of the steady-state rates of CYP3A4 with different substrates. This may be also be measured for other P450 based on the literature data, for example for CYP101 [5, 29]. This estimate is based on the absolute rate of the catalytic step Cpd I + Substrate → Product, which was reported as ∼ 1000 s-1 for CYP119 and lauric acid [30]. For CYP3A4 and TST we measured the partitioning ratio for Cpd I pathways (Product/Water) as ∼ 0.5, meaning that water is formed two times faster than TST hydroxylation. On the other hand, in CYP101 with the native substrate (camphor), this ratio is >50. In CYP101, with highly uncoupled substrates such as norcamphor, the ratio is 0.5. However, since the reactivity rates of Cpd I in CYP3A4 with TST have not been measured, the similar water production rate in CYP3A4 must be considered an estimate.
We have documented the oxidase uncoupling pathway in CYP3A4 Nanodiscs as a function of TST concentration. We found that even in the absence of substrate the futile consumption of NADPH results in substantial, irreversible formation of Cpd I. Binding of the first TST molecule at a remote high-affinity site results in steep increase of water production which is likely caused by stabilization of oxy-complex and improved coupling at the autoxidation branch point. For all three studied substrates Cpd I predominantly decays via the oxidase pathway, even at substrate saturated CYP3A4. We also found that the presence of anionic lipids, in this case 30% POPS, improves overall coupling and facilitates product formation for TST and BC by a factor of 1.5 – 2.
Highlights.
Substantial reducing equivalents are lost in human P450 CYP3A4 via an oxidase channel.
Substrate binding has a pronounced effect on uncoupling in cytochrome P450.
Anionic phospholipids improve the overall coupling in CYP3A4 Nanodiscs.
Acknowledgments
This work was supported by NIH grants GM33775 and GM31756 to S. G. Sligar.
Abbreviations
- BC
bromocryptine
- Cpd I
Compound I, Fe(IV)-oxo porphyrin pi-cation radical
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
1-palmitoyl 2-oleoyl phosphatidylserine
- TST
testosterone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Groves JT. High-valent iron in chemical and biological oxidations. J Inorg Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Koppenol WH. Oxygen activation by cytochrome P450: a thermodynamic analysis. J Am Chem Soc. 2007;129:9686–9690. doi: 10.1021/ja071546p. [DOI] [PubMed] [Google Scholar]
- 3.Oprian DD, Gorsky LD, Coon MJ. Properties of the oxygenated form of liver microsomal cytochrome P-450. J Biol Chem. 1983;258:8684–8891. [PubMed] [Google Scholar]
- 4.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:825–833. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 5.Atkins WM, Sligar SG. Deuterium isotope effects in norcamphor metabolism by cytochrome P-450cam: Kinetic evidence for the two-electron reduction of a high-valent iron-oxo intermediate. Biochemistry. 1988;27:1610–1616. doi: 10.1021/bi00405a033. [DOI] [PubMed] [Google Scholar]
- 6.Narasimhulu S. Uncoupling of oxygen activation from hydroxylation in the steroid C-21 hydroxylase of bovine adrenocortical microsomes. Arch Biochem Biophys. 1971;147:384–390. doi: 10.1016/0003-9861(71)90394-8. [DOI] [PubMed] [Google Scholar]
- 7.Ullrich V, Diehl H. Uncoupling of monooxygenation and electron transport by fluorocarbons in liver microsomes. Eur J Biochem. 1971;20:509–512. doi: 10.1111/j.1432-1033.1971.tb01420.x. [DOI] [PubMed] [Google Scholar]
- 8.Staudt H, Lichtenberger F, Ullrich V. The role of NADH in uncoupled microsomal monoxygenations. Eur J Biochem. 1974;46:99–106. doi: 10.1111/j.1432-1033.1974.tb03601.x. [DOI] [PubMed] [Google Scholar]
- 9.Gorsky LD, Koop DR, Coon MJ. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. Products of oxygen reduction. J Biol Chem. 1984;259:6812–6817. [PubMed] [Google Scholar]
- 10.Zhukov AA, Archakov AI. Stoichiometry of microsomal oxidation reactions. Distribution of redox-equivalents in monooxygenase and oxidase reactions catalyzed by cytochrome P-450. Biokhimiia. 1985;50:1939–1952. [PubMed] [Google Scholar]
- 11.Hiroya K, Ishigooka M, Shimizu T, Hatano M. Role of Glu318 and Thr319 in the catalytic function of cytochrome P450d (P4501A2): Effects of mutations on the methanol hydroxylation. FASEB J. 1992;6:749–751. doi: 10.1096/fasebj.6.2.1347023. [DOI] [PubMed] [Google Scholar]
- 12.Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry. 1997;36:370–381. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- 13.Perret A, Pompon D. Electron shuttle between membrane-bound cytochrome P450 3A4 and b5 rules uncoupling mechanisms. Biochemistry. 1998;37:11412–11424. doi: 10.1021/bi980908q. [DOI] [PubMed] [Google Scholar]
- 14.Yun CH, Kim KH, Calcutt MW, Guengerich FP. Kinetic analysis of oxidation of coumarins by human cytochrome P450 2A6. J Biol Chem. 2005;280:12279–12291. doi: 10.1074/jbc.M411019200. [DOI] [PubMed] [Google Scholar]
- 15.Locuson CW, Gannett PM, Tracy TS. Heteroactivator effects on the coupling and spin state equilibrium of CYP2C9. Arch Biochem Biophys. 2006;449:115–129. doi: 10.1016/j.abb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Denisov IG, Grinkova YV, Baas BJ, Sligar SG. The ferrous-dioxygen intermediate in human cytochrome P450 3A4: Substrate dependence of formation of decay kinetics. J Biol Chem. 2006;281:23313–23318. doi: 10.1074/jbc.M605511200. [DOI] [PubMed] [Google Scholar]
- 17.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 18.Denisov IG, Grinkova YV, McLean MA, Sligar SG. The one-electron autoxidation of human cytochrome P450 3A4. J Biol Chem. 2007;282:26865–26873. doi: 10.1074/jbc.M704747200. [DOI] [PubMed] [Google Scholar]
- 19.Grinkova YV, Denisov IG, Sligar SG. Functional reconstitution of monomeric CYP3A4 with multiple cytochrome P450 reductase molecules in Nanodiscs. Biochem Biophys Res Commun. 2010;398:194–198. doi: 10.1016/j.bbrc.2010.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen HN, Rasmussen UF. Oxygen solubilities of media used in electrochemical respiration measurements. Anal Biochem. 2003;319:105–113. doi: 10.1016/s0003-2697(03)00274-4. [DOI] [PubMed] [Google Scholar]
- 21.Baas BJ, Denisov IG, Sligar SG. Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch Biochem Biophys. 2004;430:218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Krauser JA, Voehler M, Tseng LH, Schefer AB, Godejohann M, Guengerich FP. Testosterone 1beta-hydroxylation by human cytochrome P450 3A4. Eur J Biochem. 2004;271:3962–3969. doi: 10.1111/j.1432-1033.2004.04339.x. [DOI] [PubMed] [Google Scholar]
- 23.Isin EM, Guengerich FP. Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. J Biol Chem. 2006;281:9127–9136. doi: 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- 24.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. Cooperativity in cytochrome P450 3A4: Linkages in substrate binding, spin state, uncoupling, and product formation. J Biol Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 25.Davydov DR, Rumfeldt JA, Sineva EV, Fernando H, Davydova NY, Halpert JR. Peripheral ligand-binding site in cytochrome P450 3A4 located with fluorescence resonance energy transfer (FRET) J Biol Chem. 2012;287:6797–6809. doi: 10.1074/jbc.M111.325654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denisov IG, Sligar SG. A novel type of allosteric regulation: Functional cooperativity in monomeric proteins. Arch Biochem Biophys. 2012;519:91–102. doi: 10.1016/j.abb.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das A, Grinkova YV, Sligar SG. Redox potential control by drug binding to cytochrome P450 3A4. J Am Chem Soc. 2007;129:13778–13779. doi: 10.1021/ja074864x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das A, Sligar SG. Modulation of the cytochrome P450 reductase redox potential by the phospholipid bilayer. Biochemistry. 2009;48:12104–12112. doi: 10.1021/bi9011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loida PJ, Sligar SG. Molecular recognition in cytochrome P-450: Mechanism for the control of uncoupling reactions. Biochemistry. 1993;32:11530–11538. doi: 10.1021/bi00094a009. [DOI] [PubMed] [Google Scholar]
- 30.Rittle J, Green MT. Cytochrome P450 compound I: Capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]