

Abstract
Major advances have been made in exploring the transglycosylation activity of endo-β-N-acetylglucosaminidases (ENGases) for synthetic purpose. The exploration of synthetic sugar oxazolines as donor substrates for the ENGase-catalyzed transglycosylation has expanded the substrate availability and significantly enhanced the overall transglycosylation efficiency. On the other hand, site-directed mutagenesis in combination with activity screening has led to the discovery of the first generation ENGase-based glycosynthases that can use highly active sugar oxazolines as substrates for transglycosylation but lack hydrolytic activity on the ground-state products. ENGases have shown amazing flexibility in transglycosylation and possess much broader substrate specificity than previously thought. Now the ENGase-based chemoenzymatic method has been extended to the synthesis of a range of complex carbohydrates, including homogeneous glycopeptides, glycoproteins carrying well-defined glycans, novel oligosaccharide clusters, unusually glycosylated natural products, and even polysaccharides. This article highlights recent advances related to ENGase-catalyzed transglycosylation with a focus on their synthetic potential.
Keywords: endoglycosidase, transglycosylation, sugar oxazoline, chemoenzymatic synthesis, glycoprotein
A. Introduction
Recent advances in functional glycomics have implicated essential roles of oligosaccharides and glycoconjugates in many important biological recognition processes such as intracellular signaling, cell adhesion, cell differentiation, cancer progression, host-pathogen interactions, and immune responses [1–6]. Following nucleic acids and proteins, glycans are now viewed as the third language of life. Yet, we are just beginning to unveil the codes and mysteries of this language. A major difficulty in this pursuit is the lack of homogeneous oligosaccharides and glycoconjugates as standards and probes, which are not easy to isolate from natural sources because of their structural heterogeneity and, in some cases, their natural scarcity. To address this problem, a number of laboratories worldwide have dedicated to the development of efficient chemical and enzymatic methods for synthesizing structurally well-defined complex carbohydrates of biological relevance [7–20]. In contrast to pure chemical synthesis that usually requires tedious protection/de-protection manipulations in order to achieve regio- and stereo-selectivity, enzymatic glycosylation usually provides perfect control of the anomeric configuration and high regio-selectivity without the need of protecting groups. Toward this end, both glycosyltransferases and glycosidases have been vigorously explored for synthetic purposes [21–23]. Glycosyltransferases are the natural enzymes for making glycosidic bonds, while the inherent nature of glycosidases is to break glycosidic linkages in carbohydrate metabolism. Despite the hydrolytic nature of glycosidases, there have been increasing interests in exploring their transglycosylation activity for making complex carbohydrates. In contrast to glycosyltransferase-catalyzed reaction that uses expensive sugar nucleotide as the donor substrate and usually has stringent substrate specificity, glycosidase-catalyzed transglycosylation has several advantages, including the use of readily available donor substrates, the relaxed substrate specificity for acceptors, and the relatively easy access to the enzymes. Moreover, in contrast to glycosyltransferases that extend sugar chain by adding monosaccharide one at a time, endo-β-N-acetylglucosaminidase (ENGase), which is the focus of the present review, can transfer a large oligosaccharide en bloc to an acceptor in a single step to form a new, complex glycoconjugate in a concise manner.
Endo-β-N-acetylglucosaminidases (ENGases) are a class of endoglycosidases that cleave N-glycans from glycoproteins by hydrolyzing the β-1,4-glycosidic bond in the N,N′-diacetylchitobiose core. These enzymes are widely distributed in nature and have been found in microorganisms, plants, animals, and humans [24–26]. In addition to their hydrolytic activity, some ENGases possess transglycosylation activity, i.e., the ability to transfer the released oligosaccharide to a suitable acceptor to form a new glycosidic linkage. These include: Endo-A from Arthrobactor protophormiae [27–29], Endo-M from Mucor hiemalis [30], Endo-CE from Caenorhabditis elegans [25], Endo-BH from alkaliphilic Bacillus halodurans C-125 [31], and Endo-D from streptococcus pneumoniae [32]. These enzymes show distinct substrate specificity in hydrolysis and transglycosylation. For example, Endo-A and Endo-CE are specific for high-mannose type N-glycans, while Endo-M can take both high-mannose type and complex type N-glycans. Early studies had been focused on applying the transglycosylation activity of wild type Endo-A and Endo-M for the synthesis of large homogeneous glycopeptides, including glycosylated calcitonin [33], C-glycosidic analogs of N-glycopeptides [34, 35], glycosylated fragments of the nicotinic acetylcholine receptor (nAChR) [36], glycosylated substance P [37], large HIV-1 envelope glycoprotein fragments [38, 39], and homogeneous CD52 antigens carrying full-size high-mannose and complex type N-glycans [40]. This chemoenzymatic method were also used for the synthesis of multivalent glyco-polymers [41, 42], natural and neo-glycoproteins [43, 44, 45], glycosylated cyclodextrins [46], and glycosylated insulin [47]. Despite these useful synthetic applications, the ENGase-catalyzed transglycosylation method had suffered from two major limitations: the restriction to the use of natural N-glycans or N-glycopeptides as donor substrates and the quick enzymatic hydrolysis of the products formed. Fortunately, major breakthroughs have been made to address these issues in recent years. These include the exploration of synthetic sugar oxazolines as donor substrates for the ENGase-catalyzed transglycosylation that significantly broadened the substrate availability, and the creation of glycosynthase mutants that can perform transglycosylation with the highly activated sugar oxazolines but lack product hydrolysis activity. In addition, ENGases have demonstrated much broader substrate specificity than previously thought. Now the enzymes have been successfully applied to the synthesis of various homogeneous glycoproteins, novel oligosaccharide clusters, unusually glycosylated natural products, and even polysaccharides. Indeed, the ENGases have shown amazing substrate flexibility in transglycosylation and continue to give us surprises. In this article, I would like to give an account on how we got into this field and to highlight some major discoveries related to the ENGase-catalyzed transglycosylation.
B. Initial attempt to explore sugar oxazolines as donor substrates for ENGases
I became interested in endo-β-N-acetylglucosaminidases when I was a postdoctoral fellow with Prof. Y. C. Lee at Johns Hopkins University. One of my projects was to synthesize N- and C-glycopeptides for studying another class of enzymes, the peptide:N-glycanases (PNGases). We exploited the transglycosylation activity of Endo-A for making the target glycopeptides carrying a high-mannose type N-glycan [35]. While I was amazed by the ability of Endo-A to transfer a large oligosaccharide to a GlcNAc-peptide acceptor in a single step to form a new glycopeptide in a regio- and stereo-specific manner, I was also troubled by the difficulty to control the enzymatic reaction for a high-yield synthesis. It was found that both the glycopeptide product and the Man9GlcNAc2-Asn starting material were quickly hydrolyzed by Endo-A during the reaction, and large excess of the precious natural N-glycan donor substrate should be used in order to obtain sufficient amount of the product. The overall transglycosylation efficiency was generally low, in the range of 5–20%, even with incorporation of organic solvents in the reaction medium to reduce the activity of water. There was no efficient way to address this problem at that time except a careful monitoring of the reaction process by HPLC. I got a chance to look at this issue again seriously when I joined the faculty of University of Maryland in 2000, which was after a 2-year stint at MIT with Prof. Walker, where I pursued my interest in the biochemistry of carbohydrate signals involved in the nitrogen-fixation symbiosis between bacterium rhizobium and plant alfalfa [48, 49]. My attention was turned to exploring sugar oxazolines as synthetic substrates for the ENGase-catalyzed transglycosylation for two considerations. First, the ENGase-catalyzed reaction was implicated to proceed via a substrate-assisted mechanism through the participation of the 2-acetamido group to form a 1,2-oxazolinium ion intermediate, in analogy to some GH family-18 chitinases [50–52] and some GH family-20 N-acetyl-β-hexosaminidases [53, 54] (Figure 1); secondly, there was a precedent at the time that a disaccharide oxazoline could serve as a substrate for chitinase-catalyzed polymerization to form artificial chitin [55].
Figure 1.
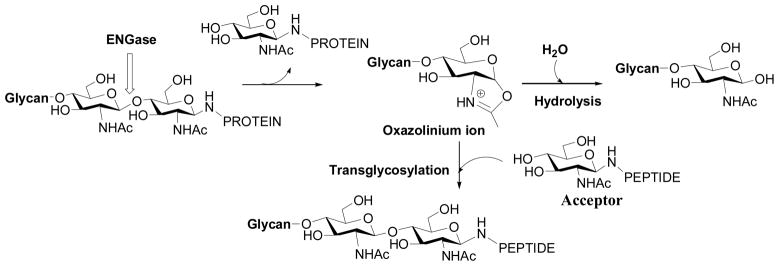
ENGase-catalyzed hydrolysis and transglycosylation
To test the feasibility of synthetic oligosaccharide oxazolines as substrates for Endo-A, we started with a semi-synthesis of a Man6GlcNAc-oxazoline. Man6GlcNAc2-Asn was prepared by digestion of chicken ovalbumin with pronase followed by chromatographic separation of the N-glycan on a carbon column [38]. The Man6GlcNAc2-Asn was cleaved by Endo-A to give Man6GlcNAc and then the free oligosaccharide was converted to sugar oxazoline by per-O-acetylation, oxazoline formation, and de-O-acetylation (Scheme 1) (unpublished results). It took about a year for us to obtain a small amount of the oligosaccharide oxazoline. Surprisingly, an initial test with Endo-A resulted in no detection of any transglycosylation product! The only net result was the hydrolysis of the oxazoline to give the free oligosaccharide Man6GlcNAc. This result was very disappointing, as we expected that the Man6GlcNAc-oxazoline would be at least comparable to Man6GlcNAc2-Asn for Endo-A catalyzed transglycosylation (Man6GlcNAc2-Asn could give about 20% yield in transglycosylation under the same reaction conditions). One plausible explanation was that the transglycosylation with Man6GlcNAc-oxazoline might also occur but, because the putative product Man6GlcNAc2-peptide was an excellent substrate for Endo-A, the product might have been hydrolyzed in situ immediately before it was released from the catalytic center.
Scheme 1.

Synthesis of Man6GlcNAc-oxazoline and its reaction with Endo-A
When we were puzzled by this unexpected observation, Prof. Shoda and co-workers published a seminal paper in 2001, showing that a simple disaccharide oxazoline derived from Manβ1,4GlcNAc could serve as a substrate of Endo-A and Endo-M for transglycosylation to form a p-nitrophenyl trisaccharide derivative, which was used for detecting the transglycosylation activity [56]. The yield was about 50% when two-fold excess of the sugar oxazoline was used, but it was found that the trisaccharide product Manβ1,4GlcNAcβ1,4GlcNAc-PNP was not hydrolyzed by Endo-A or Endo-M. This timely study yielded a very important piece of information, implicating that a sugar oxazoline corresponding to a truncated N-glycan was recognized by the enzyme for transglycosylation whereas the resulting ground-state product could be resistant to enzymatic hydrolysis because of the truncated structures. Thus, the original idea to explore sugar oxazolines as donor substrate for ENGase-catalyzed transglycosylation was not a bad idea, but we might have just picked up a “wrong” target to start with. To test this, we managed to synthesize several truncated N-glycan oxazolines, including the di- and tetra-saccharide oxazolines corresponding to the N-glycan core. Meanwhile, we synthesized several GlcNAc-peptides of the HIV-1 envelope glycoproteins that would serve as the acceptors. The hard work on the chemical synthesis of these substrates was finally paid off when we observed that indeed the truncated N-glycan oxazoline such as the Man3GlcNAc-oxazoline was an excellent substrate for Endo-A catalyzed transglycosylation to form the corresponding N-glycopeptide with the expected natural β-1,4-glycosidic linkage for the newly formed glycosidic bond (Scheme 2) [57]. The enzymatic transglycosylation proceeded quickly under very mild conditions (phosphate buffer, pH 6.5, 23 °C). While Man3GlcNAc-oxazoline was highly active for the Endo-A catalyzed transglycosylation, the resulting Man3GlcNAc2-peptide product was found to be resistant to hydrolysis by Endo-A under the reaction conditions when only a catalytic amount of enzyme was used. The transglcyosylation usually led to the formation of the corresponding glycopeptide in 75–85% yield when 2–3 fold excess of sugar oxazoline was used. The big difference in enzymatic reactivity between the highly activated sugar oxazoline and the ground-state product favors the accumulation of the transglycosylation product. It should be mentioned that the HIV-1 C34 glycopeptide corresponding to the envelope glycoprotein gp41 was a potent anti-HIV inhibitor, and an initial synthesis of a glycoform using a high-mannose type natural N-glycan (5-fold excess) as substrate resulted in only 10% yield under an optimal condition [39]. As another example, we applied this improved chemoenzymatic method to the synthesis of a large HIV-1 gp120 fragment, a 47-mer V3 domain glycopeptide carrying two core N-linked pentasaccharide [58]. The synthesis was achieved by a concise two-step approach: the solid-phase synthesis of the 47-mer polypeptide that contains two GlcNAc moieties and the subsequent high-yield double glycosylation catalyzed by Endo-A, with Man3GlcNAc-oxazoline as the donor substrate. The synthetic V3 glycopeptides were successfully used for probing the effects of glycosylation on the global conformations of the V3 domain and on the protection of the polypeptide against protease digestion [58]. These experimental data suggest that the use of sugar oxazolines as highly activated substrates for ENGase-catalyzed transglycosylation not only expanded the substrate availability, but also led to substantial enhancement of the overall synthetic efficiency, permitting a high-yield assembly of large glycopeptides that are otherwise difficult to obtain by other means.
Scheme 2.

Transglycosylation with the tetrasacharide oxazoline by Endo-A
To evaluate the donor substrate structural requirement in the enzymatic transglycosylation, we synthesized an array of further truncated and modified sugar oxazolines, including LacNAc-oxazoline, N,N′-diacetylchitobiose-oxazoline, Glcβ1,4GlcNAc-oxazoline, and 6′-O-benzyl-Manβ1,4GlcNAc-oxazoline, and tested their activity toward Endo-A [59]. This study suggested that the disaccharide moiety, Manβ1,4GlcNAc oxazoline, was the minimum structure recognized by Endo-A for an efficient transglycosylation. But selective modification on the mannose moiety such as introducing a tag or additional sugar residue at the 6′-position could be tolerated without significant loss of substrate activity toward Endo-A. On the other hand, it was observed that change the configuration of the 2′ and/or 4′-OH in the disaccharide, or replace them with other substituents, resulted in diminished substrate activity toward Endo-A.
Independently, Prof. Antony Fairbanks (then at University of Oxford) and co-workers was also pursuing the sugar oxazoline concept for ENGase-catalyzed transglycosylation. The group synthesized another array of sugar oxazolines and tested their activity for transglycosylation with Endo-M (Scheme 3) [60]. A very interesting finding was that the glc-containing disaccharide oxazoline, Glcβ1,4GlcNAc-oxazoline, showed a residual activity in Endo-M catalyzed transglycosylation, giving a 5% yield. Moreover, when the disaccharide was extended to a trisaccharide derivative with an additional α-1,3-linked mannosyl residue attached to the glucose moiety, the resulting trisaccharide oxazoline became an excellent substrate for Endo-M, giving a 91% yield of the transglycosylation product, without product hydrolysis. This result again implicated a great potential of the chemoenzymatic method for constructing novel neoglycopeptides. However, use of larger sugar oxazolines corresponding to natural high-mannose N-glycans such as Man5GlcNAc-oxazoline was problematic, as the products became excellent substrates for hydrolysis by Endo-M [61] (Scheme 3). It seemed that Endo-M hydrolyzed the Man3GlcNAc2-peptide much more quickly than Endo-A, as we did not observe any significant hydrolysis of Man3GlcNAc2-peptide by Endo-A in our previous experiments [57, 58]. However, a precise comparison of the results might not be appropriate as the amount of enzymes used for these experiments were significantly different - we usually used much less enzyme for the catalytic transglycosylation.
Scheme 3.

Endo-M catalyzed transglycosylation with modified sugar oxazolines
C. Expanding the chemoenzymatic method to the synthesis and glycosylation remodeling of glycoproteins
With the success in synthesizing large glycopeptides, we next turned our attention to the use of the chemoenzymatic method for glycoprotein synthesis and glycosylation remodeling. To test the feasibility, we selected bovine ribonuclease (RNase) B as a model, which is a natural glycoprotein consisting of 124 amino acids and carrying a heterogeneous high-mannose type N-glycan (Man5–9GlcNAc2) at the Asn-34 site. To make the GlcNAc-protein acceptor, natural RNase B was treated with Endo-H to remove the heterogeneous N-glycans, giving the homogeneous GlcNAc-RNase with the innermost GlcNAc being still attached at the Asn-24. Initial test with Man3GlcNAc-oxazoline showed that Endo-A was very efficient to transglycosylate GlcNAc-RNase to afford a homogeneous glycoprotein carrying the core N-pentasaccharide, Man3GlcNAc2-RNase, in 82% yield without the need of denaturing the protein (phosphate buffer, pH 6.5, 23 °C) [62]. Then, we synthesized and tested a series of large sugar oxazolines and examined their feasibility for glycoprotein remodeling [63]. It was found that Endo-A could tolerate various modifications at the outer mannose residues of the Man3GlcNAc core for transglycosylation, resulting in the introduction of carbohydrate ligands and meanwhile maintain the natural core N-pentasaccharide on the protein. Thus, the chemoenzymatic method provides an efficient means to construct homogeneous glycoproteins carrying large oligosaccharide ligands (Scheme 4) [62, 63]. Among others, the incorporation of an azide-tagged core pentasaccharide into glycoproteins was of particular interest, as this will permit further site-specific modifications of the glycoproteins through click chemistry involving the azide-alkyne 1,3-dipolar cycloaddition [64–66]. For example, Cu (I)-catalyzed click chemistry between an alkyne-containing α-Gal epitope and the azide-tagged RNase resulted in simultaneous introduction of two copies of α-Gal epitopes into RNase in excellent yield (Scheme 5). The resulting glycoprotein could be specifically recognized by anti-α-Gal antibodies in human serum [63].
Scheme 4.

ENGase-catalyzed transglycosylation for N-glycoprotein synthesis
Scheme 5.

Catalytic 1,3-dipolar cycloaddition to introduce αGal epitope into RNase
Building on this foundation, we initiated a program on glycosylation engineering of human IgG1-Fc using the chemoenzymatic approach. Fc glycosylation plays an essential role in modulating the interactions between Fc domain and respective Fc receptors, thus affecting the effector functions of antibodies [5, 67]. For the purpose, we first constructed the yeast expression vector encoding human IgG1-Fc and expressed the protein in yeast Pichia pastoris. The large heterogeneous high-mannose type N-glycans were then removed by Endo-H to leave only the innermost GlcNAc residue at the Asn-297 glycosylation site, giving the GlcNAc-Fc homodimer. Finally a pre-assembled glycan oxazoline was transferred by Endo-A to the GlcNAc-Fc to produce the desired homogeneous glycoform of IgG-Fc (Figure 2) [68]. An important finding was that the Endo-A catalyzed transglycosylation could proceed smoothly on the seemingly crowded GlcNAc residue at the CH2 domain of the Fc homodimer without the need of denaturing the IgG-Fc protein [68]. Thus the structural integrity of the IgG-Fc was preserved. This proof-of-concept preliminary study implicates a potentially general approach to glycosylation engineering of IgG-Fc domain for structural and functional studies.
Figure 2.

Glyco-engineering of human IgG1-Fc
D. Generation of novel glycosynthase mutants: Synthesis of homogeneous glycoproteins carrying full-size natural N-glycans
Product hydrolysis by wild type enzymes would not be avoidable if the targets are glycoproteins carry full-size natural N-glycans, as they are excellent substrates of wild type enzymes. A promising approach to addressing this problem is to generate efficient ENGase-based glycosynthases. Glycosynthases are glycosidase mutants that lack hydrolytic activity because of mutations but can still take an activated glycosyl donor such as glycosyl fluoride with an opposite anomeric configuration as substrate for transglycosylation to form a new glycosidic bond [13, 69, 70]. However, the conventional approach by mutating the nucleophilic residue of common retaining β-glycosidases at the catalytic site to create glycosynthase would not be applicable to ENGases as this class of enzymes proceeds via a substrate-assisted mechanism in which the nucleophile is the 2-acetamido group in the substrate. As the first attempt to generate ENGase-based glycosynthases, Prof. Kenji Yamamoto’s group (then at Kyoto University) and my group at University of Maryland formed a very productive collaboration focusing on Endo-M. We synthesized a Man9GlcNAc-oxazoline of the full-size natural high-mannose N-glycan and used it as the substrate to test the mutants generated by site-directed mutations on the residues in the putative catalytic region. The site-directed mutations on highly conserved residues were mainly based on sequence alignment of family GH85 glycosyl hydrolases that are proposed to proceed with a substrate-assisted mechanism via an oxazolinium ion intermediate. The screening led to the identification of two interesting mutants, Y217F and N175A, which showed superior transglycosylation activity with reduced product hydrolysis activity [71]. Notably, the EndoM-N175A mutant was almost devoid of product hydrolysis activity while it could still take the activated sugar oxazoline for transglycosylation. Therefore, EndoM-N175A became the first ENGase-based glycosynthase. On the basis of these experimental observations, a substrate-assisted mechanism was proposed, in which the Asn-175 residue plays a crucial role in promoting oxazoline ring formation upon glycosidic bond activation by residue Glu-177, while the Tyr-217 may be involved in the activation of a water molecule for hydrolysis [71]. Thus a mutation at N175 would inactivate its ability to promote oxazolinium ion formation for hydrolysis, but the resulting mutant could still take the pre-formed oxazoline for transglycosylation (Figure 3). More recently, a systematic examination of 19 mutants at N175 was performed, in which the Asn residue was replaced with the other 19 natural amino acid residues [72]. Interestingly, several new N175 mutants showed transglycosylation activity comparable or superior to the original N175A mutant. In particular, a N175Q mutant was shown to have both glycosynthase and transglycosidase activity that could also take natural N-glycan for transglycosylation with diminished hydrolytic activity.
Figure 3.
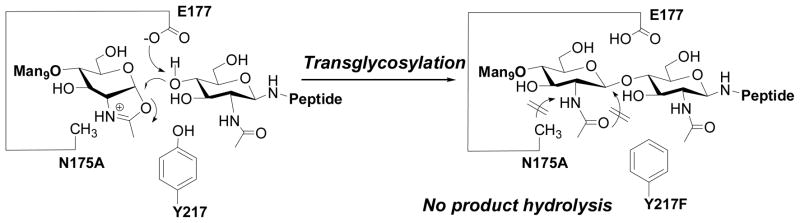
Transglycosylation by glycosynthase mutant EndoM-N175A
Based on sequence alignment, we created a N171A mutant of Endo-A, which was viewed as an equivalent to the EndoM-N175A [73]. We found that EndoA-N171A was a glycosynthase that could efficiently perform transglycosylation with natural high-mannose type N-glycan oxazoline without product hydrolysis. We also found that when wild type enzyme Endo-A was used with Man9GlcNAc-oxazoline, no transglycosylation was observed during the reaction, except a quick hydrolysis of the sugar oxazoline. This experimental data was consistent with our initial observation on Man6GlcNAc-oxazoline with wild type Endo-A, which showed only hydrolysis. The usefulness of the EndoM-N175A and EndoA-N171A mutants were exemplified for the synthesis of the pure ribonuclease B glycoforms carrying natural high-mannose type and complex type N-glycans [73]. More recently, the glycosynthase mutants were successfully used for the synthesis of homogeneous glycoforms of CD52 antigens carrying natural N- and O-glycans [74]. As a separate study, Prof. Fairbanks and co-workers reported that the E173H and E173Q mutants of Endo-A also showed some transglcyosyaltion activity with Man3GlcNAc-oxazoline [75]. But the specific activity of the E173H and E173Q mutants seemed to be low, as a relatively large amount of enzymes needed to be used for the transglycosylation. There were also important progresses in the crystallographic studies of GH family 85 ENGases. Three papers were recently published on the crystal structures of Endo-A and Endo-D [76–78]. In the case of Endo-A, the structural studies have confirmed the essential roles of several key residues, including N171, E173, and Y205, which were within hydrogen bonding distance to the substrate [76, 77]. In this pursuit, the use of a sugar thiazoline, a non-hydrolyzable sugar oxazoline analog that we synthesized [79], facilitated the crystallization of Endo-A and helped to reveal the potential roles of the key residues [76]. In addition, a comparison between the Endo-A and Endo-D structures indicated that the two enzymes had almost identical folding topology of the catalytic domain, suggesting that the two share a general mechanism of substrate-assisted catalysis [76–78]. These structural and mechanistic studies provide an important basis to perform enzyme engineering for improving the transglycosylation activity or for discovering new glycosynthase mutants with novel substrate specificity.
For glycoprotein synthesis via the ENGase-catalyzed transglycosylation, a key precursor is the GlcNAc-containing protein. While GlcNAc-protein may be prepared by several ways such as Endo-H cleavage of yeast expressed glycoproteins, it would be highly desirable if an Escherichia coli (E. coli) system, the biotechnology’s working horse for protein expression, could be explored for making GlcNAc-proteins. However, E. coli usually cannot produce glycoproteins as it lacks protein glycosylation machinery. To solve this problem, we collaborated with Prof. Markus Aebi at ETH and developed a method for producing homogeneous glycoproteins with humanized glycosylation by a combination of novel E. coli glycoprotein expression and in vitro glycosylation remodeling [80]. The method involves glycosylation pathway engineering and functional transfer of the Campylobacter jejuni glycosylation machinery [81] in E. coli to express a glycoprotein, in which the Asn-linked monosaccharide bacillosamine in the Asn-linked bacterial N-glycan was changed to the eukaryotic GlcNAc moiety. This was achieved by deleting the genes involved for the biosynthesis (pglD, pglE and pglF) and transfer (pglC) of bacillosamine and by taking advantage of the E. coli existing biosynthetic pathway for making the GlcNAc-diphosphate-lipid precursor. Fortunately, the C. jejuni oligosaccharyl transferase PglB was able to use the resulting GlcNAc-containing glycolipid as substrate to glycosylate the target protein. Then the bacterial N-glycans were trimmed down to the key GlcNAc-protein. Finally eukaryotic N-glycans of high-mannose type or complex type were introduced by the enzymatic transglycosylation to fulfill the homogeneous eukaryotic glycoproteins (Scheme 6). This method was applied for making glycosylated human IgG-Fc CH2 domain and a single chain antibody. With these promising proof-of-concept results, future studies should be directed to enhancing the efficiency of glycosylation of biomedically important glycoproteins through optimizing this engineered E. coli expression system.
Scheme 6.

A combined method for producing homogeneous glycoprotein
E. Introduction of N-glycans to natural products and tandem transglycosylation for polysaccharide synthesis
To investigate the scope of ENGase-catalyzed transglycosylation, we examined the feasibility of the enzymatic method to glycosylate various small molecule natural products. It was found that Endo-A had a very broad substrate specificity and was able to accommodate a wide range of glucose (Glc) or GlcNAc-containing natural products as acceptors for transglycosylation [82]. For example, N-glycan can be readily added to a steroid structure that contains a Glc or GlcNAc moiety to form a new glycoconjugate (Scheme 7). Moreover, multiple N-glycans can be introduced to natural products that have two or more Glc moieties such as some flavonoid glycosides by the Endo-A catalyzed reaction using sugar oxazoline as the donor substrate. This glycosylation method is concise, efficient, and highly convergent. Since N-glycans of glycoprotein origin can serve as ligands for various lectins and cell-surface receptors, site-specific introduction of a defined N-glycan into biologically significant natural products may bestow novel properties onto these natural products, e.g., for drug discovery and development. As to acceptor substrate specificity of ENGases, Endo-A was also able to glycosylate triazole-linked GlcNAc/Glc-peptides [83]. Although Endo-A could be forced to transfer released N-glycans to different monosaccharides and alcohols when they are present at extremely high concentrations, Prof. Inazu and co-workers recently reported that Endo-M could selectively glycosylate acyclic compounds with a 1,3-diol structure consisting of primary and secondary hydroxyl groups, and the glycosylation occurred at the secondary hydroxyl group [84]. This study points to an interesting substrate recognition mode of Endo-M.
Scheme 7.

Enzymatic introduction of N-glycans into natural products by Endo A
During the study of substrate specificity, we discovered an interesting tandem transglycosylation by Endo-A with a disaccharide oxazoline, Glcβ-1,4-GlcNAc oxazoline. We found that the terminal Glc moiety in the donor substrate could also serve as acceptor, leading to self-condensation to form novel glycopeptides carrying repeating disaccharide structure [85]. Moreover, in the absence of an external acceptor, the Endo-A catalyzed self-condensation led to the formation of polysaccharides with the disaccharide repeating units. This is the first example of Endo-A catalyzed polymerization for polysaccharide synthesis.
F. Chemoenzymatic synthesis of novel N-glycan clusters
We have recently explored the potential of ENGases for synthesizing a class of novel N-glycan clusters [86]. We found that the internal β-1,2-linked GlcNAc moieties in the N-glycan core, once exposed in the non-reducing terminus, was able to serve as acceptors for enzymatic transglycosylation catalyzed by Endo-A and EndoM-N175A. The tandem transglycosylation thus permitted a quick extension of the sugar chains to form a class of glycan clusters containing multiple N-glycan cores linked by native glycosidic linkages (Scheme 9). Moreover, we also observed that the GlcNAc moieties at the α1,3Man arm and α1,6Man arm had distinct activity, and the Endo-M mutant could discriminate the two to selectively introduce different glycans at the two arms to form novel hybrid N-glycan clusters. These synthetic glycan clusters possess unusual lectin recognition properties as revealed by lectin microarray studies [86]. As clustering effect is an important feature in carbohydrate-protein interactions, these novel synthetic glycan clusters would be useful tools for functional glycomics studies to unveil new functions of both glycans and glycan-binding proteins.
Scheme 9.
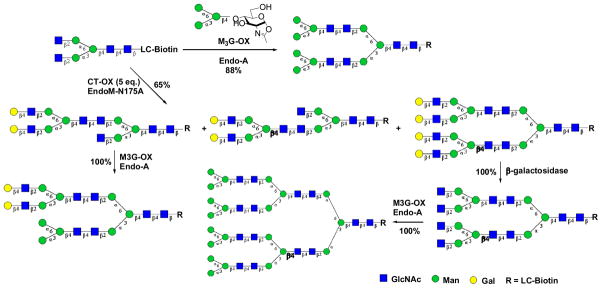
Chemoenzymatic synthesis of novel N-glycan clusters
G. Promiscuous substrate specificity of Endo-A: the ability of Endo-A to accommodate complex type N-glycan oxazolines for transglycosylation
Endo-A is an ENGase that is specific for the hydrolysis of high-mannose or hybrid type N-glycans but cannot hydrolyze complex type N-glycans. Work from our group as well as other groups has demonstrated that Endo-A was able to take truncated N-glycan oxazolines such as Man3GlcNAc-oxazoline for transglycosylation [57, 58, 61, 63, 68, 87]. However, we recently discovered that Endo-A had much broader substrate specificity than previously thought when sugar oxazolines was used as substrates. We have found that Endo-A possesses significant activity on sialylated as well as asialylated N-glycan oxazolines for transglycosylation [88]. When a relatively large amount of the enzyme was used, a high-yield synthesis of complex type N-glycopeptide could be achieved (Scheme 10). It was also confirmed that the ground-state, complex type glycopeptide product was completely resistant to hydrolysis by Endo-A. Thus the wild-type Endo-A itself acts as a glycosynthase for complex type N-glycopeptides when the corresponding sugar oxazoline was used as highly activated substrate. This novel property of Endo-A was explored for a one-pot transformation of high-mannose type RNase B to the sialylated complex type RNase C [88]. While the specific activity of Endo-A on complex type glycan oxazolines was still relatively low in comparison with Man3GlcNAc-oxazoline, the unexpected discovery of the amazing substrate promiscuity of Endo-A provides an excellent platform to apply directed evolution strategy for improving and expanding Endo-A’s transglycosylation activity. It should be also emphasized that thanks to a highly efficient one-step conversion of free N-glycan to glycan oxazoline reported by Prof. Shoda and co-workers [89], now we could readily recover the excess sugar oxazolines in the form of free oligosaccharide and convert it into glycan oxazoline for recycling in transglycosylation [88]. On the other hand, the Endo-M mutants, N175A and N175Q, were shown to be able to take sialylated N-glycan oxazoline for transglycosylation [88, 90]. These studies opened an efficient avenue to the synthesis of sialylated glycoproteins.
Scheme 10.

Endo-A catalyzed transglycosylation with complex type N-glycan oxazoline
H. Conclusion
Endo-β-N-acetylglucosaminidases are a class of glycosyl hydrolases that cleave the N-glycans from glycoproteins. Yet, the past decade has witnessed major advances in exploring their transglycosylation activity for synthetic purpose. The exploration of synthetic sugar oxazolines as donor substrates for the ENGase-catalyzed transglycosylation has expanded the substrate availability and significantly enhanced the transglycosylation efficiency. The creation of novel glycosynthase mutants has overcome a major limitation involving ENGase-catalyzed synthesis, the product hydrolysis. Original application of the ENGase-catalyzed transglycosylation was limited to the synthesis of N-glycopeptides. Now the chemoenzymatic method has been extended to the synthesis of a range of complex carbohydrates, including homogeneous glycoproteins, novel oligosaccharide clusters, unusually glycosylated natural products, and even polysaccharides. Indeed, ENGases have shown amazing flexibility in transglycosylation and possess much broader substrate promiscuity than we have previously thought. It seems that we are just beginning to unveil the full potential of ENGase-catalyzed transglycosylation. It is expected that new ENGases with novel transglycosylation properties will be discovered and novel mutants with enhanced activity or altered substrate specificity will be generated in the future.
Scheme 8.

Endo-A catalyzed tandem transglycosylation
Acknowledgments
I want to express my sincere thanks to Prof. Kaoru Takegawa and Prof. Kenji Yamamoto for their great support of our chemoenzymatic synthesis program over the years and for fruitful collaborations. I also appreciate my very able postdoctoral fellows and students and other collaborators, whose names were cited in the references, for their exceptional contributions to the research program. This work was supported by the National Institutes of Health (NIH grant R01 GM080374 and R01 GM073717).
References
- 1.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 3.Haltiwanger RS, Lowe JB. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 4.Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 5.Nimmerjahn F, Ravetch JV. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 6.Jefferis R. Nat Rev Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 7.Danishefsky SJ, Allen JR. Angew Chem Int Ed. 2000;39:836–863. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 8.Nicolaou KC, Mitchell HJ. Angew Chem Int Ed. 2001;40:1576–1624. [PubMed] [Google Scholar]
- 9.Sears P, Wong CH. Science. 2001;291:2344–2350. doi: 10.1126/science.1058899. [DOI] [PubMed] [Google Scholar]
- 10.Dziadek S, Kunz H. Chem Rec. 2004;3:308–321. doi: 10.1002/tcr.10074. [DOI] [PubMed] [Google Scholar]
- 11.Seeberger PH, Werz DB. Nat Rev Drug Discov. 2005;4:751–763. doi: 10.1038/nrd1823. [DOI] [PubMed] [Google Scholar]
- 12.Kajihara Y, Yamamoto N, Miyazaki T, Sato H. Curr Med Chem. 2005;12:527–550. doi: 10.2174/0929867310504050527. [DOI] [PubMed] [Google Scholar]
- 13.Hancock SM, Vaughan MD, Withers SG. Curr Opin Chem Biol. 2006;10:509–519. doi: 10.1016/j.cbpa.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda Y, Totani K, Matsuo I, Ito Y. Curr Opin Chem Biol. 2009;13:582–591. doi: 10.1016/j.cbpa.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Wang LX, Huang W. Curr Opin Chem Biol. 2009;13:592–600. doi: 10.1016/j.cbpa.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamblin DP, Scanlan EM, Davis BG. Chem Rev. 2009;109:131–163. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- 18.Hinou H, Nishimura S. Curr Top Med Chem. 2009;9:106–116. doi: 10.2174/156802609787354298. [DOI] [PubMed] [Google Scholar]
- 19.Andre S, Kozar T, Kojima S, Unverzagt C, Gabius HJ. Biol Chem. 2009;390:557–565. doi: 10.1515/BC.2009.072. [DOI] [PubMed] [Google Scholar]
- 20.Boltje TJ, Buskas T, Boons GJ. Nat Chem. 2009;1:611–622. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crout DH, Vic G. Curr Opin Chem Biol. 1998;2:98–111. doi: 10.1016/s1367-5931(98)80041-0. [DOI] [PubMed] [Google Scholar]
- 22.Bennett CS, Wong CH. Chem Soc Rev. 2007;36:1227–1238. doi: 10.1039/b617709c. [DOI] [PubMed] [Google Scholar]
- 23.Shaikh FA, Withers SG. Biochem Cell Biol. 2008;86:169–177. doi: 10.1139/O07-149. [DOI] [PubMed] [Google Scholar]
- 24.Tarentino AL, Plummer TH., Jr Methods Enzymol. 1994;230:44–57. doi: 10.1016/0076-6879(94)30006-2. [DOI] [PubMed] [Google Scholar]
- 25.Kato T, Fujita K, Takeuchi M, Kobayashi K, Natsuka S, Ikura K, Kumagai H, Yamamoto K. Glycobiology. 2002;12:581–587. doi: 10.1093/glycob/cwf073. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Yano K, Sugimoto S, Kitajima K, Lennarz WJ, Inoue S, Inoue Y, Emori Y. Proc Natl Acad Sci U S A. 2002;99:9691–9696. doi: 10.1073/pnas.152333599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takegawa K, Yamaguchi S, Kondo A, Iwamoto H, Nakoshi M, Kato I, Iwahara S. Biochem Int. 1991;24:849–855. [PubMed] [Google Scholar]
- 28.Takegawa K, Yamaguchi S, Kondo A, Kato I, Iwahara S. Biochem Int. 1991;25:829–835. [PubMed] [Google Scholar]
- 29.Fan JQ, Takegawa K, Iwahara S, Kondo A, Kato I, Abeygunawardana C, Lee YC. J Biol Chem. 1995;270:17723–17729. doi: 10.1074/jbc.270.30.17723. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto K, Kadowaki S, Watanabe J, Kumagai H. Biochem Biophys Res Commun. 1994;203:244–252. doi: 10.1006/bbrc.1994.2174. [DOI] [PubMed] [Google Scholar]
- 31.Fujita K, Takami H, Yamamoto K, Takegawa K. Biosci Biotechnol Biochem. 2004;68:1059–1066. doi: 10.1271/bbb.68.1059. [DOI] [PubMed] [Google Scholar]
- 32.Parsons TB, Patel MK, Boraston AB, Vocadlo DJ, Fairbanks AJ. Org Biomol Chem. 2010;8:1861–1869. doi: 10.1039/b926078a. [DOI] [PubMed] [Google Scholar]
- 33.Mizuno M, Haneda K, Iguchi R, Muramoto I, Kawakami T, Aimoto S, Yamamoto K, Inazu T. J Am Chem Soc. 1999;121:284–290. [Google Scholar]
- 34.Wang LX, Fan JQ, Lee YC. Tetrahedron Lett. 1996;37:1975–1978. [Google Scholar]
- 35.Wang LX, Tang M, Suzuki T, Kitajima K, Inoue Y, Inoue S, Fan JQ, Lee YC. J Am Chem Soc. 1997;119:11137–11146. [Google Scholar]
- 36.O’Connor SE, Pohlmann J, Imperiali B, Saskiawan I, Yamamoto K. J Am Chem Soc. 2001;123:6187–6188. doi: 10.1021/ja010094s. [DOI] [PubMed] [Google Scholar]
- 37.Haneda K, Inazu T, Mizuno M, Iguchi R, Tanabe H, Fujimori K, Yamamoto K, Kumagai H, Tsumori K, Munekata E. Biochim Biophys Acta. 2001;1526:242–248. doi: 10.1016/s0304-4165(01)00135-0. [DOI] [PubMed] [Google Scholar]
- 38.Singh S, Ni J, Wang LX. Bioorg Med Chem Lett. 2003;13:327–330. doi: 10.1016/s0960-894x(02)01025-9. [DOI] [PubMed] [Google Scholar]
- 39.Wang LX, Song H, Liu S, Lu H, Jiang S, Ni J, Li H. ChemBioChem. 2005;6:1068–1074. doi: 10.1002/cbic.200400440. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Singh S, Zeng Y, Song H, Wang LX. Bioorg Med Chem Lett. 2005;15:895–898. doi: 10.1016/j.bmcl.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 41.Fan JQ, Quesenberry MS, Takegawa K, Iwahara S, Kondo A, Kato I, Lee YC. J Biol Chem. 1995;270:17730–17735. doi: 10.1074/jbc.270.30.17730. [DOI] [PubMed] [Google Scholar]
- 42.Makimura Y, Watanabe S, Suzuki T, Suzuki Y, Ishida H, Kiso M, Katayama T, Kumagai H, Yamamoto K. Carbohydr Res. 2006;341:1803–1808. doi: 10.1016/j.carres.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 43.Takegawa K, Tabuchi M, Yamaguchi S, Kondo A, Kato I, Iwahara S. J Biol Chem. 1995;270:3094–3099. doi: 10.1074/jbc.270.7.3094. [DOI] [PubMed] [Google Scholar]
- 44.Fujita K, Takegawa K. Biochem Biophys Res Commun. 2001;282:678–682. doi: 10.1006/bbrc.2001.4631. [DOI] [PubMed] [Google Scholar]
- 45.Fujita K, Yamamoto K. Biochim Biophys Acta. 2006;1760:1631–1635. doi: 10.1016/j.bbagen.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 46.Yamanoi T, Yoshida N, Oda Y, Akaike E, Tsutsumida M, Kobayashi N, Osumi K, Yamamoto K, Fujita K, Takahashi K, Hattori K. Bioorg Med Chem Lett. 2005;15:1009–1013. doi: 10.1016/j.bmcl.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 47.Tomabechi Y, Suzuki R, Haneda K, Inazu T. Bioorg Med Chem. 2010;18:1259–1264. doi: 10.1016/j.bmc.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez JE, Semino CE, Wang LX, Castellano-Torres LE, Walker GC. Proc Natl Acad Sci U S A. 1998;95:13477–13482. doi: 10.1073/pnas.95.23.13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang LX, Wang Y, Pellock B, Walker GC. J Bacteriol. 1999;181:6788–6796. doi: 10.1128/jb.181.21.6788-6796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terwisscha van Scheltinga AC, Armand S, Kalk KH, Isogai A, Henrissat B, Dijkstra BW. Biochemistry. 1995;34:15619–15623. doi: 10.1021/bi00048a003. [DOI] [PubMed] [Google Scholar]
- 51.Tews I, Terwisscha van Scheltinga AC, Perrakis A, Wilson KS, Dijkstra BW. J Am Chem Soc. 1997;119:7954–7959. [Google Scholar]
- 52.Brameld KA, Shrader WD, Imperiali B, Goddard WA., 3rd J Mol Biol. 1998;280:913–923. doi: 10.1006/jmbi.1998.1890. [DOI] [PubMed] [Google Scholar]
- 53.Mark BL, Vocadlo DJ, Knapp S, Triggs-Raine BL, Withers SG, James MN. J Biol Chem. 2001;276:10330–10337. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 54.Williams SJ, Mark BL, Vocadlo DJ, James MN, Withers SG. J Biol Chem. 2002;277:40055–40065. doi: 10.1074/jbc.M206481200. [DOI] [PubMed] [Google Scholar]
- 55.Kobayashi S, Kiyosada T, Shoda S. J Am Chem Soc. 1996;118:13113–13114. [Google Scholar]
- 56.Fujita M, Shoda S, Haneda K, Inazu T, Takegawa K, Yamamoto K. Biochim Biophys Acta. 2001;1528:9–14. doi: 10.1016/s0304-4165(01)00164-7. [DOI] [PubMed] [Google Scholar]
- 57.Li B, Zeng Y, Hauser S, Song H, Wang LX. J Am Chem Soc. 2005;127:9692–9693. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]
- 58.Li H, Li B, Song H, Breydo L, Baskakov IV, Wang LX. J Org Chem. 2005;70:9990–9996. doi: 10.1021/jo051729z. [DOI] [PubMed] [Google Scholar]
- 59.Zeng Y, Wang J, Li B, Hauser S, Li H, Wang LX. Chem Eur J. 2006;12:3355–3364. doi: 10.1002/chem.200501196. [DOI] [PubMed] [Google Scholar]
- 60.Rising TW, Claridge TD, Moir JW, Fairbanks AJ. ChemBioChem. 2006;7:1177–1180. doi: 10.1002/cbic.200600183. [DOI] [PubMed] [Google Scholar]
- 61.Rising TW, Heidecke CD, Moir JW, Ling Z, Fairbanks AJ. Chem Eur J. 2008;14:6444–6464. doi: 10.1002/chem.200800365. [DOI] [PubMed] [Google Scholar]
- 62.Li B, Song H, Hauser S, Wang LX. Org Lett. 2006;8:3081–3084. doi: 10.1021/ol061056m. [DOI] [PubMed] [Google Scholar]
- 63.Ochiai H, Huang W, Wang LX. J Am Chem Soc. 2008;130:13790–13803. doi: 10.1021/ja805044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kolb HC, Sharpless KB. Drug Discov Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 65.Best MD. Biochemistry. 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- 66.Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jefferis R. Biotechnol Prog. 2005;21:11–16. doi: 10.1021/bp040016j. [DOI] [PubMed] [Google Scholar]
- 68.Wei Y, Li C, Huang W, Li B, Strome S, Wang LX. Biochemistry. 2008;47:10294–10304. doi: 10.1021/bi800874y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perugino G, Trincone A, Rossi M, Moracci M. Trends Biotechnol. 2004;22:31–37. doi: 10.1016/j.tibtech.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 70.Faijes M, Planas A. Carbohydr Res. 2007;342:1581–1594. doi: 10.1016/j.carres.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 71.Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang LX, Yamamoto K. J Biol Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]
- 72.Umekawa M, Li C, Higashiyama T, Huang W, Ashida H, Yamamoto K, Wang LX. J Biol Chem. 2010;285:511–521. doi: 10.1074/jbc.M109.059832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, Wang LX. J Am Chem Soc. 2009;131:2214–2223. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang W, Zhang X, Ju T, Cummings RD, Wang LX. Org Biomol Chem. 2010;8:5224–5233. doi: 10.1039/c0ob00341g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heidecke CD, Ling Z, Bruce NC, Moir JW, Parsons TB, Fairbanks AJ. ChemBioChem. 2008;9:2045–2051. doi: 10.1002/cbic.200800214. [DOI] [PubMed] [Google Scholar]
- 76.Yin J, Li L, Shaw N, Li Y, Song JK, Zhang W, Xia C, Zhang R, Joachimiak A, Zhang HC, Wang LX, Liu ZJ, Wang P. PLoS One. 2009;4:e4658. doi: 10.1371/journal.pone.0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ling Z, Suits MD, Bingham RJ, Bruce NC, Davies GJ, Fairbanks AJ, Moir JW, Taylor EJ. J Mol Biol. 2009;389:1–9. doi: 10.1016/j.jmb.2009.03.050. [DOI] [PubMed] [Google Scholar]
- 78.Abbott DW, Macauley MS, Vocadlo DJ, Boraston AB. J Biol Chem. 2009;284:11676–11689. doi: 10.1074/jbc.M809663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li B, Takegawa K, Suzuki T, Yamamoto K, Wang LX. Bioorg Med Chem. 2008;16:4670–4675. doi: 10.1016/j.bmc.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwarz F, Huang W, Li C, Schulz BL, Lizak C, Palumbo A, Numao S, Neri D, Aebi M, Wang LX. Nat Chem Biol. 2010;6:264–266. doi: 10.1038/nchembio.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kowarik M, Numao S, Feldman MF, Schulz BL, Callewaert N, Kiermaier E, Catrein I, Aebi M. Science. 2006;314:1148–1150. doi: 10.1126/science.1134351. [DOI] [PubMed] [Google Scholar]
- 82.Huang W, Ochiai H, Zhang X, Wang LX. Carbohydr Res. 2008;343:2903–2913. doi: 10.1016/j.carres.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang W, Groothuys S, Heredia A, Kuijpers BH, Rutjes FP, van Delft FL, Wang LX. Chembiochem. 2009;10:1234–1242. doi: 10.1002/cbic.200800741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomabechi Y, Odate Y, Izumi R, Haneda K, Inazu T. Carbohydr Res. 2010;345:2458–2463. doi: 10.1016/j.carres.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 85.Ochiai H, Huang W, Wang LX. Carbohydr Res. 2009;344:592–598. doi: 10.1016/j.carres.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang W, Wang D, Yamada M, Wang LX. J Am Chem Soc. 2009;131:17963–17971. doi: 10.1021/ja9078539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parsons TB, Moir JW, Fairbanks AJ. Org Biomol Chem. 2009;7:3128–3140. [Google Scholar]
- 88.Huang W, Yang Q, Umekawa M, Yamamoto K, Wang LX. Chembiochem. 2010;11:1350–1355. doi: 10.1002/cbic.201000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda SI. J Org Chem. 2009;74:2210–2212. doi: 10.1021/jo8024708. [DOI] [PubMed] [Google Scholar]
- 90.Umekawa M, Higashiyama T, Koga Y, Tanaka T, Noguchi M, Kobayashi A, Shoda S, Huang W, Wang LX, Ashida H, Yamamoto K. Biochim Biophys Acta. 2010;1800:1203–1209. doi: 10.1016/j.bbagen.2010.07.003. [DOI] [PubMed] [Google Scholar]