

Abstract
A newly recognized primary cause of the obesity epidemic is the developmental programming effects of infants born to mothers with obesity or gestational diabetes, intrauterine growth restricted newborns, and offspring exposed to environmental toxins including Bisphenol A. The mechanisms which result in offspring obesity include the programming of the hypothalamic appetite pathway and adipogenic signals regulating lipogenesis. Processes include nutrient sensors, epigenetic modifications, and alterations in stem cell precursors of both appetite/satiety neurons and adipocytes which are modulated to potentiate offspring obesity. Future strategies for the prevention and therapy of obesity must address programming effects of the early life environment.
Obesity represents a public health crisis, contributing importantly to morbidity and mortality throughout the United States and the developed world. Obesity is central to the development of metabolic syndrome, which includes a constellation of abnormalities comprised of insulin resistance, elevated triglycerides, hypertension and atherosclerosis. Among US adults, 66% are overweight (BMI 25 to <30 kg/m2) and 33% are obese (BMI≥30 kg/m2), while 20% of children are obese, and thus at increased risk of adult obesity. Among childbearing women, there has been a continued increase in prevalence of obesity, with a 25–36% increase in maternal BMI over the last decade. In conjunction, we have witnessed a spectrum of pregnancy complications and a 25% increase in the incidence of high birth weight babies, which itself represents a risk factor for childhood obesity. Although much attention has been focused on the role of environmental factors, including the availability of calorie-dense foods and lifestyles involving less physical work, this review will present evidence that the predisposition to obesity may be programmed, or predetermined in utero. The underlying mechanisms that result in obesity include a dysregulation of appetite/satiety and adipogenesis/lipid metabolism.
Programmed Obesity
Humans
Human epidemiological studies have confirmed that both low and high birth weights result in an increased risk for childhood and adult obesity (review1). Intrauterine growth restricted (IUGR) or low birth weight infants, particularly those with rapid catch-up growth in the first several years of life, have higher risk of adult obesity and metabolic syndrome.1;2 Whereas IUGR infants develop in a state of relative “undernutrition”, fetal “overnutrition” also has consequence for offspring health. Specifically, maternal obesity during pregnancy, or increased weight gain in pregnancy, is associated with higher birth weight newborns3 and an increased risk of obesity and diabetes risk in later life.
There may well be an optimal newborn weight (potentially specific to an individual mother) at which the programming of obesity potential is minimized. However, within ranges of lower to higher birth weights, studies indicate a gradation of increased propensity to program offspring obesity. Thus, alterations from “optimal” in utero growth, be it from limited or excess nutrition, increase the relative risk of adult metabolic syndrome.
Animal Models
Animal models of IUGR, using a variety of methods, such as maternal nutrient restriction, placental uterine ligation or glucocorticoid exposure, have demonstrated that IUGR offspring are at an increased risk of adult adiposity,4 particularly among those who evident rapid catch-up growth.5 More recent animal models of maternal overnutrition, including maternal obesity and Western, high-fat (HF) diets similarly replicate the human experience in that offspring are predisposed to adult obesity.5
Mechanisms of Nutrition-induced Programmed Obesity
As noted above, increased appetite and enhanced adipogenesis represent critical pathways promoting obesity.
Appetite
Studies have demonstrated that programmed appetite dysregulation contributes to obese phenotype in IUGR and HF offspring.6 We will briefly describe the development of appetite/satiety neurons to demonstrate potential mechanisms by which a programmed enhanced appetite and/or reduced satiety can develop.
Appetite is primarily controlled by a complex circuit of hypothalamic nuclei involved in synthesis of appetite/satiety signals, action areas where messengers act and regulatory sites. The predominant appetite regulatory site, the hypothalamic arcuate nucleus (ARC) receives input from peripheral (brain, pancreas, and adipocytes) and central sources. The ARC contains at least two populations of neurons with opposing actions on food intake: primarily medial orexigenic (NPY; neuropeptide Y and AgRP; agouti-related protein) and primarily lateral anorexigenic (POMC; pro-opiomelanocortin and CART; cocaine- and amphetamine-regulated transcript) neurons. The most critical ARC “appetite-regulatory” projections are to the paraventricular nucleus which ultimately regulates ingestive behavior.
The hypothalamic regulation of appetite and satiety function develops in utero in precocial species in order to prepare for newborn life, though there is continued neural development and maturation during the neonatal period.7 Thus, nutritional effects during the fetal and/or newborn period may permanently “program” ARC structure and function and influence appetite and development of obesity. During this period of development, hypothalamic neural stem cells (NSC) proliferate and ultimately differentiate into neurons, astrocytes or oligodendrocytes which migrate and populate hypothalamic nuclei. Once NSCs differentiate to neurons, those cells destined for the ARC appetite center further differentiate to express orexigenic or anorexigenic peptides. NSC differentiation to neurons or glial cells and the ultimate differentiation to appetite or satiety neurons is regulated by a complex spatial/temporal interplay of pathways, which may be significantly altered by the nutrient environment or select toxins.
In our laboratory studies, we have confirmed that maternal undernutrition results in IUGR pups, which when nursed by control dams demonstrate significantly increased food intake with rapid catch-up growth and adult obesity.5;8 The obese phenotype is a result of dysfunction at several levels of the appetite/satiety pathway, as evidenced by reduced satiety responses to leptin,9 impaired ARC signaling responses to leptin,9 and increased responses to appetite stimulatory factors (i.e, ghrelin).6 Our studies and others have confirmed that in response to maternal undernutrition, offspring may express an increased ratio of appetite/satiety gene expression.10;11;12;13 We have further explored the mechanisms which contribute to the programmed upregulation of appetite, using a model of hypothalamic NSCs which form the appetite neurons. We have demonstrated that NSCs from IUGR fetuses exhibit reduced migration in vivo, and reduced proliferation and neuronal differentiation in vitro.14 Importantly, even in culture media outside the fetal environment, IUGR NSCs are programmed to preferentially differentiate to appetite as compared to satiety neurons (unpublished data). Maternal obesity/HF diet also results in pups which demonstrate increased food intake and adult obesity.15 Thus, both maternal under- or over- malnutrition may program offspring hyperphagia and metabolic syndrome.
Adipose Tissue
In addition to programmed appetite/satiety, evidence indicates that mechanisms regulating adipose tissue development and function (lipogenesis) may be a key factor in the development of programmed obesity. Adipogenesis is the process of cell differentiation by which preadipocytes become adipocytes and requires highly organized and precisely controlled expression of a cascade of transcription factors which, similar to neural development, may be influenced by the nutrient environment.
Adipogenesis and Lipogenesis
The cellular development associated with adipose tissue growth involves both cellular hyperplasia (increase in cell number) and hypertrophy (increase in cell size). Hyperplasia (adipogenesis) involves the proliferation and differentiation of preadipocytes, whereas hypertrophy is the result of excess triglyceride accumulation in existing adipocytes due to a positive energy balance. Mechanistic studies on enhanced adipogenesis or alteration in function/response of adipocytes in IUGR offspring are limited. However, our laboratory studies indicate that IUGR offspring specifically demonstrate hypertrophic adipocytes and increased de novo fatty acid synthesis,16;17 both predictive of increased propensity for fat storage. We have shown that at 1 day of age, IUGR male offspring have upregulated adipogenic signaling cascade16 as evident by an increased expression of enzymes promoting adipocyte lipid synthesis and storage. As these changes are evident early in life prior to the onset of obesity, it suggests a programmed pathway of increased adipocyte differentiation and lipogenesis which likely promotes the development of obesity and metabolic abnormalities in IUGR offspring.5;16
We have further explored whether the increased adipogenic potential of IUGR adipocytes is due to an intrinsic cellular change by utilizing a primary adipocyte cell culture. IUGR adipocytes in culture retain the phenotype of enhanced adipogenesis, evidenced by growth in culture and also enzyme/signaling expression.17 Thus, the adipocyte itself exhibits “programmed” adipogenesis/lipogenesis, independent of the IUGR hormonal milieu.
Our initial mechanistic studies of offspring of obese HF mothers similarly demonstrate an enhanced adipogenesis, akin to IUGR newborns. Thus both under- and overnutrition programs increased adipogenesis. Though the downstream adipogenic signaling factors are similar in both IUGR and HF offspring, there are marked differences in the nutrient/energy sensors responses, dependent upon the primary nutrient stress.18
Environmental Obesogens
Increasing human exposure to a wide range of industrial/agricultural chemicals has been well recognized. The CDC reported significant human exposure to endocrine-disrupter chemicals (EDCs), including those acting via estrogen receptors (eEDC). Both adults and children are routinely exposed to Bisphenol A (BPA), as there is measurable BPA in breast milk, maternal and fetal serum, amniotic fluid and placental tissues 19 Importantly, the level of BPA is higher in infants and children than in adults.
There is accumulating data that endogenous estrogens and eEDCs are involved in both body weight and energy regulation. Estrogen treatment of hamsters decreases body weight and fat, and deficiency of estrogens in animals or women results in fat accrual.20 In contrast to adult effects, developmental eEDC exposure may impact offspring body weight via changes in adipocyte or NSC growth and differentiation and long term epigenetic modifications. Offspring of rats exposed perinatally to low dose BPA exhibit an increased body weight, while mice exposed to low BPA from mid to late gestation have increased body mass at birth and weaning. Notably, low rather than high dose of maternal BPA has been reported to be effective in promoting offspring weight gain. Importantly, these adverse effects seen in animals at “low dose” are well-within the range of human exposure to BPA.
In humans, epidemiologic studies support the association of developmental EDC exposure and obesity in later life. Prenatal and early life polychlorinated bisphenyl (PCB) exposure is associated with increased male and female weight at puberty, in utero exposure to hexachlorobenzene is linked to overweight children at age 6, and organochlorine pesticides are positively associated with BMI.
Mechanisms of BPA-induced Programmed Obesity
Similar to mechanisms by which under- and overnutrition program appetite/satiety and adipogenesis, BPA exposure may result in adult obesity. Pro-adipogenic effects of environmental obesogens have been well documented, with recent studies demonstrating effects on adipocyte generation, differentiation, and lipogenic function, with potential epigenetic effects traversing generations. In addition to adipogenic effects, low dose maternal BPA exposure has been shown to accelerate neurogenesis and neuronal migration in mice, and result in aberrant neuronal network formation. Prenatal/neonatal BPA exposure induces dysfunction of the hippocampal cholinergic system. As a consequence of accelerated neurogenesis, maternal BPA reduces the fetal (e14.5) neural stem/progenitor cell (NPC) population.
In our preliminary studies, pregnant dams were administered oral BPA from gestational day 10 to term showed increased body weights and food intake. Offspring of BPA dams were heavier at 1 day and 3 weeks of age, indicating early onset obesity. We further explored the effects of BPA on preadipocyte and NSC proliferation. Preadipocytes and hypothalamic NSCs treated with BPA for 5 days demonstrated increased proliferation. These findings suggest that environmental toxin, particularly endocrine disrupter compounds such as BPA, may have marked effects on fetal neurogenesis and adipogenesis, potentially contributing to the current obesity epidemic.
Epigenetics and Programming
The essential concept of ‘gestational programming’ signifies that the nutritional, hormonal, and metabolic environment provided by the mother permanently alters organ structure, cellular responses and gene expression that ultimately impact metabolism and physiology of her offspring (Figure 1). Further, these effects vary dependent upon the developmental period, and as such, rapidly growing fetuses and neonates are more vulnerable. The programming events may have immediate effects, for example, impairment of organ growth at a critical stage, whereas other programming effects are deferred until expressed by altered organ function at later age. In this instance, the question is how the memory of early events is stored and later expressed, despite continuous cellular replication and replacement. This may be mediated through epigenetic control of gene expression which involves modification of the genome without altering the DNA sequence itself.
Figure 1. In Utero Programming of Obesity.
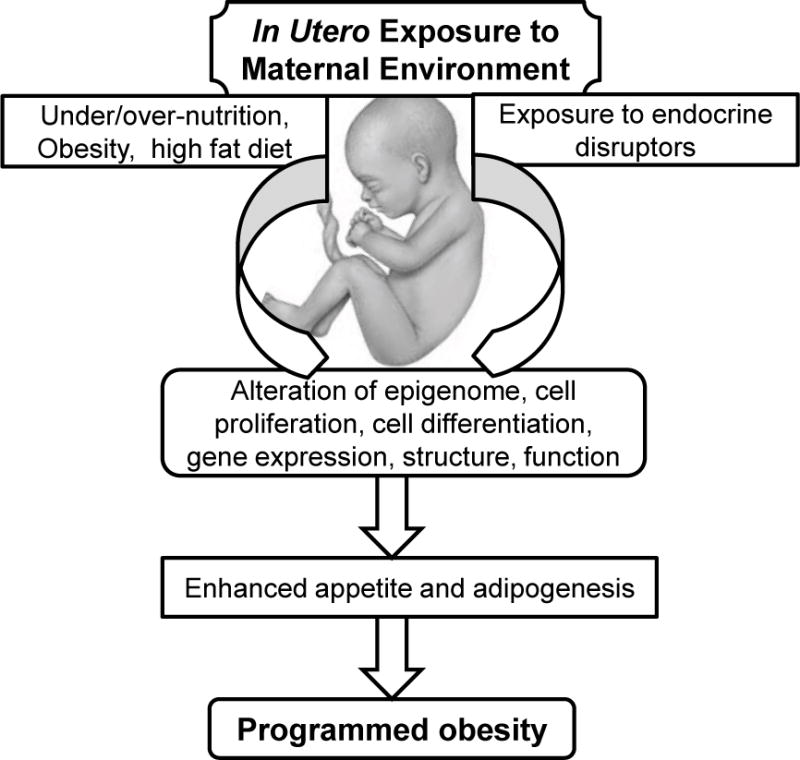
Maternal nutritional and environmental chemical exposure may alter fetal epigenome, stem cell proliferation/differentiation and/or organ structure that ultimately impact appetite and adipose tissue function of her offspring.
Epigenetic phenomena are a fundamental feature of mammalian development that cause heritable and persistent changes in gene expression without altering DNA sequence. Epigenetic regulation includes changes in the DNA methylation pattern, and/or modifications of chromatin packaging via posttranslational histone changes. DNA methylation represents a primary epigenetic mechanism. The DNA of the early embryo is hypomethylated, and with progressive increases in DNA methylation in response to environmental signals, organogenesis and tissue differentiation occurs. Increased methylation is associated with transcriptional silencing. Anomalous DNA methylation may be associated with inappropriate gene silencing. As such, changes in epigenetic marks are associated with multiple human diseases, including many cancers, neurological disorders, and even inflammation. As methylation involves the supply and enzymatic transfer of methyl groups, it is plausible that in utero nutritional, hormonal, or other metabolic cues alter the timing and direction of methylation patterns during fetal development.
Another essential mechanism of gene expression and silencing is the packaging of chromatin into open (euchromatic) or closed (heterochromatic) states, respectively. Chromatin consists of DNA packaged around histones. Post-translational modification of histone tails may potentiate (e.g, acetylation) or suppress (e.g, deacetylation) gene expression.
Finally, microRNAs are emerging as a potential third epigenetic mediator. While these noncoding RNAs are usually associated with regulation of gene expression at the translational level, recent work suggests they may be involved in DNA methylation as well, thereby further regulating transcription of their targets.
Via these series of epigenetic mediators, in utero nutrition, environmental exposures and likely other factors (e.g, stress) may permanently alter offspring gene expression, and the structure and function of cells and organs (Figure 2).
Figure 2. Epigenetics and Programming.

Epigenetic regulation includes changes in the (i) DNA methylation pattern, (ii) modifications of chromatin packaging via posttranslational histone acetylation/deacetylation, and (iii) noncoding RNAs (miRNA) at translation level. Altered nutrition or environmental exposures can modify any of the three epigenetic regulatory factors.
Therapies, Applications and Conclusions
The obesity epidemic represents one of the major public health challenges in the 21st century. Devising effective policy and practice to combat childhood obesity is a high priority for many governments and health professionals. There is irrevocable evidence that departures from optimal growth in utero, whether from limited or excess nutrition, and new evidence that exposure to select environmental toxins, increase the relative risk of adult obesity. Alterations in epigenomics may be a key mechanism by which environmental exposures can influence gene expression, and therefore, phenotype. Future strategies for the prevention and therapy of obesity will ideally address programming effects of the early life environment. Most importantly, it is critical that we recognize that obese individuals truly may have an enhanced appetite and a predisposition to lipid storage and adipogenesis.
Acknowledgments
Our work reported is supported by the National Institutes of Health Grants R01DK081756 and R01HD054751.
Reference List
- 1.Gluckman PD, Hanson MA, Morton SM, Pinal CS. Life-long echoes–a critical analysis of the developmental origins of adult disease model. Biol Neonate. 2005;87(2):127–139. doi: 10.1159/000082311. [DOI] [PubMed] [Google Scholar]
- 2.Simmons R. Perinatal programming of obesity. Semin Perinatol. 2008;32(5):371–374. doi: 10.1053/j.semperi.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115(3):e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 4.Seckl JR, Meaney MJ. Glucocorticoid programming. Ann N Y Acad Sci. 2004;1032:63–84. doi: 10.1196/annals.1314.006. [DOI] [PubMed] [Google Scholar]
- 5.Desai M, Gayle D, Babu J, Ross MG. Programmed obesity in intrauterine growth-restricted newborns: modulation by newborn nutrition. Am J Physiol Regul Integr Comp Physiol. 2005;288(1):R91–R96. doi: 10.1152/ajpregu.00340.2004. [DOI] [PubMed] [Google Scholar]
- 6.Jia Y, Nguyen T, Desai M, Ross MG. Programmed alterations in hypothalamic neuronal orexigenic responses to ghrelin following gestational nutrient restriction. Reprod Sci. 2008;15(7):702–709. doi: 10.1177/1933719108316982. [DOI] [PubMed] [Google Scholar]
- 7.Grove KL, Sekhon HS, Brogan RS, Keller JA, Smith MS, Spindel ER. Chronic maternal nicotine exposure alters neuronal systems in the arcuate nucleus that regulate feeding behavior in the newborn rhesus macaque. J Clin Endocrinol Metab. 2001;86(11):5420–5426. doi: 10.1210/jcem.86.11.8033. [DOI] [PubMed] [Google Scholar]
- 8.Desai M, Gayle D, Babu J, Ross MG. The timing of nutrient restriction during rat pregnancy/lactation alters metabolic syndrome phenotype. Am J Obstet Gynecol. 2007;196(6):555–557. doi: 10.1016/j.ajog.2006.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai M, Gayle D, Han G, Ross MG. Programmed hyperphagia due to reduced anorexigenic mechanisms in intrauterine growth-restricted offspring. Reprod Sci. 2007;14(4):329–337. doi: 10.1177/1933719107303983. [DOI] [PubMed] [Google Scholar]
- 10.Remmers F, Verhagen LA, Adan RA, Delemarre-van de Waal HA. Hypothalamic neuropeptide expression of juvenile and middle-aged rats after early postnatal food restriction. Endocrinol. 2008;149(7):3617–3625. doi: 10.1210/en.2007-1388. [DOI] [PubMed] [Google Scholar]
- 11.Garcia AP, Palou M, Priego T, Sanchez J, Palou A, Pico C. Moderate caloric restriction during gestation results in lower arcuate nucleus NPY- and alphaMSH-neurons and impairs hypothalamic response to fed/fasting conditions in weaned rats. Diabetes Obes Metab. 2010;12(5):403–413. doi: 10.1111/j.1463-1326.2009.01174.x. [DOI] [PubMed] [Google Scholar]
- 12.Plagemann A, Harder T, Rake A, Melchior K, Rohde W, Dorner G. Hypothalamic nuclei are malformed in weanling offspring of low protein malnourished rat dams. J Nutr. 2000;130(10):2582–2589. doi: 10.1093/jn/130.10.2582. [DOI] [PubMed] [Google Scholar]
- 13.Fukami T, Sun X, Li T, Desai M, Ross MG. Mechanism of Programmed Obesity in Intrauterine Fetal Growth Restricted Offspring: Paradoxically Enhanced Appetite Stimulation in fed and Fasting States. Reprod Sci. 2012 doi: 10.1177/1933719111424448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desai M, Li T, Ross MG. Hypothalamic neurosphere progenitor cells in low birth-weight rat newborns: Neurotrophic effects of leptin and insulin. Brain Res. 2011;1378:29–42. doi: 10.1016/j.brainres.2010.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desai M, Li T, Han G, Guberman C, Ross MG. Maternal obesity and increased risk of offspring metabolic syndrome. Reprod Sci Suppl. 2010;17:104A–139. [Google Scholar]
- 16.Desai M, Guang H, Ferelli M, Kallichanda N, Lane RH. Programmed upregulation of adipogenic transcription factors in intrauterine growth-restricted offspring. Reprod Sci. 2008;15(8):785–796. doi: 10.1177/1933719108318597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yee JK, Lee WN, Ross MG, Lane RH, Han G, Vega J, et al. Peroxisome proliferator-activated receptor gamma modulation and lipogenic response in adipocytes of small-for-gestational age offspring. Nutr Metab (Lond) 2012;9(1):62. doi: 10.1186/1743-7075-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desai M, Han G, Li T, Ross MG. Transcriptional regulation of adipogenesis in newborns exposed to maternal obesity. Reprod Sci Suppl. 2010;17:217A–532. [Google Scholar]
- 19.Padmanabhan V, Siefert K, Ransom S, Johnson T, Pinkerton J, Anderson L, et al. Maternal bisphenol-A levels at delivery: a looming problem? J Perinatol. 2008;28(4):258–263. doi: 10.1038/sj.jp.7211913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JQ, Brown TR, Russo J. Regulation of energy metabolism pathways by estrogens and estrogenic chemicals and potential implications in obesity associated with increased exposure to endocrine disruptors. Biochim Biophys Acta. 2009;1793(7):1128–1143. doi: 10.1016/j.bbamcr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]