

Summary
There is well-established variability in the numbers of lipid bodies (LB) in macrophages, eosinophils, and neutrophils. Similarly to the steatosis observed in adipocytes and hepatocytes during hyperinsulinemia and nutrient overload, immune cell LB hyper-accumulate in response to bacterial and parasitic infection and inflammatory presentations. Recently we described that hyperinsulinemia, both in vitro and in vivo, drives steatosis and phenotypic changes in primary and transformed mast cells and basophils. LB reach high numbers in these steatotic cytosols, and here we propose that they could dramatically impact the transcytoplasmic signaling pathways. We compared calcium release and influx responses at the population and single cell level in normal and steatotic model mast cells. At the population level, all aspects of FcεRI-dependent calcium mobilization, as well as activation of calcium-dependent downstream signalling targets such as NFATC1 phosphorylation are suppressed. At the single cell level, we demonstrate that LB are both sources and sinks of calcium following FcεRI cross-linking. Unbiased analysis of the impact of the presence of LB on the rate of trans-cytoplasmic calcium signals suggest that LB enrichment accelerates calcium propagation, which may reflect a Bernoulli effect. LB abundance thus impacts this fundamental signalling pathway and its downstream targets.
Keywords: Lipid bodies, calcium signaling, steatosis, inflammation
1. Introduction
Adipocytes, hepatocytes, skeletal and cardiac myocytes, and pancreatic beta cells accumulate cytoplasmic lipid droplets during obesity and endocrine imbalance or nutrient overload [1-3]. In addition to this pathophysiology, there is also well-established plasticity in the numbers of lipid bodies in macrophages and other immunocytes. Here, lipid bodies (LB) hyper-accumulate in response to bacterial and parasitic infection [4, 5] and other inflammatory presentations, such as arthritic joint leukocytes [6], lavage of patients with acute respiratory distress [7], peripheral blood leukocytes in sepsis [8, 9] and sites of mycobacterial infection [10, 11]. There is also evidence that endocrine inputs relevant to metabolic syndrome and obesity can alter lipid storage in immunocytes. The best-characterized example of this is the accumulation of dietary lipids in foamy macrophages during atherosclerosis, with these lipid droplet pools being sensitive to hormones such as resistin and leptin [12]. Recently we described sensitivity of mast cell lipid body numbers, both in vitro and ex vivo, to insulin, with chronic insulin exposure driving steatosis and marked phenotypic changes in these cells ([13], WEG and HT, manuscript submitted).
There are distinctions at several levels between immunocyte LB and the lipid droplets (LD) of cells such as adipocytes and hepatocytes [14-16]. LB are storage and synthesis sites for bioactive lipid mediators and their precursors [6, 12, 17-20]. LD predominantly store free fatty acids, triacylglycerol and cholesterol for use in oxidative phosphorylation, and their abundance reflects the porting of dietary lipids (palmitate, oleate, etc.) into cells for storage [21, 22]. LB in immunocytes have some commonalities in terms of protein content and lipidome with LD, but also manifest important differences [12, 13, 23-25]. Our studies suggest that even in response to an endocrine input (chronic hyperinsulinemia), the expansion in LB content in mast cells is predominantly comprised of LB that store inflammatory precursors, alongside energy substrates that may also function as precursors for bioactive lipids [13, 24]. This expansion is, unsurprisingly, linked to an elevated potency in antigen mediated de novo synthesis and release of bioactive lipids such as LTC4 [13]. However, other consequences of steatosis in mast cells are less predictable, notably the induction of ER stress and remodeling and the marked suppression of degranulation responses ([13], WEG and HT, unpublished data). Steatotic mast cells are phenotypically different than their normal counterparts, creating a need to more fully understand the impact of steatosis on mast cell signaling.
LB numbers in the steatotic cytosol are striking, and we propose that they could dramatically impact the transcytoplasmic signaling pathways that are necessary for cellular function. The presence of such large numbers of lipid structures seems likely to cause dramatic remodeling of the cytoplasm, with subsequent effects on the integrity of cellular signaling pathways. There are few studies directly addressing this issue, but in steatotic hepatocytes and adipocytes there is intriguing evidence of cytoskeletal remodeling, [26-30] altered calcium dynamics and uncharacterized signaling changes that result in altered functional responses. While cells that exhibit this steatosis have altered functional phenotypes, the mechanistic links between cytosolic LD/LB accumulation and altered cellular signaling and functional responses have not been explored. In the current study we tested the hypothesis that mast cell steatosis would impact calcium signaling dynamics in mast cells.
In mast cells, the generation of a calcium signal is an essential requirement for an array of physiological functions including the production of eicosanoids, the optimal induction of cytokine gene transcription and degranulation in response to antigens or other stimulants [31-34]. A relationship between calcium signalling and steatosis has only been marginally explored in the literature, with one study suggesting altered calcium-dependent contractile signalling in skeletal myocytes with ectopic lipid deposition (ELD), and a study in the porcine system suggesting that ovarian follicle LB act as reservoirs of stored calcium [35, 36]. Moreover, intriguing recent data in the eosinophil system demonstrate that there are ER lamellae within LB, which may imply that the calcium storage functionality of the ER may be transferred, along with the physical structures, to the LB [37]. However, since calcium is central to so many downstream cellular activation events, it seems reasonable to study whether alterations in functional responses could be attributable to LB-mediated disruption of this fundamental second messenger.
In the current study, we performed a comparative analysis of calcium release and influx responses at the population and single cell level in normal and steatotic model mast cells (RBL2H3). At the population level, all aspects of FcεRI-dependent calcium mobilization, as well as activation of calcium dependent downstream signalling targets such as NFATC1 phosphorylation are suppressed. Reflecting either general or targeted disruption of protein synthesis associated with accumulation of lipid in the ER, we note altered expression of calcium handling proteins that will play a role in, in turn, altered shaping of calcium responses. We extended our studies to assess the impact of LB accumulation on calcium dynamics and response characteristics within a single cell, demonstrating that LB can act as both sources and sinks of calcium during an FcεRI-induced response. We document that there is a strong association of LB with long term calcium sinks that emerge in RBL2H3 after FcεRI activation. We performed an unbiased analysis of the impact of the presence of LB on the rate of progress of a transcytoplasmic calcium signal. Cytosol that is heavily occluded with LB displays accelerated calcium waves, which we attribute to a Bernoulli effect. Taken together, these data support the hypothesis that a steatotic and non-steatotic immunocyte display non-equivalent calcium signals in terms of both magnitude and character. LB abundance thus impacts this fundamental signalling pathway and its downstream targets.
2. Materials and Methods
2.1. Cell culture
RBL2H3 were grown at 37 °C, 5% CO2, and 95% humidity in Dulbecco's Modified Eagle's Medium (Mediatech Inc., Herndon, VA) with 10% heat-inactivated Fetal Bovine Serum (Mediatech) and 2mM Glutamine [38].
2.2. Chemicals, Reagents and Stimulations
General chemicals were from VWR (West Chester, PA) and Sigma Aldrich (St. Louis, MO). PMA and Ionomycin were from Calbiochem (Gibbstown, NJ). IgE anti-DNP is from Sigma and KLH-DNP was from Calbiochem. Antibodies were from the following: anti-NFATC1, anti-ITPR1, anti-ITPR2, anti-ITPR3 (InsP3R types I, II and III), anti-SERCA 2a and 2b, anti-PMCA1, Abcam (Cambridge, MA); anti-NFATC1 S54, GeneTex (Irvine, CA); anti-Grb2, Cell Signalling (Danvers, MA); anti-CRACM1, ProSci (Poway, CA). Nile Red, Oil Red O and hematoxylin were from EMD Chemicals (Gibbstown, NJ) and ScyTek Laboratories (Logan, UT) respectively. Alexa- and HRP conjugated secondary antibodies were from Invitrogen (Temecula, CA) and Amersham (Piscataway, NJ). FcsRI stimulation used 0.1μg/ml IgE anti-DNP for 16 hours at 37°C, followed by three washes and the addition of 250ng/ml KLH-DNP for the indicated times. PMA and ionomycin were both used at 500nM. Lipogenesis was induced by incubating RBL2H3 with insulin, dexamethasone and isobutylmethylxanthine (IBMX) at 10μg/ml, 0.01μg/ml, 0.25μM and 2.5μM, respectively for 6 days [39].
2.3. Cell Lysis and Western blot
Cells were pelleted (2000g, 2 min) and washed once in ice cold PBS. For lysates, approximately 107 cells were lysed (ice/30 minutes) in 350μl of lysis buffer (50mM Hepes pH 7.4, 250mM or 75m (see below) NaCl, 20mM NaF, 10mM iodoacetamide, 0.5%(w/v) Triton X100, 1mM PMSF (phenylmethylsulfonylfluoride), 500 mg/ml aprotinin, 1.0 mg/ml leupeptin and 2.0 mg/ml chymostatin). Nuclear extraction was performed as follows: Lysates were prepared in 250mM NaCl-containing lysis buffer and lysates were clarified at 17,000g for 20 min at 4°C, pelleting nuclear material and using the high salt concentration to extract nuclear protein. Cytosolic lysates were prepared as follows: Lysates were prepared in 75 mM NaCl-containing lysis buffer and lysates were clarified at 10,000g for 5 min at 4°C. Lysates were acetone precipitated (1.4 volumes acetone for 1h at −20°C, followed by 10,000g for 5 min). Protein samples were resolved by 10% SDS-PAGE under reducing conditions. Resolved proteins were electro-transferred to PVDF in 192mM glycine, 25mM Tris (pH 8.8). For Western blotting, membranes were blocked using 5% non-fat milk in PBS for 1 hour at RT. Primary antibodies were dissolved in PBS/0.05% Tween-20/0.05% NaN3 and incubated with membranes for 16 hours at 4°C. Developing antibodies comprised anti-rabbit or anti-mouse IgGs conjugated to horseradish peroxidase (Amersham) diluted to 0.1μg/ml in PBS/0.05% Tween-20 and incubated with membranes for 45 minutes at RT. Four washes (5 min PBS/0.1% Tween-20) were performed between primary and secondary antibodies and following secondary antibody. Signal was visualized using enhanced chemiluminescence (Amersham) and exposure to Kodak BioMax film.
2.4. Oil Red O Staining
Cells (50,000 per cm2) coverslips were fixed on coverslips (0.4% (w/v) paraformaldehyde 1h, RT), washed twice with tap water and stained with Oil Red (0.35% in 6:4 EtOH:water, 15 min at RT followed by two dH2O washes). Coverslips were mounted in Crystal-Mount (Electron Microscopy Sciences, Hatfield, PA) for imaging. Live cell experiments comprised 30 min staining at 37°C in media with 4μM Fluo-4 and 0.05% ORO from 5% stock in 70:30 water:EtOH.
2.5. Imaging
Bright field and fluorescence imaging of cells in MatTek dishes (50,000 cells per cm2) were performed on a Nikon Ti Eclipse C1 epi-fluorescence and confocal microscopy system, equipped with heated stage. Available laser lines in FITC, TxRed and Cy5 were supplied by a 488nm 10mW solid state laser, a 561nm 10mW diode pump solid state (DPSS) laser and a 638nm 10mW modulated diode laser. Z stack sizes ranged from 3-8 microns depending on the cell being imaged. Each z disc (optical section) ranged from 0.15-1 micron. Pinhole size for all images was 60 microns. Images were analysed in NIS Elements (Nikon, Melville, NY). Microapplication was accomplished with Eppendorf CellTram Vario and Femtojet microapplicator systems controlled by an Eppendorf micromanipulator (Eppendorf, Hamburg, Germany). Unless otherwise stated images were acquired through a Plan Apo VC 100X 1.40 oil objective (Nikon). Excitation and emission of dyes used are as follows: ORO, Ex 561 nm/Ex 590/50 nm; Fluo-4, Ex 488 nm/Ex 515/30 nm.
2.6. Calcium assay (bulk method)
RBL2H3 were washed and incubated with 0.2μM Fluo-4 [40] for 30 minutes at 37°C in a standard modified Ringer's solution of the following composition (in mM): NaCl 145, KCl 2.8, CsCl 10, CaCl2 10, MgCl2 2, glucose 10, Hepes·NaOH 10, pH 7.4, 330 mOsm. Cells were transferred to 96-well plates at 50,000 cells/well and stimulated as indicated. Calcium signals were acquired using a Flexstation 3 (Molecular Devices, Sunnydale, USA). Data was analyzed using SoftMax® Pro 5 (Molecular Devices). Where indicated, nominally calcium-free external conditions were achieved by the preparation of 0mM CaCl2 Ringer solution containing 1mM EGTA.
2.7. Calcium assay (single cell method)
RBL2H3 were plated on glass coverslip dishes (MatTek, Ashland, MA) and incubated with 1μM Fluo-4 for 30 minutes at 37°C in a standard modified Ringer's solution as described above. After washing, cells were stimulated as indicated on a 37° C heated stage. Calcium signals were acquired using a Nikon Ti Eclipse confocal microscopy system, using EZ C1 software for acquisition and NIS Elements software (Nikon) for analysis. Where indicated, nominally calcium-free external conditions were achieved by the preparation of 0mM added CaCl2 Ringer solution containing 1mM EGTA.
2.8. mRNA analysis
RNA was extracted from RBL2H3 (Qiagen, Hilden, Germany). Agilent RNA Spike-In, One Color Mix (Agilent, Santa Clara, USA) was prepared as instructed by the manufacturer and 100ng RNA was converted into cRNA and labeled with cyanine-3 with Low Input Quick Amp Labelling Kit (Agilent Technologies). Samples were purified using RNeasy mini columns (Qiagen) and cRNA was assessed using Nanodrop (ND-2000C). Using the one-color protocol, equal amounts of Cy3-labeled cRNA (1500 ng) from stimulated and non-treated cells were hybridized in duplicates to Agilent Whole Rat Genome Microarray 4×44K (G4131F) for 17 hrs at 65 °C. The microarray comprised of 45,000 probes, representing >41,000 transcripts. Hybridized microarrays were washed, scanned and analyzed (Agilent G2565CA, Agilent Genespring v11.5). Intensities were scaled to the median of all samples and no baseline normalization was performed. Fold changes with associated p-values were calculated. For statistical analyses, an unpaired t-test was performed, using Benjamini-Hochberg for multiple testing amongst the duplicates.
2.9. Analysis
Results are shown as the mean ± standard deviation. Statistical significance was determined based on Student's t-test or ANOVA. Adjacent to data points in the respective graphs, significant differences were recorded as follows: single asterisk, p < 0.05; double asterisk, p < 0.01; triple asterisk, p < 0.001; no symbol, p > 0.05. Experiments are all n of at least 3.
2.10. Ordination
Principal component analysis (PCA) is the unconstrained ordination technique used to help explain the multiple variables measured during cell stimulation. The graticules are assigned positions on the biplot based a distance matrix calculated using Euclidean distances between graticules. The arrangements of the sites are based on the first two principal components and are the best representation of the distances between the sites as defined in the matrix. The amount of variance shown in each plot is defined by the goodness-of-fit (GOF) of each axis. In addition to the PCA analysis two other generalized additive models (GAM) were overlaid on the biplot; vectors were fitted to each of the independent variables using the envfit function provided in the vegan package in R [41, 42]. The vectors show the direction of maximal correlation and to which direction the vectors most rapidly change from the origin of the biplot. The second GAM uses a contour plot consisting of thin plate splines using the ordisurf function also in the vegan package in R [41], which fits more closely the values in a smoothing function and is linear in the PCA. Each spline is assigned a value of magnitude corresponding to the influence of the explanatory variable in the ordination space.
3. Results
3.1. Suppression of FcεRI-induced calcium fluxes in steatotic mast cells
We have previously established conditions for expansion of the lipid body (LB) pool in primary bone marrow derived mast cells, peripheral blood basophils and in the rat basophilic mast cell line RBL2H3 [38]. The latter are an established context for the study of FcεRI-induced calcium signalling, exhibiting store release from diverse thapsigargin-sensitive and –insensitive stores, store-operated calcium entry via Orai family members and non selective cation channel-mediated calcium influx through TRP family members [32]. Chronic exposure to insulin is a convenient method to induce a steatosis-like accumulation of LB in the cytosol [13], which can be confirmed using Oil Red O (ORO) staining (Figure 1A-C). At the population level (Figure 1D), FcεRI-induced bulk calcium fluxes are suppressed in cells treated with chronic insulin to induce steatosis. Calcium-dependent downstream responses such as nuclear translocation and both serine regulatory region (SRR) [43-45] and Ser54 [46, 47] phosphorylation of the transcription factor NFAT (Figures 1E and F) are also suppressed. ERK phosphorylation, which integrates both kinase and calcium signalling pathways initiated by antigenic cross-linking of FcεRI, is also suppressed in the steatotic cell population (Figure 1G).
Figure 1. Induced lipid body accumulation alters calcium responses and downstream signaling pathways in a RBL2H3.
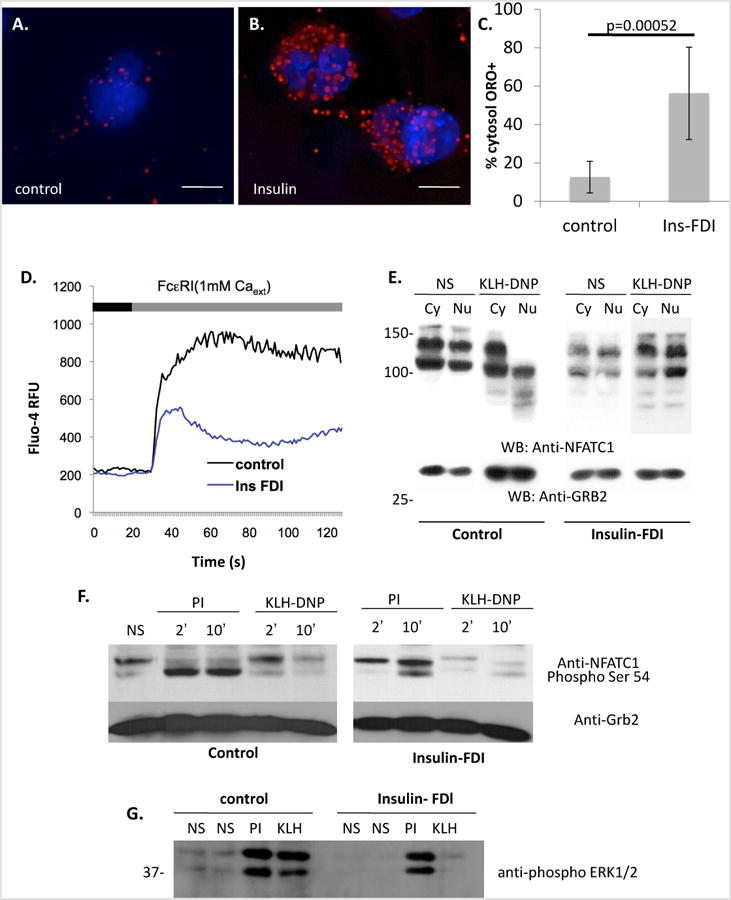
A-C. Induction of lipid body accumulation by chronic insulin exposure. Extended depth of focus (EDF) projections of deconvolved epi-fluorescently imaged z stacks of unstimulated (A) and 6d insulin-exposed (B) RBL2H3 stained with DAPI and Oil Red O (ORO). C. Percentage of cytosolic volume of control and insulin-treated RBL2H3 occupied by ORO-postitive LB structures. Z-stacks of 14 control and 14 treated cells were images and a binary thresholds were placed on the reconstructed images to estimate cell volume, nuclear volume and total volume of ORO-positive structures. Percentage of cellular volume (excluding nucleus) occupied by LB was calculated and is shown here. D. Population-based calcium assay of control and insulin-treated cells (6d) stimulated with IgE and KLH-DNP (200ng/ml) to cross-link the FcεRI. Antigen (black bar) was added after 30s establishment of baseline Fluo-4 signal (grey bar). E. Decreased calcium-dependent dephosphorylation and nuclear localization of NFATC1 in antigen stimulated steatotic cells. NFATC1 phosphorylation status was assessed by Western blot of cytoplasmic and nuclear lysates. NFATC1 phospho-form is lower mobility (higher apparent MW) band while dephospho-form is higher mobility (lower apparent MW) band. These data primarily reflect phopshorylation events occurring within the serine regulatory region (SRR). Calcium-dependent dephosphorylation and nuclear import of NFATC1 was stimulated by FcεRI crosslinking to a lesser degree in Insulin-treated than control cells. Anti-Grb2 Western blot is included as a loading control. F. Comparison of Ser-54 NFATC1 in control and 6d insulin exposed cells. Calcium-dependent phosphorylation of Ser54 was initiated pharmacologically (PMA/Ionomycin, PI) or via FcεRI crosslinking (KLH-DNP). Ser 54 phosphorylation is visible in both the SRR-phospho and SRR-dephospho forms, reflected as different mobility bands. G. Comparison of ERK1/2 phosphorylation in control and 6d insulin exposed cells. Calcium-dependent phosphorylation of ERK1/2 was initiated pharmacologically (PMA/Ionomycin, PI) or via FcεRI crosslinking (KLH-DNP). Each lane contains 10 μg protein confirmed by BCA assay. Steatosis in >80% of the cell population was confirmed in parallel with each experiment microscopically. All experiments n of at least 3.
In the presence of external calcium, FcεRI-induced calcium responses reflect both store release and influx components. We isolated store release responses in normal and steatotic cell populations using nominally calcium-free external solution. Store release responses averaged from cell populations (Figure 2A) exhibit an approximately 37% slower initial rate of development, a 50% decrease in peak magnitude and an integrated area under curve, (AUC, representing total stored calcium) decreased by approximately 66% (Figure 2B). Since the exposure to hyperinsulinemic conditions necessary for induction of steatosis spans multiple days, we asked whether altered expression of major calcium handling proteins could be responsible for altered shaping of calcium responses. While few expression changes were seen at the mRNA level (Figure 2C), Plasma membrane calcium ATPase 1 (PMCA1) transcripts were highly upregulated, possibly in a compensatory effort following down-regulation at the protein level (Figure 2D). SERCA 2b was also downregulated. Significant, possibly compensatory, upregulation in CRACM1 protein (Orai1) is occurring, despite marked suppression of influx responses. Alterations in sarcoplasmic endoplasmic reticulum calcium ATPases (SERCA) and Ins(1,4,5)P3 receptor isotype balances [48] in steatotic cells have been shown to alter the shaping of release responses. Taken together, our data indicate that populations of steatotic cells exhibit markedly suppressed calcium signals, but do not differentiate between altered response shaping due to expression changes in major calcium handing proteins and the biophysical consequences of LB accumulation or occlusion of the cytosol. There is also complexity inherent in altered post-translational modification (i.e. altered mobility in SDS-PAGE of proteins such as INSP3RI II) of calcium handling proteins that is induced by insulin. We further explored the possibility that occlusion of the cytosol with LB is affecting response-shaping.
Figure 2. Suppression of ER store calcium release in insulin-exposed RBL2H3.
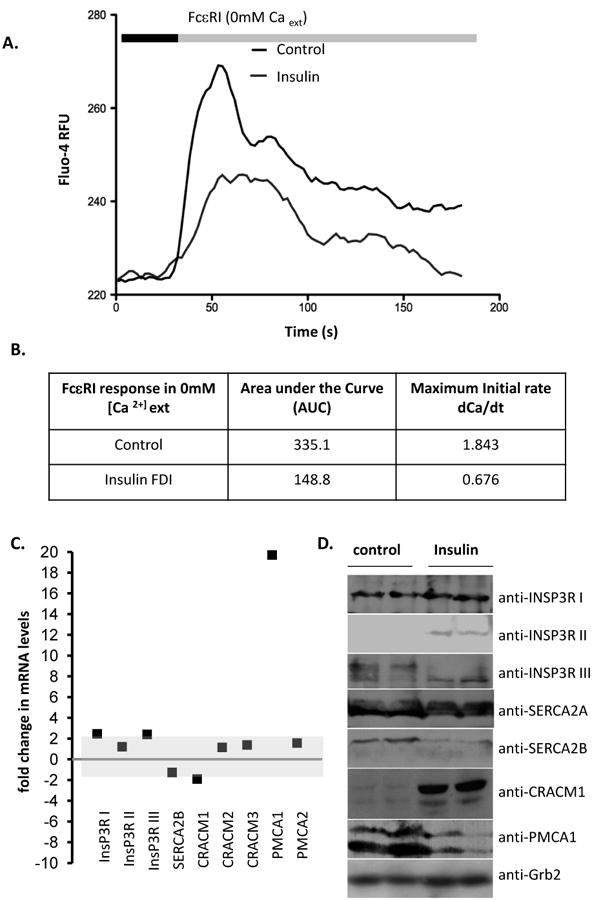
A. Population-based calcium assay of control and insulin-treated cells (6d) stimulated with IgE and KLH-DNP (200ng/ml) to cross-link the FcεRI. Antigen (black bar) was added after 30s establishment of baseline Fluo-4 signal (grey bar). Experiment was performed under nominally calcium free external conditions to isolate store release from influx responses. B. Analysis of AUC and initial rates of calcium mobilization in control and insulin-FDI treated RBL2H3. Values are shown as mean of 3 experiments. P values (control, insulin)=0.0037 (AUC), 0.00098 (rate). RFU, Relative fluorescence units; dCa/dt, rate of change in calcium signal over time. C. Microarray analysis of transcript levels for major calcium handling proteins. Fold changes between control and 6d insulin exposed cell populations are shown. INSP3R I, II, III, inositol (1,4,5) trisphosphate receptors types I, II and III; SERCA, sarcoplasmic endoplasmic reticulum calcium ATPase; CRAC, calcium release activated channel; PMCA, plasma membrane calcium ATPase. D. Western blot analysis of calcium handling protein levels in control and 6d insulin-exposed RBL2H3. Grb2 Western is presented as a loading control.
3.2. Simultaneous live cell imaging of lipid bodies and calcium at the single cell level
We established a methodology to visualize both calcium responses and LB in live cells. ORO accumulation in discrete LB in live cells was achieved after a period of optimization of staining conditions (see Methods). Figure 3A shows that discrete ORO+ areas of cytoplasm may be visualized in cells co-stained with Fluo-4. At higher magnifications, a single confocal z disc through an ORO+ area of cytosol shows the distinctive lobed/spherical droplet appearance of LB clusters [12, 13, 25, 37, 49] (Figure 3A, inset). ORO-rich areas corresponding to LB may display significant Fluo-4 associated signals (Figure 3B) but this is not uniformly the case. Under conditions of cellular stimulation, while Fluo-4 fluorescence intensity increase as expected, ORO intensity remains stable (Figure 3C), suggesting that this method can be used to examine ongoing cellular responses.
Figure 3. Live cell concurrent imaging of Fluo-4 and Oil red O.
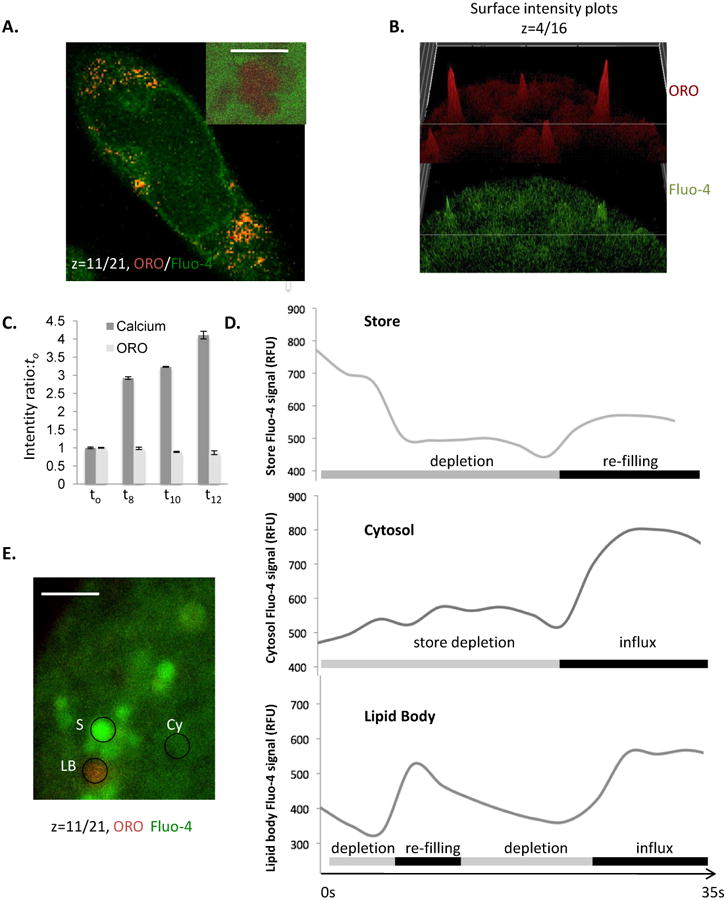
A. Confocal z disc (main panel) of Fluo-4/ORO co-staining in live cells, demonstrating simultaneous imaging of LB and intracellular free calcium levels. Scale bar is 10 microns. Inset shows high resolution image of large lipid body cluster (scale bar is 0.5 microns) where a single z-disc has bisected the LB cluster, again co-stained with ORO and Fluo-4 in a live cell. B. Intensity surface projections of ORO (upper panel) and Fluo-4 (lower panel) fluorescence intensity of a diverse set of LB in RBL2H3. Panels are presented to illustrate that LB may show intense staining in one or both of these fluorescence channels, indicating that some but not all LB are Fluo-4/Calcium enriched relative to others and the cytosol. C. Intensity ratio of Fluo-4 (calcium) and ORO fluorescence signals normalized to the t0 timepoint and charted at the indicated times (in s) after addition of antigen to the cells. Averaged signals from ROI corresponding to z discs consistently taken at 50% of the maximum cell height of the cell bodies of 55 individual cells were used to calculate these averages prior to normalization. D and E. Averaged Fluo-4 fluorescence intensity profiles for 25 ROI per trace of 1 micron diameter drawn around either apparent calcium stores, cytosol or ORO-positive LB during the first 35s of cellular exposure to FcεRI cross-linking (D). Store (S), Cytosol (CY) and lipid body (LB); exemplar ROI are shown in E.
We examined the Fluo-4 fluorescence over time in regions of interest (ROI) selected to characterize the calcium flux dynamics of normal cytosol, pre-existing calcium stores and the lipid bodies themselves (Figure 3D). The traces in Figure 3D represent 25 ROI of matched area that were placed over structures defined as follows: Pre-existing stores (upper panel) were punctate/vesicular structures having baseline Fluo-4 intensity at least 5 fold elevated over the bulk cytosol; Cytosol (middle panel) was defined as extra-nuclear ROI with a starting Fluo-4 intensity less than 50% of the whole cell starting average intensity; Lipid body (lower panel) were defined as ORO+ structures. Figure 3E shows an example of these different compartment ROIs; store, cytosol and lipid body in one z-plane. Figure 3D shows that, following antigen application, across a 35s time course, averages of 25 ROI show depletion and some refilling in store ROI, and cytosol ROI display slight elevations during the phase of store depletion then large increases in Fluo-4 intensity once store-operated influx occurs. LB ORO+ areas, averaged across 25 ROI, display a complex profile of net depletion, followed by refilling and a second phase of depletion. During the influx phase these structures tend to reach a sustained high level of Fluo-4 intensity. Taken together these data indicate that LB do have a pattern of calcium response that is regulated by antigen receptor signalling pathways and that does not simply mirror that of the bulk cytosol or conventional stores. However, we noted that each individual LB response was highly variable, creating a need to widen the analyses beyond the ROI-based approach.
3.3. Whole cell correlation of calcium response characteristics with lipid body location
We developed a whole cell-based, unbiased approach for studying the relationship between LB and calcium flux. Single cell imaging experiments, under conditions of antigen stimulation, were conducted using our live cell ORO protocol. Figure 4A shows Fluo-4 and ORO localization in a representative cell at rest and after 300s stimulation. During analysis, the field of view (comprising one 150nm z disc at 100x resolution of a single cell) was divided into several hundred graticule ROI of equal size (1 square micron), shown as an example in the right panel of Figure 4A. Each of these graticules was analysed for its fluorescence profile in both the Texas Red (ORO) and FITC (Fluo-4) channels over time. This approach essentially divides this single cell into 756 distinct calcium assays performed with data collected over 232 timepoints. Analyses of this type were conducted for 6 cells. Figure 4B looks at the overall trend of Fluo-4 response as the mean of ∼3000 graticules across 6 cells. Graticules were scored as OROlo (in the lowest 25% of initial ORO fluorescence intensity) or ORO hi (in the highest 25% of initial ORO fluorescence intensity), and the mean Fluo-4 intensity of these selected graticules were plotted over time. OROlo cytosol tends to exhibit a classic antigen-induced calcium response. However (Figure 4B), across this large data set OROhi areas of cytosol tend to exhibit slowed and attenuated initial calcium responses in the 0-200s time frame. However, sustained Fluo-4 intensity (from 3000 graticules across 6 cells) is significantly (p<0.0005 from ANOVA) higher in OROhi versus OROlo areas of cytosol, leading us to hypothesize that LB may act as long term sinks of calcium similar to the functions postulated for the Golgi apparatus and mitochondria in previous studies [50-52].
Figure 4. Unbiased analysis of calcium signalling in ORO-positive and ORO-negative subcellular locations.


A. Example of graticule ROI positioning over a single live cell (pre-treated with insulin at 10 μg/ml for 6d to induce LB formation) stained with both Fluo-4 and ORO. Left panel is at 0s and right panel at 300s after stimulation. B. Mean Fluo-4 intensity changes over a 450s time course following antigen stimulation at 20s. Black trace. Averaged signal from graticule ROI within 0- 25th percentile ORO fluorescence intensity (ORO lo). Grey trace. Averaged signal from graticule ROI within 75th-100th percentile ORO fluorescence intensity (ORO hi). C. Conditional formatting visualization of 3 exemplar (A, B and C) cells with ORO and Fluo-4 fluorescence intensities scored over time (vertical) for individual graticules (horizontals). OROhi graticule ROI (see above) were formatted red, Fluo-4 signals in excess of 2 fold over initial intensity were formatted blue. D. Time series of Fluo-4 (488nm) and ORO (568nm) imaging either merged (upper panels) or Fluo-4 alone (lower panels) at the indicated times (in s). Antigen stimulus was added at 20s. Sustained high intensity Fluo-4 signal is notable in areas corresponding to high ORO staining. Each panel is one confocally-acquired z disc and is 12 microns in width. Arrow indicate areas of long term elevation in Fluo-4 fluorescence. E. Multivariate analysis of graticules during three different phases of FcεRI-induced calcium signaling. Principal component analysis (PCA) biplots are shown for 18sec, 84sec, and 374sec that represent pre-stimulated, peak, and sustained phases of calcium signal, respectively. Individual circles represent each graticule (ROI) mapped over the model cell during the stimulation the time course. Species vectors and thin plate splines are mapped across the plot in order to visualize the signal gradients (red = Oil Red O, blue = Fluo-4). The magnitude of the splines is rendered with each smoothing line and the directional magnitude of the vectors follows from the biplot origin to the direction of highest influence. Non-cell graticules (GR191 & GR512), border graticules (GR200 & GR382), cellular non-lipid body graticules (GR350 & GR490) and cellular lipid body graticules (GR119, GR150 & GR524) are labeled in all three time points in order to follow the relationships in those different regions. We expect to see graticules with higher ORO signals to have corresponding higher Fluo-4 signals and for those signals to be maintained after stimulation which would show evidence of a calcium sink. We also expect graticules from similar regions to be plotted closer to each other in the biplots.
Figure 4C looks at three cells, with data presented as ORO and Fluo-4 intensities over time (each row is a 2s timepoint) with each vertical column representing one graticule square (500-800 in total). Conditional formatting reflects the highest 25% of ORO intensity (red) and Fluo-4 intensity exceeding 2 fold of baseline levels (blue). In this representation of the data set, there are notable instances where high ORO staining correlates with sustained elevation in Fluo-4 intensity, suggesting that some, but not all, LB structures sequester calcium following FcεRI-induced flux. Figure 4D represents this idea spatially. Antigen-induced influx peaks at 30-60s, declining to a sustained level at 200-400s (cf Figure 4B). However, some areas of the cytoplasm, corresponding to ORO-stained LB, maintain bright Fluo-4 staining from 200-400s, suggesting that calcium levels may remain high in those regions after PMCA and SERCA have depleted the bulk of the cytosol.
Unconstrained ordination analysis (principal component analysis (PCA), see Methods) was performed in R [41] to test and visualize the relationships of each graticule linked to the multivariate ORO and Fluo-4 data. Each graticule was treated as a dependent variable linked to the independent variables of Fluo-4 and ORO intensities (explanatory variables). Figure 4E is representative of results gathered from the other analyses. Data from 756 graticules mapped on Cell 6 (Figure 4A) were used for calculating ordination biplots. As Figure 4A illustrates, graticules are mapped over diverse regions imaged during the cell stimulation experiments. The non-cell and border regions mapped within the micrograph served as a control for the observations within the cell. These outlying graticules were expected to plot in distinctly different spaces and show little relationship or be influenced by the explanatory variables.
Three different phases of the calcium response were analysed using the ordination model described in Methods: pre-stimulation, peak, and sustained. The phases for a representative cell were measured at time points 18 sec, 84 sec, and 374 sec for pre-stimulation, peak, and sustained, respectively (cf Figure 4B). Each circle in the plot represents a graticule that has a Fluo-4 and ORO value associated with it. At 18 sec the Oil Red O signal has the greatest influence over the cellular graticules in the lower left quadrant of the plot. Component 1 explains 82% and component 2 explains 18% of the variance in this ordination model with a P<0.001 after 999 permutations. Oil Red O stains the lipid bodies inside the cell and is independent of the stimulus; this signal is expected to be consistent during the entire time course. The low background of free calcium is apparent in the splines and might be influential in the lower left side (e.g. GR119, GR150, GR524) positioning of the cellular graticules (e.g. GR350, GR490). The non-cellular graticules (e.g. GR191, GR512) fall to the right side quadrants and the origin, whereas border graticules (e.g. GR200, GR382) are most likely influenced by proximate signal from the edge of the cell. These border graticules plot to the left of the non-cellular sites in the pre-stimulated cell.
At 84 sec after antigen exposure the calcium signal peaks. The influence of this signal with the ordination of the graticules when compared to the pre-stimulated cell is pronounced. The biplot is spread across the left side quadrants compared to the more compact pre-stimulated biplot. The graticules with the greatest ORO (GR150, GR150, GR524) and Fluo-4 signals are maintained in the lower left quadrant and are lightly influenced along the Fluo-4 signal gradient. The border graticules (GR200 and GR382) become more closely associated with the non-cellular (GR191 and GR512) graticules as the fluorescent signals from the cell are increased. The non-cellular graticules remain constant on the opposite side of the plot as the cellular ones as there is no significant signal associated with them (component 1 explains 99% and component 2 explains 1% of the variance, P<0.001 after 999 permutations).
At 374 sec the sustained phase is observed. There is a larger diversity in the two different fluorescent signals, which is defined by a broader spread of points to the left of the origin in the ordination space. The graticules to the left of the origin have spread as the influence of the Fluo-4 signal sinks and sustains with the ORO signal (component 1 explains 98% and component 2 explains 2% of the variance, P<0.001 after 999 permutations). GR150 and GR524 are demonstrative of a calcium sink in this system. Comparatively, GR490 and GR350 are cellular sites with a diminishing Fluo-4 signal. Interestingly GR119 is a site with a small ORO signal and did not exhibit calcium sink, which is consistent with other analyses (Figure 4C), and so is plotted with the other cellular graticules. In all three time points the trajectories of the ORO and Fluo-4 vectors were nearly synonymous, which infers a relationship between these two signals and the influence over the positions of the graticules. Following the diminution of the generalized Fluo-4 signal in the cellular graticules, several of the left quadrant points are maintained in a position of high ORO and Fluo-4 signal that strongly suggests a correlation to calcium retention and LB as indicated by the high value plate splines and vector trajectories.
3.4. Influence of LB enrichment upon calcium signal progression through the cytosol
LB occur in large clusters in steatotic cells, and their enrichment would seem likely to affect the progress of a small highly mobile second messenger as it moves through the soluble phase of the cytosol [13, 14, 25, 26, 41, 53]. We chose to examine the influence of LB enrichment on the progress of a calcium signal under carefully selected conditions. We used a micromanipulator controlled micropipette for pulsed directional application of ∼ 1 μl of a sub-optimal concentration of antigen to a discrete region of membrane in the cell body. The stimulus was applied perpendicularly to the cell (Figure 5A left panel). We examined calcium signal progression in a process emanating from the cell body, where the cytosol is at its thinnest (∼1 micron depth). We were able to resolve images exemplified by Figure 5A, where Fluo-4 intensity progressively increases along the cell process between 0 and 8s of antigen exposure. For analysis, the cellular process was placed under an ROI graticule and 16 horizontal vectors were defined, each 2 graticule squares deep. As in Figure 4, conditional formatting was used to display the intensity profile of the process in terms of Fluo-4 and each graticule was scored for ORO abundance based on the sum of OROhi pixels in the vector. The rate of the front (defined as attaining >2 fold the initial intensity) of calcium progression was measured for each vector, and a range of rates from 1.96-3.57 μm•s-1 were noted. The vectors were rank-ordered for ORO/LB abundances and rate of calcium progression (Figure 5B), showing a high level of apparent agreement between a higher velocity along a vector and a higher abundance of LB. This relationship is plotted in Figure 5C.
Figure 5. Influence of LB enrichment on progress of a cytosolic calcium signal.
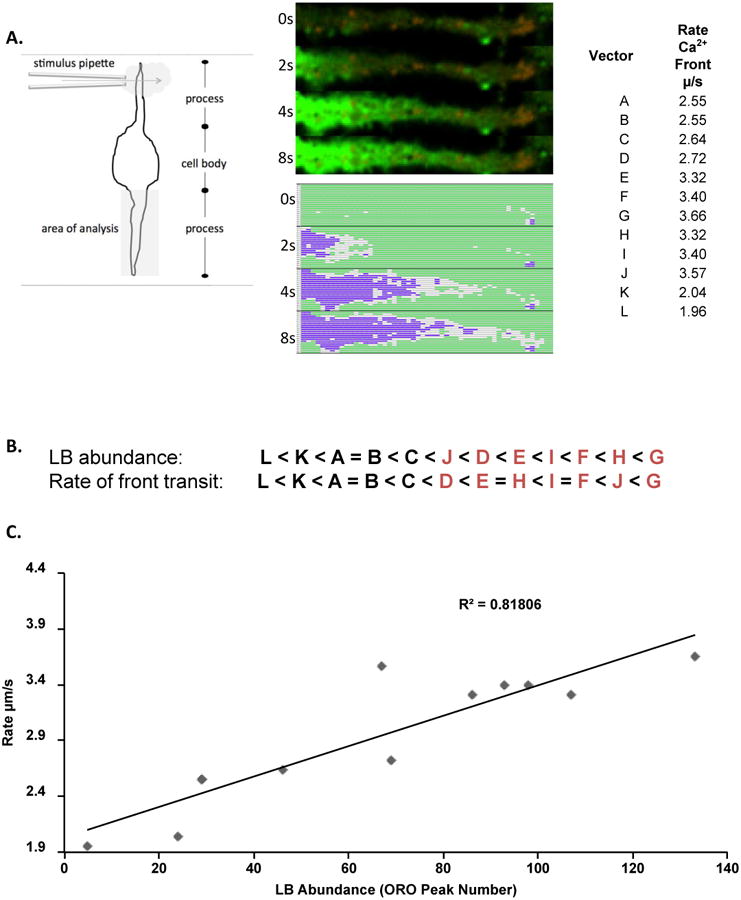
A. Vector-based analysis of calcium signal propagation. Left panel. Schematic of experimental and analysis set-up. Center Panel. Upper panel shows single z disc of cellular process concurrently imaged for Fluo-4 and ORO in a live cell format. Sequential images at 0, 2, 4, and 8s after cessation of antigen application are shown vertically as raw images or conditionally formatted as in Figure 4. For analysis image was divided into longitudinal vectors (designated A-L). Right panel. Measured rates of calcium signal progression. B. Rank order of vectors based on LB abundance (averaged ORO intensity) and rate of calcium signal progression. Letters in red indicate vectors with the highest ranked prevalence of ORO-positive pixels. C. Correlation analysis of LB abundance and rate of signal progression in individual vectors.
4. Discussion
Lipid droplets are conserved structures used for energy storage across multiple phyla [1, 12, 17, 54]. Their numbers are variable, responding to hormonal and nutrient inputs. Lipid droplets have also been co-opted for more specialized purposes, including hormone and lipid soluble cofactor storage, and as cytoplasmic reaction vessels for lipophilic biochemistry. Lipid bodies (LB) are common in immune system cells, for both energy storage and the storage and synthesis of bioactive lipid precursors [12, 17, 19, 20, 55, 56]. Specifically in granulocytes, the LB plays various roles, as sites for storage and synthesis of eicosanoids, prostanoids and their precursors, as sites for protein synthesis and as inducible organelles [12, 20, 23, 57, 58]. In the former role, the number of LB is a clear determinant of the intensity of receptor-induced release of bioactive lipid mediators such as the pro-inflammatory eicosanoids.
Lipid droplets are implicated in pathology, with the nutrient overload that underlies epidemic obesity in Western societies causing their supra-physiological accumulation both in terms of abundance and in terms of their build-up in non-physiological, ectopic locations [1, 21, 59, 60]. This steatosis has a physiological counterpart, with accumulations in granulocyte LB occurring in response to infection and other immune stimuli [12]. Thus there are multiple physiological and pathophysiological situations in which LB accumulate to high levels in the cytosol. This paper explores one consequence of that accumulation.
We used an insulin-induced steatosis model [13] in a mast cell line to explore the consequences of LB accumulation for a signalling pathway that is central to immune cell activation; the mobilization of intracellular free calcium. Both calcium release from Ins (1,4,5) P3 dependent intracellular stores and store-operated influx responses are markedly suppressed in populations of steatotic cells. At the population level, it is not straightforward to distinguish whether some characteristics of the steatosis-associated calcium response are due to alterations in expression of calcium handling proteins that we observe. However, alterations in the initial rate of store release, given that InsP3R expression levels are largely unaffected by the pro-steatotic stimulus may reflect an effect of LB accumulation. Similarly, the decreased calcium storage capacity of the ER has been postulated in the heavily steatotic obese liver. The Hotamisligil laboratory described that the ER, from which lipid droplets in adipocytes and hepatocytes may be formed, becomes lipid engorged in obesity, and that this engorgement while increasing its physical size, diminishes the capacity of the ER to store calcium [61, 62]. We also observed diminished calcium storage capacity in the ER (decrease area under the curve of antigen-induced calcium responses in nominally calcium free media, Figure 2A) and its lipid overload (WEG and HT, manuscript in submission) and suggest that the wide-ranging perturbations in ER function documented in obesity are reshaping cellular responses to calcium-mobilising stimuli [61-63].
The impact of large scale occlusion of the cytosol with LB is likely to be best studied at the single cell and subcellular level. We developed a protocol for concurrent imaging of Fluo-4 and LB in live cells. Live cell staining with ORO has a higher background and more structural variation than in PFA fixed cells, and the clear spherical architecture of the ORO-stained bodies is less apparent [13, 25, 53]. In the absence of fixation, these LB appear to present different surface tension characteristics, and their diversity in shape agrees more closely with our EM observations (HT and WEG, manuscript in submission) and those of other groups published recently [37]. A further consideration is that ORO does not stain all the LB in a mast cell, since we have recently shown that ORO+ LB and those that stain positively with reagents such as filipin, lipidtox and nile red are not completely overlapping populations (WEG and HT, manuscript submitted), nevertheless, live cell ORO staining does differentiate a clear LB population and provides a counterpoint for the analysis of calcium signalling in and around these structures.
Under conditions of cellular stimulation, Fluo-4 in LB is able to detect alterations in localized free calcium levels. While we have no way of knowing whether the kinetics of calcium binding to Fluo-4 are altered by the lipidic environment [40], we do not note marked changes in Fluo-4 baselines between LB and cytosol. Moreover, our limited studies of the effect of ORO mixing and bulk lipid on Fluo-4 excitation/emission spectra and calcium-responsiveness in a plate reader assay system do not suggest that Fluo-4 is behaving drastically differently in a lipid or ORO-enriched environment. However, it is not possible to actually mimick the true LB composition in this context. We do note that Fluo-4 signals dynamically change in LB in response to stimuli such as antigen (Figure 3), thapsigargin (data not shown) and ionomycin (data not shown). These data suggest that the basic function of Fluo-4 as a reporter of calcium increase is intact in these environments, but we do not have a way of calibrating its responsiveness to absolute changes in free calcium concentration. Similarly Fluo-4 is sensitive to large fluctuations in its microenvironmental pH, which is an unknown in the context of an LB. With these caveats, we discern a range of Fluo-4 characteristics in ORO+ structures, including those that that appear to have lower, equivalent or higher Fluo-4 signals at rest than the surrounding cytosol. There are some ORO positive structures from which Fluo-4/Ca appear excluded at rest, and conversely there are instances where at rest OROhi structures have higher Fluo-4 fluorescence than the surrounding cytosol. Similarly, during the initial phases of a calcium response following antigen stimulation, LB may respond with net depletion or apparent accumulation. Once the influx phase of the calcium response is underway, LB appear flooded with calcium with the bulk of the cytosol, and their behaviour is not separable from the averaged signal in ORO- regions. However, after the decline of the release/influx phases of calcium mobilization, the behaviour of ORO+ regions again strongly segregates itself from the bulk of the cytosol. Some, but again not all, ORO+ regions show strong, sustained, retention of high Fluo-4 fluorescence intensity following FcεRI stimulation.
The sustained elevation in Fluo-4 fluorescence in ORO+ LB regions may reflect them acting as long-term calcium sinks. This idea has functional and mechanistic implications. First, if LB do store calcium relative to basal cytosolic levels, then their abundance in the cytosol would be predicted to impact basal and initial calcium responses. If they act to absorb calcium from the cytosol then their increased abundance would tend to modify calcium gradients between stores and cytosol, and contribute to the reabsorption of calcium from cytosol into stores/sinks that follows depletion or influx. Second, assuming that their stored calcium can be accessed, the presence of loaded sinks after an initial stimulus would be predicted to change and shape subsequent responses to repeat stimuli. Long term sinks of calcium have been previously characterised as mitochondria and Golgi vesicles, and LB may now be added to that group [50-52]. Barba et al examined cardiomyocytes in which ectopic lipid deposition was induced and the subsequent effects upon myocyte calcium signals and contractility [35]. Interestingly, they noted that after ischemia-reperfusion, there was a net accumulation of calcium in lipid droplet positive regions, supporting the idea of these structures as sinks [35].
Mechanistically, these data raise intriguing questions. We considered how LB might store or sequester calcium. The unilamellar phospholipid monolayer that is postulated to encapsulate LB might bind calcium (e.g. 2 calcium ions per phosphatidylcholine [64]), but we do not see evidence that calcium binding is localized to the LB periphery. Moreover, a study in ovarian follicles suggested that calcium phosphate deposits are localized within the core of lipid droplets [36]. A recent study suggests another intriguing possibility: Melo et al suggest that ER cisternae are present internally to eosinophil LB, reflecting the biogenesis of the LB from the ER itself [37]. If these ER cisternae retain calcium storage capacity, then they may contribute to the phenomena we observe. This is an attractive possibility also to explain how LB calcium stores might exhibit apparent depletion and uptake characteristics that are reminiscent of bona fide calcium stores (data not shown), which could argue for the presence of pump and channel molecules that it would be hard to visualize residing in the unilamellar phosphatidylcholine shell that encapsulates LB. Proteomic data support the presence of such proteins and it may be premature to dismiss them as reflecting contamination of LB preparations [23, 65-68].
Our data suggest that one manner in which LB accumulation would affect calcium signalling is through the sudden appearance of an abundant new population of calcium sinks. However, the possibility that a cytosol occluded with LB may display altered calcium signal propagation should also be considered. We approached this question by examining calcium signal propagation in z discs imaged along thin (∼1 micron depth) cellular processes, following acute localized stimulation of the cell using a micropipette positioned at the farthest point of the cell from the process. For analysis we defined longitudinal vectors along the process, and we examined the behavior of a calcium signal front in vectors with higher and lower levels of LB along their axis. Interestingly, the apparent rate of propagation was faster (r2 = 0.82) in LB-enriched areas relative to the unenriched cytosol. The functional significance of this level of change cannot be known at this point and these rates cannot be taken as literal because they reflect also the kinetics of the Fluo-4 dye binding to calcium. Mechanistically, these data could reflect a Bernoulli effect where the cytosol is compressed and under higher pressure in the areas between LB, leading to faster fluid dynamics in those narrowed conduits for calcium ions behaving as a liquid moving through an aqueous phase.
In summary, LB hyper-accumulation in immunocytes is both a physiological adaptation to immunological challenge and a feature of nutrient overload. This accumulation of LB affects both the intensity and characteristics of calcium responses stimulated by antigen exposure, and alters downstream, functional responses in these cells. LB may variously act as sources and sinks of calcium, adding complexity to our understanding of calcium dynamics.
Highlights.
Obesity and hyperinsulinemia are significant health burdens globally
Lipid body accumulation at the cellular level characterizes these disorders
The effect of lipid body accumulation on transcytoplasmic signal transduction is studied here
The presence of large numbers of lipid bodies alters calcium signaling dynamics in the cytoplasm of an immunocyte
Acknowledgments
This work was funded by the Victoria and Bradley Geist Foundation of the Hawaii Community Foundation (grant 45408), the National Institutes of Health P20MD006084, the NIH INBRE 2P20GM103466 and the NSF EPSCOR EPS-0903833 (all to HT) and the NIMHD-RCMI- BRIDGES G12 MD007601 (AJS).
Footnotes
Authorship. WEG, HT, LMNS, NP and CS performed experiments. MS designed and implemented analytical models. HT, MS, and AJS wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149:942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- 2.Garbarino J, Sturley SL. Saturated with fat: new perspectives on lipotoxicity. Current opinion in clinical nutrition and metabolic care. 2009;12:110–116. doi: 10.1097/MCO.0b013e32832182ee. [DOI] [PubMed] [Google Scholar]
- 3.Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annual review of biochemistry. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrade ZA, Reed SG, Roters SB, Sadigursky M. Immunopathology of experimental cutaneous leishmaniasis. American Journal of Pathology. 1984;114:137–148. [PMC free article] [PubMed] [Google Scholar]
- 5.Keyel PA, Tkacheva OA, Larregina AT, Salter RD. Coordinate stimulation of macrophages by microparticles and TLR ligands induces foam cell formation. Journal of immunology. 2012;189:4621–4629. doi: 10.4049/jimmunol.1200828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bozza PT, Payne JL, Morham SG, Langenbach R, Smithies O, Weller PF. Leukocyte lipid body formation and eicosanoid generation: Cyclooxygenase-independent inhibition by aspirin. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11091–11096. doi: 10.1073/pnas.93.20.11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Triggiani M, Oriente A, de Crescenzo G, Rossi G, Marone G. Biochemical functions of a pool of arachidonic acid associated with triglycerides in human inflammatory cells. Int Arch Allergy Immunol. 1995;107:261–263. doi: 10.1159/000236997. [DOI] [PubMed] [Google Scholar]
- 8.Pacheco P, Bozza FA, Gomes RN, Bozza M, Weller PF, Castro-Faria-Neto HC, Bozza PT. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular Loci involved in eicosanoid metabolism. Journal of immunology. 2002;169:6498–6506. doi: 10.4049/jimmunol.169.11.6498. [DOI] [PubMed] [Google Scholar]
- 9.Pacheco P, Vieira-de-Abreu A, Gomes RN, Barbosa-Lima G, Wermelinger LB, Maya-Monteiro CM, Silva AR, Bozza MT, Castro-Faria-Neto HC, Bandeira-Melo C, Bozza PT. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. Journal of immunology. 2007;179:8500–8508. doi: 10.4049/jimmunol.179.12.8500. [DOI] [PubMed] [Google Scholar]
- 10.Mattos KA, D'Avila H, Rodrigues LS, Oliveira VG, Sarno EN, Atella GC, Pereira GM, Bozza PT, Pessolani MC. Lipid droplet formation in leprosy: Toll-like receptor-regulated organelles involved in eicosanoid formation and Mycobacterium leprae pathogenesis. J Leukoc Biol. 2010;87:371–384. doi: 10.1189/jlb.0609433. [DOI] [PubMed] [Google Scholar]
- 11.Mattos KA, Oliveira VG, D'Avila H, Rodrigues LS, Pinheiro RO, Sarno EN, Pessolani MC, Bozza PT. TLR6-driven lipid droplets in Mycobacterium leprae-infected Schwann cells: immunoinflammatory platforms associated with bacterial persistence. Journal of immunology. 2011;187:2548–2558. doi: 10.4049/jimmunol.1101344. [DOI] [PubMed] [Google Scholar]
- 12.Melo RCN, D'Avila H, Wan HC, Bozza PT, Dvorak AM, Weller PF. Lipid Bodies in Inflammatory Cells: Structure, Function, and Current Imaging Techniques. Journal of Histochemistry & Cytochemistry. 2011;59:540–556. doi: 10.1369/0022155411404073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greineisen WE, Shimoda LM, Maaetoft-Udsen K, Turner H. Insulin-containing lipogenic stimuli suppress mast cell degranulation potential and up-regulate lipid body biogenesis and eicosanoid secretion in a PPARgamma-independent manner. J Leukoc Biol. 2012;92:653–665. doi: 10.1189/jlb.0811406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beller M, Thiel K, Thul PJ, Jackle H. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett. 2010;584:2176–2182. doi: 10.1016/j.febslet.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 15.Brown DA. Lipid droplets: proteins floating on a pool of fat. Curr Biol. 2001;11:R446–449. doi: 10.1016/s0960-9822(01)00257-3. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Cordes KR, Farese RV, Jr, Walther TC. Lipid droplets at a glance. Journal of cell science. 2009;122:749–752. doi: 10.1242/jcs.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bozza PT, Magalhaes KG, Weller PF. Leukocyte lipid bodies - Biogenesis and functions in inflammation. Biochimica et biophysica acta. 2009;1791:540–551. doi: 10.1016/j.bbalip.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bozza PT, Melo RC, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther. 2007;113:30–49. doi: 10.1016/j.pharmthera.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Dvorak AM, Dvorak HF, Peters SP, Shulman ES, MacGlashan DW, Jr, Pyne K, Harvey VS, Galli SJ, Lichtenstein LM. Lipid bodies: cytoplasmic organelles important to arachidonate metabolism in macrophages and mast cells. Journal of immunology. 1983;131:2965–2976. [PubMed] [Google Scholar]
- 20.Weller PF, Dvorak AM. Lipid bodies - Intracellular sites for eicosanoid formation. Journal of Allergy and Clinical Immunology. 1994;94:1151–1156. doi: 10.1016/0091-6749(94)90325-5. [DOI] [PubMed] [Google Scholar]
- 21.Badimon L, Storey RF, Vilahur G. Update on lipids, inflammation and atherothrombosis. Thrombosis and haemostasis. 2011;105(Suppl 1):S34–42. doi: 10.1160/THS10-11-0717. [DOI] [PubMed] [Google Scholar]
- 22.Ouimet M, Marcel YL. Regulation of Lipid Droplet Cholesterol Efflux From Macrophage Foam Cells. Arteriosclerosis Thrombosis and Vascular Biology. 2012;32:575–581. doi: 10.1161/ATVBAHA.111.240705. [DOI] [PubMed] [Google Scholar]
- 23.Cermelli S, Guo Y, Gross SP, Welte MA. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr Biol. 2006;16:1783–1795. doi: 10.1016/j.cub.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 24.Dichlberger A, Kovanen PT, Schneider WJ. Mast cells: from lipid droplets to lipid mediators. Clinical science. 2013;125:121–130. doi: 10.1042/CS20120602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dichlberger A, Schlager S, Lappalainen J, Kakela R, Hattula K, Butcher SJ, Schneider WJ, Kovanen PT. Lipid body formation during maturation of human mast cells. Journal of lipid research. 2011;52:2198–2208. doi: 10.1194/jlr.M019737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brasaemle DL, Wolins NE. Packaging of fat: an evolving model of lipid droplet assembly and expansion. The Journal of biological chemistry. 2012;287:2273–2279. doi: 10.1074/jbc.R111.309088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spandl J, White DJ, Peychl J, Thiele C. Live cell multicolor imaging of lipid droplets with a new dye, LD540. Traffic. 2009;10:1579–1584. doi: 10.1111/j.1600-0854.2009.00980.x. [DOI] [PubMed] [Google Scholar]
- 28.Takenouchi T, Miyashita N, Ozutsumi K, Rose MT, Aso H. Role of caveolin-1 and cytoskeletal proteins, actin and vimentin, in adipogenesis of bovine intramuscular preadipocyte cells. Cell biology international. 2004;28:615–623. doi: 10.1016/j.cellbi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Yang W, Guo X, Thein S, Xu F, Sugii S, Baas PW, Radda GK, Han W. Regulation of adipogenesis by cytoskeleton remodelling is facilitated by acetyltransferase MEC-17-dependent acetylation of alpha-tubulin. The Biochemical journal. 2013;449:605–612. doi: 10.1042/BJ20121121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JE, Kim HT, Lee S, Lee YS, Choi UK, Kang JH, Choi SY, Kang TC, Choi MS, Kwon OS. Differential expression of intermediate filaments in the process of developing hepatic steatosis. Proteomics. 2011;11:2777–2789. doi: 10.1002/pmic.201000544. [DOI] [PubMed] [Google Scholar]
- 31.Di Capite J, Parekh AB. CRAC channels and Ca2+ signaling in mast cells. Immunol Rev. 2009;231:45–58. doi: 10.1111/j.1600-065X.2009.00808.x. [DOI] [PubMed] [Google Scholar]
- 32.Ma HT, Beaven MA. Regulators of Ca(2+) signaling in mast cells: potential targets for treatment of mast cell-related diseases? Advances in experimental medicine and biology. 2011;716:62–90. doi: 10.1007/978-1-4419-9533-9_5. [DOI] [PubMed] [Google Scholar]
- 33.Marshall JS. Mast-cell responses to pathogens. Nature Reviews Immunology. 2004;4:787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 34.Sismanopoulos N, Delivanis DA, Alysandratos KD, Angelidou A, Therianou A, Kalogeromitros D, Theoharides TC. Mast cells in allergic and inflammatory diseases. Curr Pharm Des. 2012;18:2261–2277. doi: 10.2174/138161212800165997. [DOI] [PubMed] [Google Scholar]
- 35.Barba I, Chavarria L, Ruiz-Meana M, Mirabet M, Agullo E, Garcia-Dorado D. Effect of intracellular lipid droplets on cytosolic Ca2+ and cell death during ischaemia-reperfusion injury in cardiomyocytes. The Journal of physiology. 2009;587:1331–1341. doi: 10.1113/jphysiol.2008.163311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rozinek J, Rajmon R, Petr J, Rohlik J, Jeseta M, Sedmikova M, Rehak D, Jilek F. Ultrastructural localisation of calcium deposits in pig ovarian follicles. Animal reproduction science. 2006;91:123–132. doi: 10.1016/j.anireprosci.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Melo RC, Paganoti GF, Dvorak AM, Weller PF. The internal architecture of leukocyte lipid body organelles captured by three-dimensional electron microscopy tomography. PloS one. 2013;8:e59578. doi: 10.1371/journal.pone.0059578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Passante E, Frankish N. The RBL-2H3 cell line: its provenance and suitability as a model for the mast cell. Inflamm Res. 2009;58:737–745. doi: 10.1007/s00011-009-0074-y. [DOI] [PubMed] [Google Scholar]
- 39.Galton DJ, Wallis S. The regulation of adipose cell metabolism. Proc Nutr Soc. 1982;41:167–173. doi: 10.1079/pns19820027. [DOI] [PubMed] [Google Scholar]
- 40.Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD. Chemical calcium indicators. Methods. 2008;46:143–151. doi: 10.1016/j.ymeth.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.V. R Core Team, Austria. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2013 [Google Scholar]
- 42.Oksanen J, Kindt R, O'Hara RB. vegan: Community Ecology Package. 2005 [Google Scholar]
- 43.Gwack Y, Feske S, Srikanth S, Hogan PG, Rao A. Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell calcium. 2007;42:145–156. doi: 10.1016/j.ceca.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 45.Rao A. Signaling to gene expression: calcium, calcineurin and NFAT. Nature immunology. 2009;10:3–5. doi: 10.1038/ni0109-3. [DOI] [PubMed] [Google Scholar]
- 46.Okamura H, Aramburu J, Garcia-Rodriguez C, Viola JP, Raghavan A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Molecular cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 47.Thuille N, Lutz-Nicoladoni C, Letschka T, Hermann-Kleiter N, Heit I, Baier G. PKCtheta and Itk functionally interact during primary mouse CD3+ T cell activation. Immunology letters. 2009;126:54–59. doi: 10.1016/j.imlet.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 48.Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. Embo J. 1999;18:1303–1308. doi: 10.1093/emboj/18.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dvorak AM, Dvorak HF, Galli SJ. Ultrastructural criteria for identification of mast cells and basophils in humans, guinea pigs, and mice. Am Rev Respir Dis. 1983;128:S49–52. doi: 10.1164/arrd.1983.128.2P2.S49. [DOI] [PubMed] [Google Scholar]
- 50.Andrade FH, McMullen CA, Rumbaut RE. Mitochondria are fast Ca2+ sinks in rat extraocular muscles: a novel regulatory influence on contractile function and metabolism. Investigative ophthalmology & visual science. 2005;46:4541–4547. doi: 10.1167/iovs.05-0809. [DOI] [PubMed] [Google Scholar]
- 51.Graier WF, Frieden M, Malli R. Mitochondria and Ca(2+) signaling: old guests, new functions. Pflugers Archiv : European journal of physiology. 2007;455:375–396. doi: 10.1007/s00424-007-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pezzati R, Bossi M, Podini P, Meldolesi J, Grohovaz F. High-resolution calcium mapping of the endoplasmic reticulum-Golgi-exocytic membrane system. Electron energy loss imaging analysis of quick frozen-freeze dried PC12 cells. Molecular biology of the cell. 1997;8:1501–1512. doi: 10.1091/mbc.8.8.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Digel M, Ehehalt R, Fullekrug J. Lipid droplets lighting up: Insights from live microscopy. Febs Letters. 2010;584:2168–2175. doi: 10.1016/j.febslet.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 54.Chapman KD, Dyer JM, Mullen RT. Biogenesis and functions of lipid droplets in plants. Journal of lipid research. 2012;53:215–226. doi: 10.1194/jlr.R021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujimoto T, Ohsaki Y, Cheng J, Suzuki M, Shinohara Y. Lipid droplets: a classic organelle with new outfits. Histochemistry and cell biology. 2008;130:263–279. doi: 10.1007/s00418-008-0449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weller PF, Ackerman SJ, Nicholson-Weller A, Dvorak AM. Cytoplasmic lipid bodies of human neutrophilic leukocytes. The American journal of pathology. 1989;135:947–959. [PMC free article] [PubMed] [Google Scholar]
- 57.Bozza PT, Bakker-Abreu I, Navarro-Xavier RA, Bandeira-Melo C. Lipid body function in eicosanoid synthesis: an update. Prostaglandins, leukotrienes, and essential fatty acids. 2011;85:205–213. doi: 10.1016/j.plefa.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 58.Dvorak AM, Morgan ES, Weller PF. RNA is closely associated with human mast cell lipid bodies. Histology and histopathology. 2003;18:943–968. doi: 10.14670/HH-18.943. [DOI] [PubMed] [Google Scholar]
- 59.Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 60.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. Jama. 2001;286:1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 61.Fu SN, Watkins SM, Hotamisligil GS. The Role of Endoplasmic Reticulum in Hepatic Lipid Homeostasis and Stress Signaling. Cell Metabolism. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 62.Hotamisligil GS. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seimiya T, Ohki S. Ionic structure of phospholipid membranes, and binding of calcium ions. Biochimica et biophysica acta. 1973;298:546–561. doi: 10.1016/0005-2736(73)90073-4. [DOI] [PubMed] [Google Scholar]
- 65.Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. The Journal of biological chemistry. 2004;279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 66.Hodges BD, Wu CC. Proteomic insights into an expanded cellular role for cytoplasmic lipid droplets. Journal of lipid research. 2010;51:262–273. doi: 10.1194/jlr.R003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krahmer N, Hilger M, Kory N, Wilfling F, Stoehr G, Mann M, Farese RV, Walther TC. Protein Correlation Profiles Identify Lipid Droplet Proteins with High Confidence. Molecular & Cellular Proteomics. 2013;12:1115–1126. doi: 10.1074/mcp.M112.020230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wan HC, Melo RC, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:167–178. doi: 10.1096/fj.06-6711com. [DOI] [PMC free article] [PubMed] [Google Scholar]