

Abstract
Extracellular matrix stiffness induces focal adhesion assembly to drive malignant transformation and tumor metastasis. Nevertheless, how force alters focal adhesions to promote tumor progression remains unclear. Here, we explored the role of the focal adhesion protein vinculin, a force-activated mechano-transducer, in mammary epithelial tissue transformation and invasion. We found that extracellular matrix stiffness stabilizes the assembly of a vinculin-talin-actin scaffolding complex that facilitates PI3-kinase mediated phosphatidylinositol (3,4,5)-triphosphate phosphorylation. Using defined two and three dimensional matrices, a mouse model of mammary tumorigenesis with vinculin mutants and a novel super resolution imaging approach, we established that ECM stiffness, per se, promotes the malignant progression of a mammary epithelium by activating and stabilizing vinculin and enhancing Akt signaling at focal adhesions. Our studies also revealed that vinculin strongly co-localizes with activated Akt at the invasive border of human breast tumors, where the ECM is stiffest and we detected elevated mechano-signaling. Thus, extracellular matrix stiffness could induce tumor progression by promoting the assembly of signaling scaffolds; a conclusion underscored by the significant association we observed between highly expressed focal adhesion plaque proteins and malignant transformation across multiple types of solid cancer.
Introduction
The extracellular matrix (ECM) is remodeled and stiffened in tandem with the malignant transformation of tissues (1-5). Enhanced collagen cross-linking stiffens the ECM in vivo and in vitro, increases phosphorylation of focal adhesion kinase (p397FAK), and promotes mammary tumor progression; whereas preventing ECM cross-linking and decreasing tissue tension reduces FAK activity, and prevents tumor cell invasion and metastasis (1,6). Although these findings implicate force-induced focal adhesions in malignant progression the molecular mechanisms remain unclear. Moreover, ECM concentration and organization also influence focal adhesion assembly, and ECM density differs in tumors, while the invasive front of a transformed epithelium frequently contains tracts of perpendicularly oriented, collagen fibers (1,7-10). These data suggest that the altered ECM topology and density in tumor tissue could also promote integrin focal adhesion assembly to drive malignant progression. Along these lines Kubow and colleagues argue that adhesion-mediated cell migration in three dimensions (3D) is determined not only by myosin activity, but also by the architecture and density of the local ECM (11). Indeed, whether a cell can even assemble a focal adhesion in a 3D ECM and in tissues has recently been debated (12-14).
Focal adhesions are the conduits through which cells process extrinsic mechanical signals (15). Vinculin is a critical component of focal adhesions and has been shown to regulate cell spreading and stabilize focal adhesions (16-19). The phosphorylation and mechanical unmasking of a cryptic auto-inhibited binding site in vinculin favors a conformational change that permits binding to talin and actin (17,20-22). Vinculin also mechanically couples actin retrograde motion and increases force transmission at focal adhesions (18,23,24). Thus, vinculin is a mechanically-activated mechano-transducer that is ideally posed to mediate force-dependent cell invasion. Consistent with this hypothesis, vinculin is up-regulated in primary invasive human cancers, mediates single cell invasion within a 3D collagen hydrogel, and its loss compromises cell migration during embryonic development (25-27). Nevertheless, whether and how vinculin mediates force-induced tumor cell invasion has yet to be determined.
ECM adhesion is critical for cell growth and survival and integrins can directly and indirectly enhance growth factor receptor signaling (28,29). Integrins are a major molecular constituent of focal adhesions and focal adhesions facilitate actin assembly and actomyosin stimulation and regulate growth factor-dependent ERK and phosphoinositide 3-kinase (PI3K) activation (30,31). We showed that ECM stiffness promotes growth factor-dependent PI3K activation of Akt in culture (1). Using a mouse model of HER2/Neu induced mammary cancer we also demonstrated that inhibiting collagen cross-linking and reducing tissue tension prevents malignant transformation and represses tumor cell invasion while simultaneously decreasing PI3K signaling through Akt (1). These findings suggest that ECM stiffness could promote malignant progression and tumor cell invasion by enhancing growth factor receptor signaling. Here we asked whether vinculin, as a major mechanically sensitive focal adhesion molecule that binds actin and talin and transmits mechanical cues, could translate ECM stiffness to drive malignant transformation by potentiating growth factor receptor signaling through PI3K. We found that ECM stiffness stabilizes a mechanosensitive vinculin-talin-actin adhesion complex that facilitates PI3K-mediated Phosphatidylinositol (3,4,5)-triphosphate (PIP3) production. Thus by driving the assembly of a molecular scaffold at the focal adhesion, ECM stiffness potentiates oncogenic signaling through PI3K to drive malignant transformation.
Methods
Cell Culture and reagents
MECs including non-malignant MCF10A (ATCC, 2007; verified by epithelial cellular morphology in 2D, ability to form hollow polarized acini in 3D rBM gels, and expression of epithelial markers including E-Cadherin and cyotkeratins 8 and 14) and pre-malignant Ha-ras transformed MCF10AT (Karmanos Cancer Institute, 2008; verified by epithelial cell morphology in 2D) were cultured (7) and 3D multicellular spheroids were generated in recombinant basement membrane (rBM; Matrigel) suspension cultures (32). MCF10A vinculin knockdown (KD) and wild-type recovered MECs were generated using vinculin shRNA and vinculin-GFP retrovirus (gifted by Dr. Kris DeMali 2013; verified by epithelial cell morphology in 2D) (33). Vinculin homozygous null mouse embryonic fibroblasts were cultured as previously described (gifted by Dr. Andreas Garcia, 2013; verified by mesenchymal cell morphology in 2D)(18). The following small molecule inhibitors were used: Y27632 ROCK inhibitor at 10μM (Cayman Chemical), FAK Inhibitor 14 at 1μM (Tocris), GDC-0941 PI3K inhibitor at 1μM (Selleckchem), NSC 23766 Rac1 inhibitor at 50μM (Cayman Chemical), ML7 MLCK inhibitor at 10μM (Sigma Aldrich), and U0126 MEK1/2 inhibitor at 10μM (LC Laboratories).
Expression constructs
N-terminus GFP tagged vinculin (Gallus) constructs (gifted by Dr. Susan Craig)(24), including pEGFPC1/ Gg V1-258 WT, pEGFPC1/ Gg V1-1066 T12, pEGFPC3/ Gg V884-1066, pEGFPC1/ Gg V1-1066 A50I, pEGFPC1/ Gg V1-851. Vinculin tension sensor components 26019, 26020, and 26021 were acquired from Addgene. Other plasmid constructs used included N and C terminus tagged vinculin-mEmerald, paxillin-mCherry, farnesyl-mEmerald, farnesyl-mCherry. Transient transfection was performed using the Lonza Nucleofector Kit V, with program T024 for MECs and program U024 for fibroblasts. Lentiviral construct PGK-H2BeGFP was used in combination with Clontech mCherry-CAAX and mEmerald CAAX vectors and the pLV MCS vector to generate plV hPGK mCherry-CAAX. Stable expression of paxillin-mEmerald, vinculin-mCherry, mEmerald-Farnesyl and mCherry-Farnesyl recombinant lentivirus was prepared as described (Sup Methods). The phosphatidylinositol 4,5-bisphosphate, PIP2, (GFP-PH-PLCδ) and phosphatidylinositol (3,4,5)-triphosphate, PIP3, (mKO2-PH-Grp1) activity probes were prepared as described (gifted by Keith Mostov)(34).
Substrate and hydrogel preparation
Collagen gels were fabricated by diluting 3.8 mg/ml acid solubilized rat tail collagen (BD) in 1:1 DMEM-F12 medium (Invitrogen), neutralizing to pH 7.4 with 1M NaOH. SAP gels were fabricated by suspending cells in pre-sonicated 0.8mg/ml or 5.0mg/ml SAP (BD PuraMatrix™ Peptide Hydrogel), with 10% sucrose, 0.1% bovine serum albumin, human plasma fibronectin (Millipore), and growth medium (32). 2D polyacrylamide (PA) gels (height ~200um) were fabricated by varying the acrylamide and cross-linker concentration and assessing stiffness by Atomic Force Microscopy or using a shear rheometer (3,35). N-type [100]-orientation silicon wafers with 1933nm silicon oxide (Addison Engineering; 1 cm × 1 cm) and borosilicate glass coverslips (~150μm thick) were cleaned by successive sonication in acetone and 1M KOH (20 min) followed by chemical activation with silane (0.5% (3-aminopropyl)trimethoxysilane) and glutaraldehyde (0.5% in water). The substrates were then UV and ethanol sterilized and incubated overnight (4°C) in extracellular matrix (human fibronectin; 10 μg/ml). Prior to use, substrates were washed in PBS (5 times; pH 7.4), treated with sodium borohydride (20mg/ml) and washed with PBS. SAIM calibration wafers were prepared by sonicating carboxylate-modified red fluorescent spheres (100nm; Invitrogen), following by bead deposition (5X108 beads per ml) in NaCl (100mM) PBS solution. 3D
Immunofluorescence
Cultures were fixed with 4% paraformaldehyde (2D: 20 minutes, room temperature; 3D: overnight, 4°C) and staining was performed as described (1). Primary antibodies against vinculin (hVIN-1, Sigma Aldrich; 700062, Invitrogen; V284, Santa Cruz Biotechnology), β1 integrin (AIIB2, isolated from rat hybridoma), β4 integrin (3E1, Millipore MAB1964), p397FAK (44625G,Invitrogen), p473Akt (Cell Signaling 9271S), Akt(pan)-Alexa488 (Cell Signaling C67E7), E-cadherin (610181; BD Transduction),β-catenin (610153, BD), pan-laminin (L9393, Sigma), laminin 5 (P3H9, isolated from mouse hybridoma), α6 integrin (GoH3, eBiosciences), phospho-myosin light chain kinase 2 - Thr18/Ser19 (3674, Cell Signaling), talin (T3287, Sigma), zyxin (BD, 610521), and AlexaFluor phalloidin (633-conjugate, Invitrogen) were used. Secondary antibodies used include AlexaFluor goat anti-mouse, anti-rabbit, and anti-rat (488, 568 and 633 conjugates).
Microscopy and Analysis
Imaging was performed on a motorized TIRF inverted microscope system (Ti-E Perfect Focus System; Nikon) controlled by Metamorph software (Molecular Devices), equipped with 488nm, 561nm, and 640nm lasers, 350/50 epifluorescence, a CSU-X1 spinning disc confocal unit (Yokogawa Electric Company), electronic shutters, motorized stage, a scientific sCMOS camera (Zyla 5.5 megapixel, Andor), and an electron-multiplying charged-coupled device camera (QuantEM 512; Photometrics). Temperature and CO2 was controlled by an environmental chamber and PID controlled heater (In-Vivo Scientific). Samples were imaged with a 100x-1.49NA TIRF oil-immersion, 40x-1.2NA Long-Working-Distance (LWD) water-immersion, 20x-0.75NA air, and 10x-0.45NA air objectives. For live-cell imaging, phenol red-free growth media with 10 mM Hepes (pH 7.4) was used. For 3D imaging, confocal image slices were taken at 0.2μm or 1.0μm slices, and the individual planes were maximum intensity z-projected for 2D visualization. A custom software package for image analysis was written in Python, using ImageJ and the Eclipse Development Environment in Linux. All images were subject to a Gaussian blur of 1.0 pixel to smooth background noise. DAPI images were subject to a local background subtraction of ~5μm to reduce fringing from epifluorescence imaging through the spinning disc unit. Intensity and colocalization measurements of single confocal planes were calculated on a pixel-by-pixel basis. 3D image rendering was performed in Imaris (Bitplane). Cell projections and adhesions were counted manually. Acini cross-sectional area was quantified with the freehand selection tool in ImageJ. Photobleaching FRET (pbFRET) and Scanning Angle Interference Microscopy (SAIM) were performed as described (36) (Sup Methods), using data analysis and 3D visualization programs written in Matlab® and Python using libraries distributed by Enthought, Inc.
Mouse and Human Studies
Cohorts of PyMT (polyoma middle T) and FVB mice were maintained in accordance with University of California IACUC guidelines under protocol AN092125. Starting at 4 weeks of age, mice were treated with a LOX function-blocking polyclonal antibody (3mg/kg; OpenBiosystems) injected intraperitoneally twice per week (1). Mice were sacrificed at 80-95 days of age at which time the 4th mammary gland was paraformeldahyde fixed. Under protocol 10-05046 and in accordance with the UCSF Committee on Human Research, formalin-fixed and paraffin-embedded human breast cancer biopsies containing normal, DCIS (ductal-carcinoma-in-situ) and estrogen receptor and progesterone receptor positive invasive cancer tissue were obtained. Mouse mammary tissue (10μm) and human breast biopsy (6μm) sections were analyzed for histology (H&E) and parallel tissue was stained for the focal adhesion proteins vinculin, p397FAK, and β1 integrin and for the PI3K downstream target p473Akt.
Statistics
Statistical analysis for 2 groups was performed with an unpaired, two-tailed student’s t-test. For multiple comparisons, an ANOVA test and Holm–Bonferroni t-test method were performed. Calculations were implemented in Python.
Results
ECM stiffness and ligand density regulate focal adhesions to promote tumor cell invasion in three dimensions
Most studies linking ECM stiffness to focal adhesion assembly and cell motility have been conducted using single transformed cells or mesenchymal fibroblasts on 2D substrates (37,38). Here we examined the importance of tension-induced focal adhesions in nonmalignant versus transformed epithelial cell invasion both as single cells and tissue-like structures in 3D. We transplanted 5 day old, rBM pre-assembled, proliferating nonmalignant MCF10A and premalignant Ha-ras MCF10AT transformed mammary epithelial cell (MEC) spheroids into collagen-rBM gels with compliances calibrated to match normal (0.5kPa, 0.5mg/ml), premalignant (1.5kPa; 2.0mg/ml) and malignant (2.5kPa; 5.0mg/ml) mammary tissue (1,3,7). Phase contrast (Fig 1A, top 2 left panels) and confocal immunofluorescence imaging (Fig 1A, bottom left 2 panels) revealed that the transplanted nonmalignant mammary spheroids retained their integrity,even 24 hours after embedment within the compliant collagen/rBM gels, as indicated by the maintenance of spherical acini, intact adherens junctions, and tissue polarity, as revealed by basally-localized laminin and cell-cell localized β-catenin (Fig 1A, left column). However, immunostaining revealed that after 48 hours in the stiffer gels the basal polarity and cell-cell junctions in the nonmalignant structures were severely compromised (Fig 1A, center column; Sup Fig 1A). Importantly, the nonmalignant cells at the periphery of the colonies in the stiffer gels probed the local ECM, as indicated by prominent cell protrusions (39), although they never invaded into the gel (Fig 1A, left column). By contrast, while the Ha-Ras premalignant mammary spheroids retained a semblance of tissue polarity when embedded within the softest collagen/rBM gels (Fig 1A, center column) indicated by retention of cell-cell localized β-catenin and minimal protrusions; in the stiffer gels the structures completely collapsed and the transformed MECs invaded both collectively and individually (Fig 1A, center column)(1). Interestingly, two photon imaging revealed that Ha-ras transformed MCF10AT MECs invaded collectively and as single cells along collagen bundles that appeared to project perpendicularly from the colonies (Sup Fig 2A), and atomic force microscopy illustrated that collagen bundles can be quite stiff under compression (Sup Fig 2B and C) (1,40). Thus, although these findings do not rule out the possibility that ECM invasion into these collagen gels was also induced by the topological features of the collagen, the data do strongly implicate ECM stiffness.
Figure One. In 3D, ECM cues promote vinculin recruitment to focal adhesions, projection formation, and cell invasion.
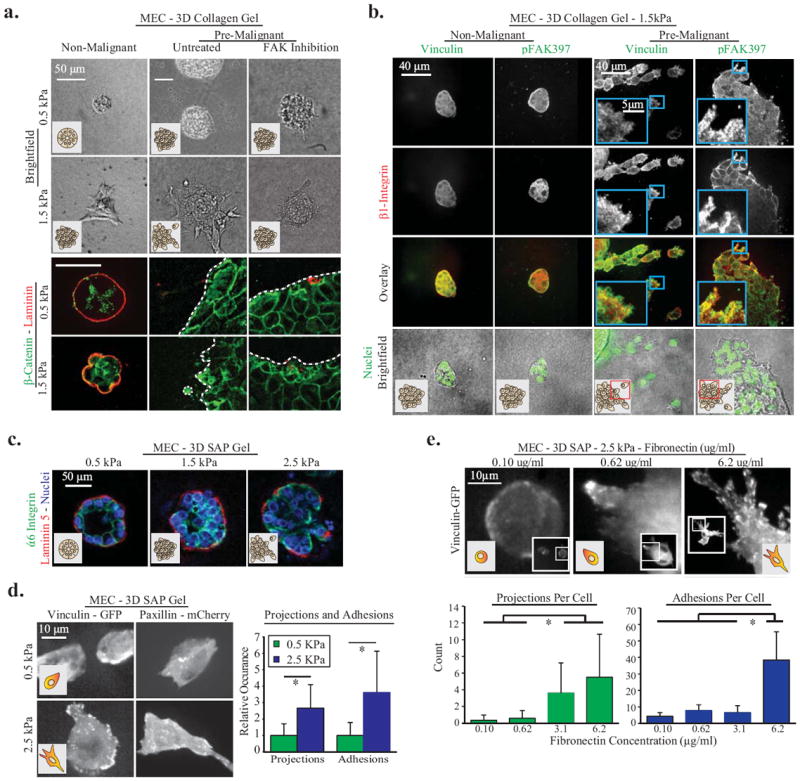
A. MEC spheroids composed of MCF10A cells in 0.5 kPa or 2 mg/ml 1.5 kPa 3D collagen gels for 24hrs. Invasion is increased by matrix stiffness and Ha-ras malignant transformation (MCF10AT cell line), and decreased via inhibition of integrin mediated FAK signaling (FAK Inhibitor 14 at 1μM). To document loss of normal 3D cell organization in stiff 3D collagen, cell polarity was studied 48 hours after seeding in collagen via immunostaining for β-catenin (cell-cell junctions) and pan-laminin (basal surface). Dotted white lines indicate spheroid edge. B. Pre-malignant and non-malignant MEC 1.5kPa 3D collagen gels for 48 hours, immunostained for integrin β1, vinculin and pFAK39. Images are maximum intensity z-projections of 1μm increment confocal image slices, and demonstrate focal adhesion formation at cell-ECM borders. C. Non-malignant MEC spheroids in 3D SAP gels with 0.8, 2.2 or 5mg/ml RADA-16 polymers; stained for polarity markers α6 Integrin (cell-cell junctions) and laminin-5 (basal polarity). D. Non-malignant MEC adhesions and projections in soft and stiff 3D SAP gels; with quantification of projections and adhesions per cell (p < .01; ±SD, n>18 cells per condition). E. Non-malignant MEC expressing vinculin-GFP In 3D SAP gels with controlled ECM fibronectin concentration. Images are maximum intensity z-projection of 1.0um confocal image planes. Cellular projections and adhesions per cell were significantly upregulated by ECM stiffness and fibronectin (p < .01; ±SD, n>12 cells per condition).
To explore functional links among focal adhesions, ECM stiffness, and tumor cell invasion, we stained 3D multi-cellular structures for the transmembrane protein β1 integrin which connects the ECM to cell adhesions, and the focal adhesion proteins vinculin and p397FAK, as well as fluorescently labelled phalloidin to image filamentous actin. Confocal imaging revealed that MECs at the periphery of the Ha-ras premalignant colonies, as well as the individual transformed MECs invading into the stiffer collagen/rBM gels, stained robustly for all three focal adhesion proteins (Fig 1B, right columns)as well as F-actin (data not shown). Moreover, treatment of the premalignant spheroids with 1μM FAK-Inhibitor 14 ablated the invasive behavior of these transformed mammary tissues within the stiffer gels (Fig 1A; right column). However, we also detected abundant levels of these adhesion proteins arranged as punctate structures reminiscent of focal adhesions in the nonmalignant MECs at the periphery of the spheroids where the cells interact directly with the stiff ECM (Fig 1A; left columns). Thus, although these findings suggest ECM stiffness activates focal adhesions to promote MEC invasion in 3D, the data also indicate that focal adhesions are not by themselves sufficient, and MEC invasion also requires the activation of pathways engaged by oncogenic transformation (1,7).
To directly test the role of ECM stiffness on tumor cell invasion in 3D, in the absence of changes in ECM pore size, topology, density or composition we used 3D RADA-16 self-assembling peptide (SAP) gels doped with saturating concentrations of laminin 111 or fibronectin (32). By increasing polymer concentration, the compliance of these SAP gels can be varied across a range of stiffness (0.5-5kPa) appropriate for mammary and other epithelial tissues, with negligible effects on pore size and ECM topology, and we and others showed that cells embedded in these gels ligate the ECM protein(s) passively adhered to the peptides and respond to the stiffness of the gel (32,41). Consistently, we observed that MECs embedded in SAP gels with saturating concentrations of ECM ligand (fibronectin; 6.2 mg/ml) increased their protrusive activity in proportion to the stiffness of the gel (Fig 1C; Sup Fig 1B); similar to what we observed in 3D collagen gels (Sup Fig 1D). Confocal imaging also revealed that MECs co-expressing GFP-tagged vinculin and mCherry paxillin assembled small punctae reminiscent of focal contacts in the softest SAP gels, and larger focal adhesion-like structures in the stiffer SAP gels (Fig 1D). Furthermore, the size and number of these adhesions as well as the number of cellular protrusions, which is a proxy for invasive behavior, increased in proportion to ligand concentration (Fig 1E, Sup Fig 1C). Importantly, after embedment into the stiffer SAP gels immunofluorescence imaging revealed that the integrity and polarity of the nonmalignant MEC organoids was severely compromised, as indicated by loss of basally-localized α6 integrin and basally deposited laminin 5 (Fig 1C). Moreover, in marked contrast to their behavior in the collagen/rBM gels where pore size limited invasion (Sup Fig 1B, bottom row, 4th column), phase contrast imaging revealed that the invasive behavior of the premalignant mammary colonies increased further in the stiffest SAP gels (Sup Fig 1B). These observations show that ECM stiffness and ligand density regulate focal adhesions to permit the invasion of an oncogenically-transformed epithelium in 3D.
ECM stiffness activates vinculin to promote an invasive phenotype
Vinculin is a major focal adhesion plaque protein whose structure-function is exquisitely sensitive to mechanical force, and vinculin can act as a mechanical clutch to stabilize adhesions (18,23). This prompted us to ask if ECM stiffness promotes tumor cell invasion by activating vinculin to stabilize focal adhesions. Consistently, we noted that MECs expressing a wild-type vinculin (vinculin WT)that were plated on a soft fibronectin-conjugated polyacrylamide gel (PA gel) assembled small focal contacts, showed only modest protrusive activity and failed to spread (Fig 2A, top left panel) (7). By contrast, parallel cultures of MECs plated on soft gels that expressed a constitutively active vinculin T12, which lacks the auto-inhibition domain, had increased adhesion area, exhibited robust protrusive activity and spread appreciably (Fig 2A, top right panel; Sup Fig 1E). Additionally, MEC expressing vinculin T12 on stiff substrates had prominent stress fibers and localized more vinculin at the focal adhesions (Fig 2B) (17). Moreover, MECs in which vinculin levels were reduced using shRNA had significantly reduced protrusive activity, reflecting invasive behavior, even when the cells were embedded within a stiff, fibronectin-saturated, SAP gel (Fig 2C). By contrast the protrusive activity of these MECs was fully restored following re-expression of an RNAi resistant vinculin (Fig 2C). In this regard, we observed that the ability of vinculin to restore the protrusive activity in vinculin null murine fibroblasts in response to ECM stiffness required a critical level of cellular vinculin, where the greatest protrusive activity was noted in cells with the highest vinculin expression (Fig 2D). Thus, fibroblasts expressing high amounts of vinculin assembled punctate adhesive-like structures analogous to focal adhesions, and increased their protrusive activity in response to a stiff SAP gel (Fig 2B)(27). These data demonstrate that ECM-induced invasion requires the engagement of a critical threshold of vinculin that stabilizes focal adhesions.
Figure Two. Vinculin activity is necessary for adhesion stabilization.
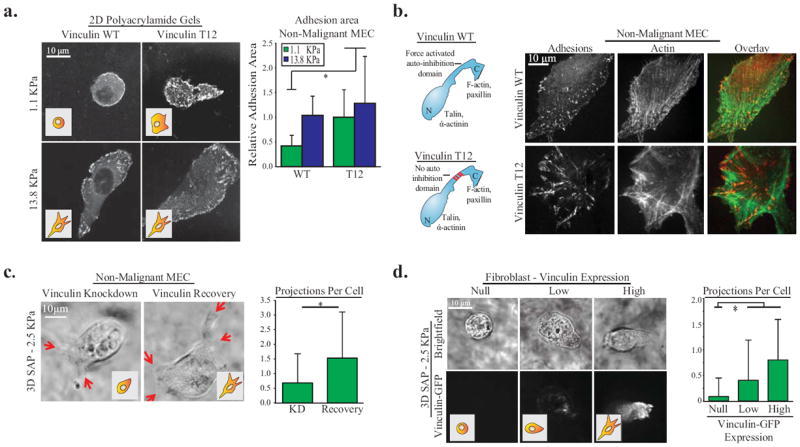
A. Non-malignant MEC expressing vinculin-WT or vinculin-T12 TFP1-mVenus tension sensors on 2D polyacrylamide (PA) gels of varying stiffness coated with fibronectin and quantification of relative adhesion area per cell (p < .01; ±SD, n>10 cells per condition). Vinculin T12 expression significantly increases cell adhesion area on soft matrices. B. Diagram of vinculin WT and vinculin T12 activity mutant prominent binding domains, and confocal images of N-terminus GFP tagged vinculin and filamentous actin stained with phalloidin in non-malignant MEC on 2D glass coated with fibronectin. MECs expressing the activated vinculin T12 show prominent stress fibers and localized more vinculin at the focal adhesions. C. Non-malignant MEC projections (indicated by red arrows) in 3D 2.5 kPa SAP gels with 6.2ug/ml fibronectin after vinculin shRNA knockdown and recovery via vinculin-GFP expression, and quantification of projections (p < .01; ±SD, n>27 cells per condition) D. Vinculin homozygous null mouse fibroblasts transiently expressing different levels of vinculin-GFP in 3D 2.5 kPa SAP gels with 6.2ug/ml fibronectin and quantification of projections (p < .01; ±SD, n>9 cells per condition). Vinculin significantly increases cell projections in stiff 3D SAP gels in MEC and fibroblasts.
Extrinsic and intrinsic force activate vinculin at focal adhesions
We next explored the relationship between force, vinculin activation, and focal adhesion stabilization. We first demonstrated that 15-45 minutes following ROCK inhibition (Y27632; 10μM), the size and number of the vinculin positive focal adhesions was significantly decreased in the non-malignant MECs expressing a GFP-tagged vinculin WT (Fig 3A, bottom left graph). By contrast, no quantifiable change in either the size or the number of adhesions was observed in the ROCK inhibitor treated MECs expressing the GFP-tagged vinculin T12 (Fig 3A, bottom left graph). These findings agree with prior data obtained in fibroblasts which established a role for actomyosin tension in vinculin-mediated focal adhesion stabilization (17,19).
Figure Three. Vinculin activity depends on extracellular and intracellular tension.
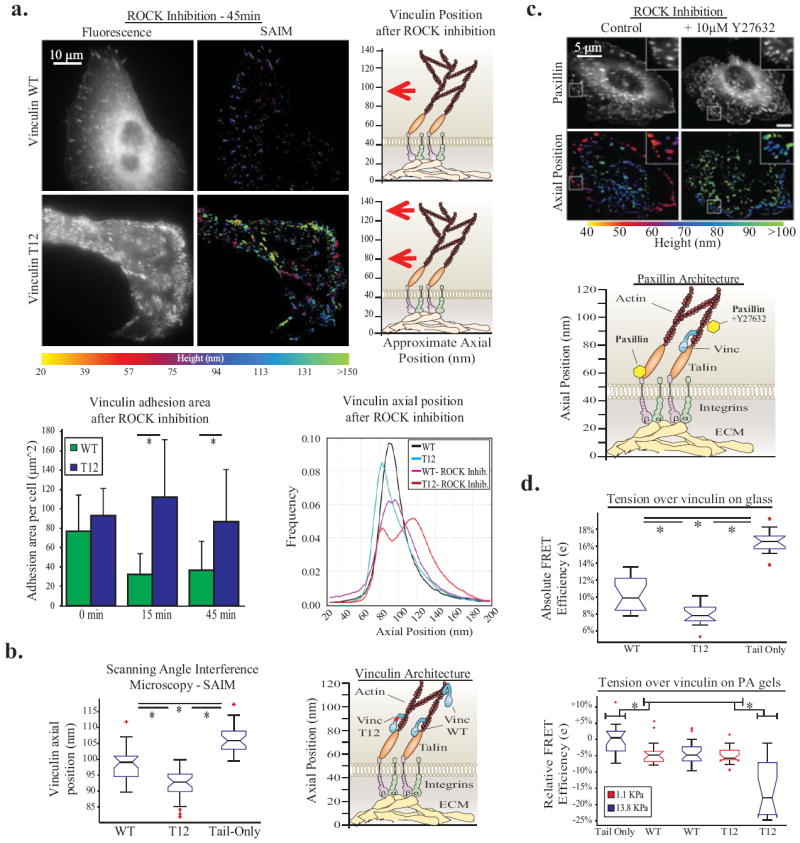
A. Non-malignant MEC plated on 2D fibronectin-coated glass 45 minutes after ROCK inhibition (10μM Y27632) Left: representative image of vinculin WT and T12 fluorescence, Middle: axial position of vinculin WT and T12 via SAIM. Right: diagram of vinculin WT and T12 after ROCK inhibition. Bottom left: Quantification of adhesion area per cell after ROCK inhibition (p < .01; ±SD, n>18 cells per condition). Bottom right: axial distribution of vinculin and vinculin T12 after ROCK inhibition. Histogram constructed from >3500 single pixel (0.012 μm2) SAIM measurements per condition (n>18 cells per condition). ROCK inhibition reduced the size of vinculin WT positive adhesions but did not perturb axial position; while vinculin T12 positive adhesions were not reduced in size, but localized to axial positions of 80nm and 120nm. B. Vinculin nanoscale axial position in non-malignant MEC plated on a 2D glass surface via SAIM measurements of whole cell averages (p < .01; ±SD, n>26 cells per condition), and diagram of vinculin axial position relative to other adhesion proteins. Vinculin T12 is at a height (95nm) indicating talin-actin binding, vinculin WT is at a height of (100nm) indicating both talin-actin and actin only interactions. C. Paxillin-GFP fluorescence and SAIM measurements in MEC plated on a 2D glass surface; paxillin axial position is increased from 60 to 100nm after ROCK inhibition. D. Tension over vinculin activity mutants in MECs plated on a 2D glass surface via an intramolecular vinculin FRET sensor using the photobleaching FRET method, measurements of all pixels in adhesions averaged over whole cell (p < .05; ±SD, n>17 cells per condition); and tension over vinculin on soft and stiff PA gels (p < .05; ±SD, n>10 cells per condition). Tension over vinculin is increased through a T12 activity mutation and ECM stiffness.
Focal adhesions are composed of over 200 proteins which are segregated into stratified functional layers (42,43). Yet, scanning angle interference microscopy (SAIM), which accurately records the nanoscale position of the molecular constituents of focal adhesions, indicates that the spatial organization of many of adhesion plaque proteins, including the scaffolding molecule paxillin change dramatically and dynamically during focal adhesion assembly and cell invasion (36). Therefore, to understand the interplay between extrinsic and intrinsic force and vinculin-mediated stabilization of focal adhesions and cell invasion we monitored vinculin dynamics during focal adhesion assembly and disassembly using SAIM. Imaging showed that vinculin WT localized at 100 nm (Fig 3B), which is a composite of the talin-actin and actin only bound molecules. Indeed, we observed that a constitutively active vinculin T12, all of which is bound to a talin-actin complex, resides at 95 nm and a vinculin tail only mutant, which binds entirely to actin, localized to a height of 105 nm (Fig 3B). To rule out the effect of cytoplasmic vinculin, we removed the dorsal cell membrane and cytoplasm in MEC and were then able to measure a significant change in vinculin T12 axial position compared to vinculin WT in the ventral cell surface (Sup Fig 3A). We also observed similar behavior of several vinculin mutants between vinc-/- mouse fibroblasts and MEC, and were able to measure the separation of vinculin’s N and C terminus (Sup Fig 4A,B). Moreover, ablating actomyosin intrinsic tension in the MECs, by treatment with a ROCK inhibitor (Fig 3A) shifted the bulk of the vinculin WT to the actin associated fraction, while vinculin T12 remained localized to the focal adhesions with altered axial localization (Fig 3A). The vinculin WT that remained at the adhesions after ROCK or myosin II inhibition (Blebbistatin; 25μM) was bound at a stable height of 100nm, which is a composite of talin-actin bound and actin-bound (Fig 3A; Sup Fig 3B). In contrast, a different adhesion plaque protein, paxillin, shifted from 60nm to a height of 90nm following myosin inhibition (Fig 3C). These findings illustrate that once engaged; the vinculin-talin-actin complex is remarkably stable.
To determine if ECM stiffness promotes cell invasion by stably increasing cell-generated force over the vinculin-actin-talin complex we used a vinculin intramolecular tension sensing probe (21) with the photobleaching Förster resonance energy transfer (pbFRET) method (Fig 3D, top panel). We then examined the response of vinculin WT and mutant expressing cells to ECM stiffness using fibronectin-conjugated PA gels. We found in MECs on either soft (1.1kPa) or stiff (13.8kPa) fibronectin-conjugated PA gels, that vinculin WT incorporated into focal adhesions was under the same tension (Fig 3D, bottom panel). However, the tension on the auto-activated vinculin T12, which forms a stable talin-actin-vinculin complex, increased significantly in MECs on the stiffer gels as compared to level on the softer gels, presumably because more force-activated talin becomes available to bind (Fig 3D). Thus, force activates vinculin to facilitate its assembly into a highly stable talin-actin complex that in turn applies actomyosin-mediated tension on the ECM that is proportional to the level of recruited vinculin.
Vinculin stabilizes focal adhesions and promotes tumor cell invasion in 3D by facilitating PI3K-dependent PIP activation
Tumor cell invasion in culture and in vivo requires PI3K activity (1,44,45) Moreover, ECM stiffness enhances epidermal growth factor receptor dependent PI3 activity in culture and inhibiting ECM cross-linking and stiffening reduces PI3K activity and tumor cell invasion (1). We therefore asked whether ECM stiffness promotes PI3K-dependent tumor cell invasion by increasing the amount of the vinculin-actin-talin complex at the focal adhesion. Consistently, we observed that more vinculin was recruited to adhesion sites when the ECM was progressively stiffened (Fig 4A). ECM stiffness also increased the quantity of activated p473Akt and to a lesser extent p397FAK to the sites of adhesion (Fig 4A, left panel), where we were able to quantify substantial co-localized vinculin to p473Akt and some vinculin and p397FAK (Fig 4A, right panel). Moreover, the quantity of signaling molecule recruited to the adhesion was significantly enhanced in vinculin null fibroblasts expressing the vinculin T12, which fosters the assembly of a stable talin-actin complex, as indicated by enhanced p397FAK and vinculin as compared to the lower levels measured at the adhesions in the WT vinculin expressing fibroblasts (Fig 4B). These findings suggest that ECM stiffness promotes the assembly of a stable vinculin-actin-talin complex at the focal adhesion that then permits the nucleation and subsequent efficient activation of signaling molecules critical for cell invasion. We observed that 10 minutes after EGF stimulation Akt was recruited to adhesions on both soft and stiff 2D PA matrices in MECs, but importantly, we were only able to document a significant increase inactivated p473Akt in vinculin-enriched focal adhesions in MECs on a stiffer substrate (Fig 4C). Moreover, treating MCF10AT premalignant mammary colonies with the PI3K inhibitor GDC-0941 (1μM) repressed their protrusive activity and invasion into a stiffened 3D collagen/rBM gel (1.5kPa) (Fig 4D, rightmost panel), while there was no reduction in vinculin and β1 integrin positive focal adhesions, even after 48 hours of treatment (Fig 4D). These data indicate that ECM stiffness stimulates tumor cell invasion by activating vinculin to assemble a stable complex with talin and actin that enhances PI3K signaling.
Figure Four. Vinculin stabilizes focal adhesions to facilitate FAK and PI3K/Akt signaling, drivers of cellular invasion in 3D.
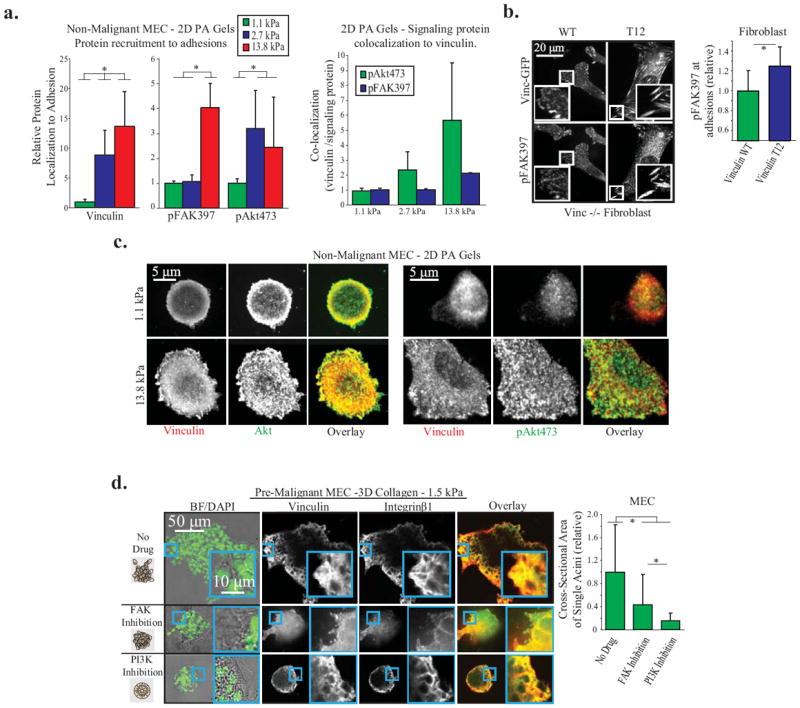
A. Vinculin, p473Akt, and p397FAK recruitment to adhesions in non-malignant MEC on 2D PA Gels of different stiffness, coated with fibronectin. Left: Protein recruitment measured by quantitative immunofluorescence at single pixels (0.011 μm2) and averaged over whole cell (p < .01; ±SD, n>18 cells per condition) Right: Quantification of signaling protein colocalization to vinculin per pixel cell, averaged over whole cell (p < .01; ±SD, n>18 cells per condition). B. p397FAK recruitment to adhesions in vinculin homozygous null mouse fibroblasts expressing vinculin WT or T12 plated on a 2D PA gel. Vinculin T12 significantly increases p397FAK; measurements made at single pixels and averaged over whole cell (p < .05; ±SD, n>9 cells per condition). C. Vinculin, Akt, and p473Akt recruitment to adhesions in non-malignant MEC on 2D PA Gels. Akt is recruited to adhesions on soft and stiff gels, while p473Akt is dramatically increased in adhesions on stiff PA gels. D. Pre-malignant MEC spheroids in 1.5 kPa 3D collagen gels for 48 hours, with FAK (1μM FAK Inhibitor 14) or PI3K (1μM GDC-0941) inhibition. Before fixation, the cross-sectional area of spheroids was measured to quantify 3D cell invasion (p < .01; ±SD, >26 spheroid per condition). Spheroids were then cryosectioned and immunostained for vinculin and integrin β1 to demonstrate that focal adhesions are not ablated after PI3K or FAK inhibition.
Force activates vinculin to increase membrane protrusions and nucleate PIP3
We next asked how the force-induced assembly of a stable vinculin-talin-actin complex potentiates PI3K signaling. In this regard, recent findings by Wang and colleagues showed that Phosphatidylinositol (3,4,5)-triphosphate (PIP3) clusters into distinct nanodomains in the plasma membrane (46). We therefore hypothesized that vinculin-adhesions could enhance the ability of PI3K to convert PIP2 to PIP3 in the plasma membrane. Consistently, in response to ECM stiffness, MECs expressing an inert membrane probe mEmerald-farnesyl assembled more and longer membrane protrusions (Fig 5A). To directly examine if force-induced vinculin activation alters the nanoscale topography of the plasma membrane, MECs expressing vinculin WT or a vinculin T12 together with an inert membrane probe (mCherry-farnesyl) were seeded on fibronectin coated silicon wafers and SAIM was performed to visualize the nanoscale position of vinculin and the associated ventral plasma membrane (Fig 5B). Imaging studies revealed that while the height of the plasma membrane varied widely across the cell (up to +/- 70nm), the height of the membrane significantly decreased by approximately 20nm at sites of vinculin-rich focal adhesions, where vinculin remained constant at 100 nm (Fig 5C). These findings indicate that vinculin positive focal adhesions correlate to areas of altered membrane topography and suggest that the assembly of a stable vinculin-talin-actin complex likely modifies membrane topology. To directly determine whether a vinculin stabilized focal adhesion facilitates PIP3 accumulation to enhance Akt activation, MECs expressing a probe for PIP3 (mKO2-PH-Grp1) with either a probe for PIP2 (PLCδ-PH-EGFP), GFP vinculin WT, or GFP vinculin T12 were seeded on fibronectin-coated coverslips and imaged. Following growth factor stimulation, we observed that PIP2 was decreased and PIP3 was increased via PI3K activity (Fig 5D). Data revealed that enhancing the assembly of the vinculin-talin-actin complex in MECs, by expressing activated vinculin T12, significantly increased the levels of PIP3 at the focal adhesion site, as compared to MECs expressing vinculin WT (Fig 5E). These results build upon previous findings that PIP3 is localized to the cell-ECM basal border in epithelial acini embedded within a 3D ECM (34), and suggest the force-induced assembly of a stable vinculin-talin-actin complex potentiates Akt activation via PI3K by facilitating membrane changes that favor the accumulation of PIP3 at the adhesion complex. Given that many oncogenes enhance PI3K activity; this mechanism may explain why ECM stiffness specifically induces invasion in a transformed cell.
Figure Five. Vinculin positive focal adhesions are colocalized to areas of altered membrane topography and composition.
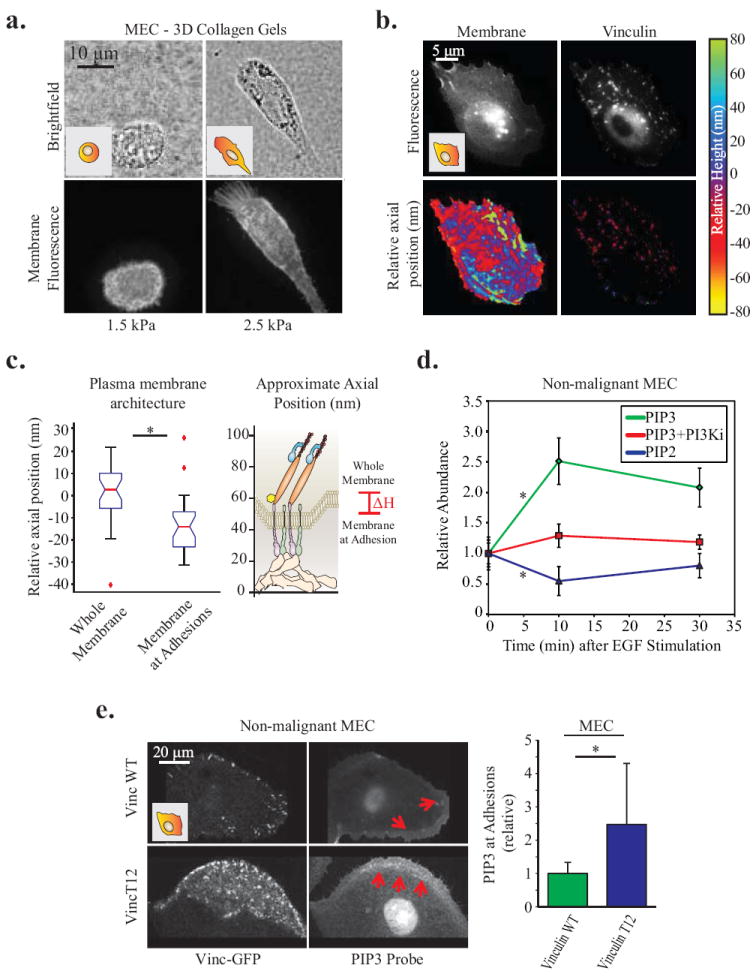
A. Non-malignant MEC in 3D collagen gels for 24hrs. Membrane protrusions are dependent on high (2.5 kPa) matrix stiffness; visualized via mEmerald-farnesyl membrane probe. B. Organization of vinculin and the ventral plasma membrane of MEC plated on a 2D glass surfaces, via SAIM measurements of vinculin-GFP and mCherry-farnesyl. Axial position is plotted relative to the mean height of the membrane and vinculin, respectively. C. Quantification of the significant decrease in membrane axial position at focal adhesions (p < .01; ±SD, n>18 cells per condition). D. Line graphs showing relative abundance of PIP2 and PIP3 in the ventral membrane of non-malignant MEC between 0-30 minutes following EGF stimulation. 10 minutes after stimulation PIP2 significantly decreases and PIP3 significantly increases. However, inhibiting PI3K activity (1μM GDC-0941) is able to abrogate this increase in PIP3 (p < .05; ±SE, n>25 cells per condition). E. Confocal images of pre-malignant MEC plated on 2D PA gels of increasing stiffness expressing vinculin-GFP activity mutants and a probe for PIP3 activity (mKO2-PH-Grp1), where red arrows indicate areas enriched in PIP3 activity. Measurements of all pixels in adhesions were averaged over whole cell; PIP3 was significantly enriched in focal adhesions of cells expressing activated vinculin T12 (p < .05; ±SD, n>7 cells per condition).
ECM stiffness regulates vinculin and p473Akt activity in experimental mouse tumors and co-localizes with vinculin-rich adhesions in invasive human breast cancer; and adhesome proteins are up-regulated in solid tumors
We and others have shown that p473Akt and p397FAK co-localize at the invasive front of experimental mammary tumors and we found that this phenotype is lost if ECM cross-linking and stiffening is prevented (1,47). Here we determined that ECM stiffness mediates the localization and levels of these signaling molecules at the invasive front by activating vinculin. Thus, while confocal immunofluorescence imaging revealed strong co-localization of vinculin and p473Akt at the invasive front of PyMT mammary tumors where the ECM is stiffest, in tissue from mice that had been treated with either a pharmacological lysyl oxidase inhibitor (data not shown) or a function-blocking antibody to inhibit lysyl oxidase activity (Fig 6A) to prevent collagen cross-linking and stiffening, p473Akt and vinculin levels were greatly reduced and little to no co-localization of these molecules was observed. The clinical relevance of these findings was illustrated by showing a progressive increase in the levels and co-localization of vinculin and p473Akt as well as β1 integrin and p397FAK (Fig 6B and C) in normal, DCIS and ER/PR positive invasive human breast cancer.
Figure Six. In vivo, vinculin is localized to cell-ECM borders and areas of high FAK and Akt signaling.
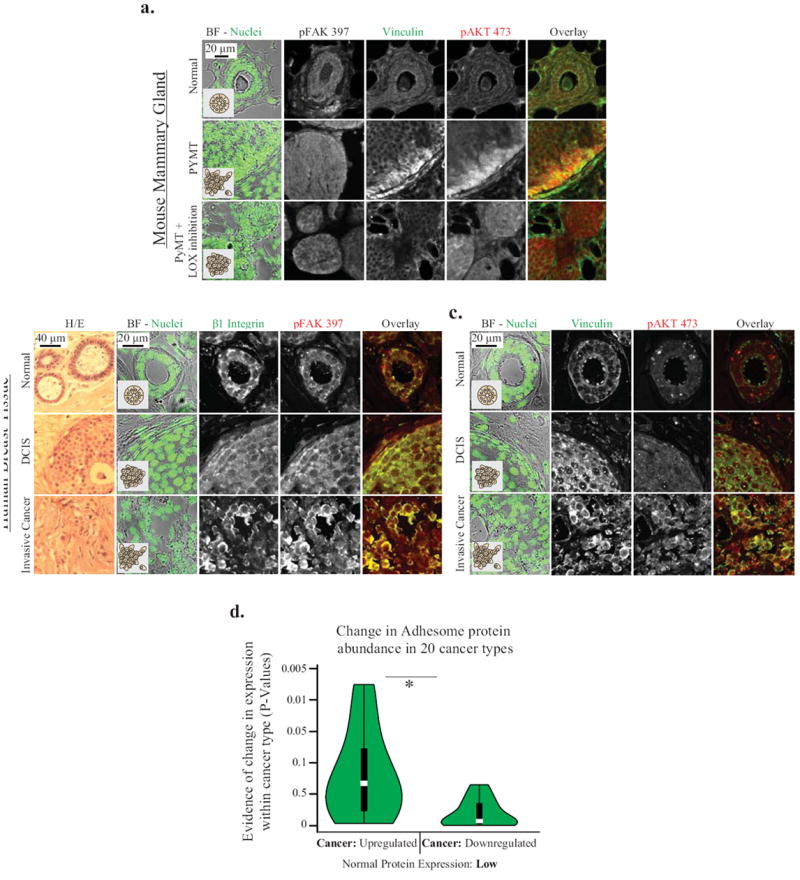
A. p397FAK and β1 integrin immunostaining of an ER/PR positive human breast cancer tumor biopsy and representative H/E staining of these tissues; B. Vinculin and immunostaining of an ER/PR positive human breast cancer tumor biopsy. 397FAK, β1 Integrin, Vinculin and 473Akt are enriched at the cell-ECM border in invasive tissue. C. Vinculin, 397FAK and 73Akt immunostaining of mammary gland tissue from FVB (normal), and PyMT background mice +/- LOX cross-linking inhibition. Cross-linking reduces vinculin and 473Akt recruitment. D. Changes in integrin adhesome protein expression in 20 cancer types as compared to normal tissue, and quantified from pathologically-scored immunohistochemical data compiled by the Human Protein Atlas database. Adhesome proteins expressed at low levels in normal tissues were significantly more likely to be upregulated within cancer type (p<.01)
Having implicated force-regulated vinculin in tumor invasion, we next asked if other adhesion proteins might also be similarly involved in malignant progression. We used a bioinformatics approach that involved the analysis of pathologically-scored immunohistochemistry data compiled by the Human Protein Atlas database (Sup Methods; Sup Table 1, and Sup Fig 5A) (48). Using an unbiased approach to pair each tumor type to its corresponding normal cell, we determined the most common expression level observed across all samples within each tumor type to generate composite profiles of “typical” protein expression levels for each tumor type. Using this approach we observed that focal adhesion proteins (42) that were normally expressed at low to negligible levels in healthy tissues were significantly likely to be over expressed in their corresponding cancer tissue (Fig 6D; Sup Fig 5B). Interestingly, we also found that many of the oncogenes implicated in cancer were also similarly up regulated (Sup Methods; Sup Fig 5C). These bioinformatics results indicate that adhesome proteins as an aggregate are up regulated at the protein level across many different solid cancer types, in a manner that is analogous to known oncogenes, suggesting they may play a more significant role in malignant progression than previously appreciated.
Discussion
The tumor microenvironment and specifically the extracellular matrix is an important regulator of malignant progression (1,3,5-7,49). Using defined 2 and 3D matrices, mouse models and human biopsies together with a series of molecular mutants and a novel super resolution imaging approach, we demonstrate for the first time in 3D and in tissues that ECM stiffness regulates the activation of vinculin and its nanoscale organization at focal adhesions. By stabilizing the vinculin-talin-actin complex our data provide the first detailed molecular mechanism to explain how tumor associated ECM cross-linking and stiffening is able to promote malignant transformation and invasion in tissue (Fig 7A and B) (1,11,27,50). Our data illustrate how the induction and stabilization of the talin-vinculin-actin scaffolding complex facilitates oncogenic signaling through pathways such as growth factor receptor induced PI3K generation of PIP3 (Fig 7B). Thereby our results emphasize vinculin’s role as a nucleator of cell signaling and highlight a vinculin-dependent signaling circuit through which cancer cells both gather information about their altered environment and alter intracellular signaling in response to this input (18,19,23,51). Indeed, by demonstrating a central role for force-regulation of a vinculin-talin-actin complex we provide the first definitive evidence for why malignant transformation and tumor cell invasion require the cooperative interplay between oncogenic transformation and a stiffened ECM (Fig 7A)(1,3,7,52).
Figure Seven. Model: MEC invasion is dependent on ECM cues, vinculin engagement and malignant transformation.
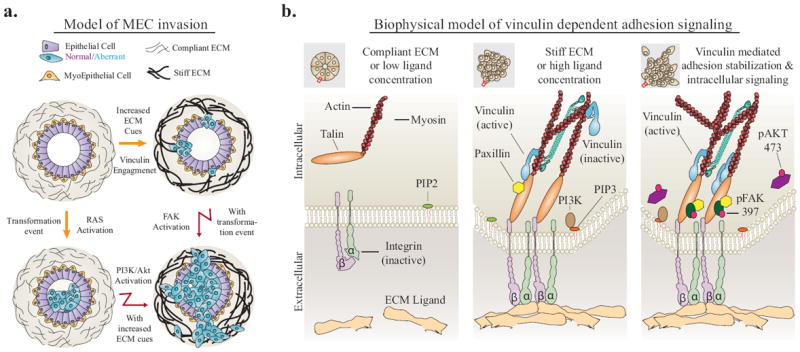
A. Model of mammary duct organization, which can be perturbed by malignant transformation or changes in ECM stiffness. Together, these events lead to cancer cell invasion through PI3K, FAK and vinculin activation. B. Biophysical model of vinculin dependent adhesion signaling. Extracellular cues drive integrin activation, talin binding, and vinculin activation. Vinculin-talin-actin stabilization facilitates PI3K conversion of PIP2 to PIP3, activation of FAK and Akt, and intracellular signaling that leads to cancer cell invasion.
While most available work on focal adhesions has been performed in 2D in vitro systems, our studies link and extend existing knowledge to cell behavior in a physiologically relevant context. We combine nanoscale characterization with 3D and in vivo analysis to provide a holistic view of how ECM stiffness changes the context in which intracellular signaling takes place. Our data show that by driving molecular scaffolding at focal adhesions, increased ECM stiffness promotes signaling in pathways that are important in both growth and invasion. On the molecular level, our data establish vinculin as a versatile adhesion molecule involved in many aspects of cell-ECM interactions. We confirm that vinculin’s auto-inhibition domain reacts in a force-dependent manner and operates as a mechanical clutch connecting the actomyosin network with the ECM. This tantalizingly suggests similar mechanisms of regulation for other plaque proteins containing cryptic binding sites, including talin and α-actinin at cell-ECM junctions, cadherins and PECAM at cell-cell junctions, and ECM proteins such as fibronectin may also play an equally important role in regulating malignancy (53-57). At the tissue level, our data suggest that cells migrating as a collective also likely employ vinculin as a force sensor at cell-cell junctions, and force responsive actin cross-linking proteins; including CAS and FERM family proteins (44,58). More broadly, our results confirm the existence of focal adhesions and focal adhesion activity in 3D and in vivo and demonstrate their functional importance by showing that they are instrumental in regulating growth factor receptor signaling (11,13,59,60). Indeed, our data showed that neither matrix stiffness nor oncogenic transformation are sufficient for malignant transformation and tumor cell invasion, suggesting ECM stiffness collaborates with key oncogenic pathways to exacerbate the potentiating mutations found in cancer. Indeed, our findings highlight vinculin in particular at the intersection of extrinsic mechanical properties and intracellular growth factor signaling (61). Moreover, our bioinformatics findings, which show amplification of adhesome molecules at the protein level across 20 solid tumor types suggests that further scrutiny of the role of up regulated adhesion proteins may be another method by which cancers may be categorized and screened (48,62,63).
The clinical relevance of our culture studies was demonstrated by our findings that there is a progressive increase in co-localized vinculin and p473Akt at the invasive tumor border in human breast cancer where the ECM is stiffest and mechanosignaling is elevated, as revealed by increased p397FAK (64) Functional links between tissue mechanics and the vinculin-PI3K phenotype was illustrated by our studies in the PyMT mouse model of mammary carcinogenesis by the loss of vinculin-p473Akt association and p397FAK when collagen crosslinking and ECM stiffening were prevented by inhibiting lysyl oxidase activity (1,65). It is therefore feasible that the concentration of vinculin at the invasive front tumors also affects the signaling of other growth and survival enhancing pathways, including enhanced Src-dependent growth or PI3K/PIP3/Akt dependent activation of mTOR and altered cell metabolism or apoptosis resistance through Bcl-2 activation of Bit1 (66-69) Accordingly, strategies to target vinculin’s scaffolding function may offer a new therapeutic approach to treat pre malignant lesions and may even help to identify high risk, non-invasive lesions (70).
Supplementary Material
Acknowledgments
We thank Kris Demali for vinculin KD MCF10A, Susan Craig for vinculin mutant plasmid constructs and vinculin null fibroblasts, Olga Ksionda and Jeroen Roose for the GDC 0941 PI3K inhibitor, Peter Marinkovich for the BM165 mouse hybridoma, Keith Mostov for the PIP activity probes, Margaret Gardel and Patrick Oakes for technical guidance, Janna Mouw for assistance with animal culture and immunohistochemistry, and Johnathan Lakins for assistance generating the lentiviral expression constructs. The work was supported by DOD NDSEG Fellowship to M.G.R, NSF GRFP to G.O., and a BCRP Scholar expansion award BC122990, NCI grants U54CA143836-01, NIH/NCI R01 CA138818-01A1, NIH/NCI RO1 CA085492-11A1, NIH/NCI U01 ES019458-01, and Susan G. Komen KG110560PP to V.M.W.
Footnotes
The authors disclose no conflicts of interest.
References
- 1.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, et al. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Molecular biology of the cell. 2009;20:3224–38. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez JI, Kang I, You W-K, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integrative biology : quantitative biosciences from nano to macro. The Royal Society of Chemistry. 2011;3:910–21. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, Oertle P, et al. Nature nanotechnology. Vol. 7. Nature Publishing Group; 2012. The nanomechanical signature of breast cancer; pp. 757–65. [DOI] [PubMed] [Google Scholar]
- 5.Egeblad M, Rasch MG, Weaver VM. Current opinion in cell biology. Vol. 22. Elsevier Ltd; 2010. Dynamic interplay between the collagen scaffold and tumor evolution; pp. 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, et al. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-β-deficient mouse mammary carcinomas. Cancer research. 2013;73:5336–46. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Oncogene. Vol. 28. Nature Publishing Group; 2009. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage; pp. 4326–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer research. 2009;69:4167–74. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shebanova O, Hammer DA. Biochemical and mechanical extracellular matrix properties dictate mammary epithelial cell motility and assembly. Biotechnology journal. 2012;7:397–408. doi: 10.1002/biot.201100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubow KE, Conrad SK, Horwitz AR. Matrix Microarchitecture and Myosin II Determine Adhesion in 3D Matrices. Current Biology. 2013;23:1607–19. doi: 10.1016/j.cub.2013.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraley SI, Feng Y, Krishnamurthy R, Kim D-H, Celedon A, Longmore GD, et al. Nature cell biology. Vol. 12. Nature Publishing Group; 2010. A distinctive role for focal adhesion proteins in three-dimensional cell motility; pp. 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubow KE, Horwitz AR. Nature cell biology. Vol. 13. Nature Publishing Group, a division of Macmillan Publishers Limited; 2011. Reducing background fluorescence reveals adhesions in 3D matrices; pp. 3–5. All Rights Reserved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harunaga JS, Yamada KM. Cell-matrix adhesions in 3D. Matrix biology : journal of the International Society for Matrix Biology. 2011;30:363–8. doi: 10.1016/j.matbio.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nature reviews Molecular cell biology. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 16.Ezzell RM, Goldmann WH, Wang N, Parashurama N, Parasharama N, Ingber DE. Vinculin promotes cell spreading by mechanically coupling integrins to the cytoskeleton. Experimental cell research. 1997;231:14–26. doi: 10.1006/excr.1996.3451. [DOI] [PubMed] [Google Scholar]
- 17.Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. The Journal of cell biology. 2007;179:1043–57. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumbauld DW, Lee TT, Singh A, Scrimgeour J, Gersbach CA, Zamir EA, et al. How vinculin regulates force transmission. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9788–93. doi: 10.1073/pnas.1216209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carisey A, Tsang R, Greiner AM, Nijenhuis N, Heath N, Nazgiewicz A, et al. Vinculin Regulates the Recruitment and Release of Core Focal Adhesion Proteins in a Force-Dependent Manner. Current Biology. 2013 doi: 10.1016/j.cub.2013.01.009. null. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golji J, Wendorff T, Mofrad MRK. Phosphorylation primes vinculin for activation. Biophysical journal. 2012;102:2022–30. doi: 10.1016/j.bpj.2012.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, et al. Nature. Vol. 466. Nature Publishing Group; 2010. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics; pp. 263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen DM, Kutscher B, Chen H, Murphy DB, Craig SW. A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. The Journal of biological chemistry. 2006;281:16006–15. doi: 10.1074/jbc.M600738200. [DOI] [PubMed] [Google Scholar]
- 23.Thievessen I, Thompson PM, Berlemont S, Plevock KM, Plotnikov SV, Zemljic-Harpf A, et al. Vinculin-actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. The Journal of cell biology. 2013;202:163–77. doi: 10.1083/jcb.201303129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Cohen DM, Choudhury DM, Kioka N, Craig SW. Spatial distribution and functional significance of activated vinculin in living cells. The Journal of cell biology. 2005;169:459–70. doi: 10.1083/jcb.200410100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lifschitz-Mercer B, Czernobilsky B, Feldberg E, Geiger B. Expression of the adherens junction protein vinculin in human basal and squamous cell tumors: Relationship to invasiveness and metastatic potential. Human Pathology. 1997;28:1230–6. doi: 10.1016/s0046-8177(97)90195-7. [DOI] [PubMed] [Google Scholar]
- 26.Xu W, Baribault H, Adamson E. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125:327–37. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- 27.Mierke CT, Kollmannsberger P, Zitterbart DP, Diez G, Koch TM, Marg S, et al. Vinculin facilitates cell invasion into three-dimensional collagen matrices. The Journal of biological chemistry. 2010;285:13121–30. doi: 10.1074/jbc.M109.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114:2553–60. doi: 10.1242/jcs.114.14.2553. [DOI] [PubMed] [Google Scholar]
- 29.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nature cell biology. 2002;4:E83–90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 30.Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. The Journal of cell biology. 2008;183:999–1005. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. The Journal of clinical investigation. 2009;119:252–66. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miroshnikova YA, Jorgens DM, Spirio L, Auer M, Sarang-Sieminski AL, Weaver VM. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Physical biology. 2011;8:026013. doi: 10.1088/1478-3975/8/2/026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng X, Cuff LE, Lawton CD, DeMali KA. Vinculin regulates cell-surface E-cadherin expression by binding to beta-catenin. Journal of cell science. 2010;123:567–77. doi: 10.1242/jcs.056432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, et al. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–97. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson KR, Leight JL, Weaver VM. Demystifying the effects of a three-dimensional microenvironment in tissue morphogenesis. Methods in cell biology. 2007;83:547–83. doi: 10.1016/S0091-679X(07)83023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paszek MJ, Dufort CC, Rubashkin MG, Davidson MW, Thorn KS, Liphardt JT, et al. Nature methods. Vol. 9. Nature Publishing Group, a division of Macmillan Publishers Limited; 2012. Scanning angle interference microscopy reveals cell dynamics at the nanoscale; pp. 825–7. All Rights Reserved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D. Substrate Compliance versus Ligand Density in Cell on Gel Responses. Biophysical Journal. 2004;86:617–28. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. Journal of cellular physiology. 2005;204:198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- 39.Meyer AS, Hughes-Alford SK, Kay JE, Castillo A, Wells A, Gertler FB, et al. 2D protrusion but not motility predicts growth factor-induced cancer cell migration in 3D collagen. The Journal of cell biology. 2012;197:721–9. doi: 10.1083/jcb.201201003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wenger MPE, Bozec L, Horton MA, Mesquida P. Mechanical Properties of Collagen Fibrils. Biophysical Journal. 2007;93:1255–63. doi: 10.1529/biophysj.106.103192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sieminski AL, Was AS, Kim G, Gong H, Kamm RD. The stiffness of three-dimensional ionic self-assembling peptide gels affects the extent of capillary-like network formation. Cell biochemistry and biophysics. 2007;49:73–83. doi: 10.1007/s12013-007-0046-1. [DOI] [PubMed] [Google Scholar]
- 42.Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nature cell biology. 2007;9:858–67. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, et al. Nature. Vol. 468. Nature Publishing Group; 2010. Nanoscale architecture of integrin-based cell adhesions; pp. 580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. The Journal of cell biology. 2010;188:11–9. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011;2:435–47. doi: 10.18632/oncotarget.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Richards DA. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biology open. 2012;1:857–62. doi: 10.1242/bio.20122071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grell P, Fabian P, Khoylou M, Radova L, Slaby O, Hrstka R, et al. International Journal of Oncology. Vol. 41. Spandidos Publications; 2012. Akt expression and compartmentalization in prediction of clinical outcome in HER2-positive metastatic breast cancer patients treated with trastuzumab; pp. 1204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Nature biotechnology. Vol. 28. Nature Publishing Group, a division of Macmillan Publishers Limited; 2010. Towards a knowledge-based Human Protein Atlas; pp. 1248–50. All Rights Reserved. [DOI] [PubMed] [Google Scholar]
- 49.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fata JE, Werb Z, Bissell MJ. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast cancer research : BCR. 2004;6:1–11. doi: 10.1186/bcr634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldmann WH, Auernheimer V, Thievessen I, Fabry B. Vinculin, cell mechanics and tumour cell invasion. Cell biology international. 2013;37:397–405. doi: 10.1002/cbin.10064. [DOI] [PubMed] [Google Scholar]
- 52.Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10334–9. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science (New York, NY) 2009;323:638–41. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roca-Cusachs P, del Rio A, Puklin-Faucher E, Gauthier NC, Biais N, Sheetz MP. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E1361–70. doi: 10.1073/pnas.1220723110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borghi N, Lowndes M, Maruthamuthu V, Gardel ML, Nelson WJ. Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13324–9. doi: 10.1073/pnas.1002662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid Shear Stress on Endothelial Cells Modulates Mechanical Tension across VE-Cadherin and PECAM-1. Current Biology. 2013:1024–30. doi: 10.1016/j.cub.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science (New York, NY) 2009;323:642–4. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 58.Friedl P, Sahai E, Weiss S, Yamada KM. Nature reviews Molecular cell biology. Vol. 13. Nature Publishing Group; 2012. New dimensions in cell migration; pp. 743–7. [DOI] [PubMed] [Google Scholar]
- 59.Starke J, Maaser K, Wehrle-Haller B, Friedl P. Mechanotransduction of mesenchymal melanoma cell invasion into 3D collagen lattices: Filopod-mediated extension-relaxation cycles and force anisotropy. Experimental cell research. 2013;319:2433–2424. doi: 10.1016/j.yexcr.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 60.Geraldo S, Simon A, Elkhatib N, Louvard D, Fetler L, Vignjevic DM. Do cancer cells have distinct adhesions in 3D collagen matrices and in vivo? European journal of cell biology. 91:930–7. doi: 10.1016/j.ejcb.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. The Biochemical journal. 2009;418:491–506. doi: 10.1042/BJ20081948. [DOI] [PubMed] [Google Scholar]
- 62.Simpson KJ, Selfors LM, Bui J, Reynolds A, Leake D, Khvorova A, et al. Nature cell biology. Vol. 10. Nature Publishing Group; 2008. Identification of genes that regulate epithelial cell migration using an siRNA screening approach; pp. 1027–38. [DOI] [PubMed] [Google Scholar]
- 63.Kohn KW, Zeeberg BR, Reinhold WC, Sunshine M, Luna A, Pommier Y. Gene expression profiles of the NCI-60 human tumor cell lines define molecular interaction networks governing cell migration processes. PloS one. 2012;7:e35716. doi: 10.1371/journal.pone.0035716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ganguly KK. Journal of Cancer Therapy. 03. Scientific Research Publishing; 2012. Studies on Focal Adhesion Kinase in Human Breast Cancer Tissue; pp. 7–19. [Google Scholar]
- 65.Kanapathipillai M, Mammoto A, Mammoto T, Kang JH, Jiang E, Ghosh K, et al. Inhibition of mammary tumor growth using lysyl oxidase-targeting nanoparticles to modify extracellular matrix. Nano letters American Chemical Society. 2012;12:3213–7. doi: 10.1021/nl301206p. [DOI] [PubMed] [Google Scholar]
- 66.Holle AW, Tang X, Vijayraghavan D, Vincent LG, Fuhrmann A, Choi YS, et al. In situ mechanotransduction via vinculin regulates stem cell differentiation. Stem cells (Dayton, Ohio) 2013;31:2467–77. doi: 10.1002/stem.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR Leads to Adaptive Resistance in Matrix-Attached Cancer Cells. Cancer Cell. 2012;21:227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park CC, Zhang H, Pallavicini M, Gray JW, Baehner F, Park CJ, et al. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer research. 2006;66:1526–35. doi: 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jan Y, Matter M, Pai J, Chen Y-L, Pilch J, Komatsu M, et al. A Mitochondrial Protein, Bit1, Mediates Apoptosis Regulated by Integrins and Groucho/TLE Corepressors. Cell. 2004;116:751–62. doi: 10.1016/s0092-8674(04)00204-1. [DOI] [PubMed] [Google Scholar]
- 70.Esserman L, Shieh Y, Thompson I. Rethinking screening for breast cancer and prostate cancer. JAMA : the journal of the American Medical Association American Medical Association. 2009;302:1685–92. doi: 10.1001/jama.2009.1498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.