

Abstract
Objective
Friedreich ataxia (FRDA) is caused by an expanded GAA triplet-repeat (GAA-TR) mutation in the FXN gene. Patients are typically homozygous for expanded alleles containing 100–1300 triplets, and phenotypic severity is significantly correlated with the length of the shorter of the two expanded alleles. Patients have a severe deficiency of FXN transcript, which is predominantly caused by epigenetic silencing of the FXN promoter. We sought to determine if the severity of FXN promoter silencing is related to the length of the expanded GAA-TR mutation in FRDA.
Methods
Patient-derived lymphoblastoid cell lines bearing a range of expanded alleles (200–1122 triplets) were evaluated for FXN transcript levels by quantitative RT-PCR. FXN promoter function was directly measured by quantitative analysis of transcriptional initiation via metabolic labeling of newly synthesized transcripts in living cells.
Results
FXN transcriptional deficiency was significantly correlated with the length of the shorter of the two expanded alleles, which was noted both upstream (R2=0.84; p=0.014) and downstream (R2=0.89; p=0.002) of the expanded GAA-TR mutation, suggesting that FXN promoter silencing in FRDA is related to repeat length. A bilinear regression model revealed that length-dependence was strongest when the shorter of the two expanded alleles contained <400 triplets. Direct measurement of FXN promoter activity in patients with expanded alleles containing <400 versus >400 triplets in the shorter of the two expanded alleles revealed a significantly greater deficiency in individuals with longer GAA-TR alleles (p<0.05).
Interpretation
FXN promoter silencing in FRDA is dependent on the length of the expanded GAA-TR mutation.
Introduction
Friedreich ataxia (FRDA),1 the most common inherited ataxia, is characterized by slowly progressive sensory ataxia, dysarthria, absent lower limb reflexes, loss of position and vibration senses, and secondary skeletal abnormalities. Two-thirds of patients have cardiomyopathy and as many as 30% develop diabetes mellitus. The age of onset is typically between 10 – 15 y, and most patients are symptomatic by 25 y. However, about 25% of patients have atypical FRDA; presenting with later age of onset (>25 y) and / or with retention of tendon reflexes.
The vast majority of patients are homozygous for an abnormally expanded GAA triplet-repeat (GAA-TR) mutation in intron 1 of the FXN gene.2 Whereas unaffected individuals have alleles with <30 triplets, patients typically have two expanded alleles, each containing 100 – 1300 triplets. Phenotypic severity, including the age of onset, rate of progression (e.g. duration until wheelchair use), prevalence and severity of cardiomyopathy, and secondary skeletal abnormalities such as scoliosis, shows a strong correlation with the length of the expanded GAA-TR mutation.3–7 The strongest genotype-phenotype correlation is seen with the size of the shorter of the two expanded alleles.
The expanded GAA-TR mutation results in severe deficiency of FXN transcript8 that ultimately leads to deficiency of the mitochondrial protein frataxin. While impeded transcriptional elongation through the expanded repeat contributes to the transcriptional deficiency,8–11 the predominant mechanism by which the expanded GAA-TR mutation results in transcriptional deficiency in FRDA is by epigenetic silencing of the FXN gene promoter that leads to a severe deficiency of transcriptional initiation.11, 12 Given that the length of the expanded GAA-TR mutation (especially the shorter of the two alleles) correlates well with phenotypic severity3–7 and the level of transcriptional deficiency in FRDA,13 we hypothesized that epigenetic silencing of the FXN gene promoter would be dependent on the length of the expanded GAA-TR mutation. Here we show that the length of the shorter of the two GAA-TR mutations correlates well with the severity of FXN promoter silencing. Therefore, variable silencing of the FXN gene promoter, modulated by the length of the expanded GAA-TR mutation, is a critical underlying molecular mechanism of the variability of phenotypic severity in FRDA.
Materials and Methods
Cell lines
Lymphoblastoid cell lines from three healthy subjects, three asymptomatic carriers, and 16 individuals with FRDA were obtained from Coriell Cell Repositories. All individuals with FRDA were homozygous for expanded GAA-TR alleles that ranged in size from 200 to 1122 triplets (shorter GAA-TR allele range: 200 – 925 triplets; longer GAA-TR allele range: 500 – 1122 triplets). The three asymptomatic carriers were heterozygous for expanded GAA-TR alleles containing 570, 760, and 1285 triplets.
Quantitative measurement of FXN transcript
This was done by reverse transcription followed by quantitative polymerase chain reaction (quantitative RT-PCR), using protocols and primer sequences as previously described.12 Briefly, reverse transcription was performed either with a mixture of random hexamers and oligo dT primers (for the exon 3-4 amplicon) or with a strand-specific RT primer (for the exon 1 amplicon) using the QuantiTect® reverse transcription kit (Qiagen). mRNA levels were quantified by real-time PCR relative to expression of the control TBP gene using the ΔΔCt method on the Eppendorf RealPlex-4 Mastercycler with SsoAdvanced™ SYBR® green supermix (BioRad).
Quantitative analysis of FXN promoter activity
This was done by metabolic labeling of nascent RNA using the Click-iT® Nascent RNA Capture Kit (Life Technologies) as previously described.12 Briefly, lymphoblastoid cell lines were incubated with 5-ethynyl uridine. Following 2h and 4h of incubation, RNA was extracted and used for biotinylation by Click reaction. Biotinylated RNA bound to streptavidin beads was reverse transcribed using the SuperScript® VILO™ cDNA synthesis kit (Life Technologies). Transcript levels were quantified by real-time PCR relative to expression of the control TBP gene from total biotinylated RNA using the ΔΔCt method on the Eppendorf RealPlex-4 Mastercycler with SsoAdvanced SYBR green supermix (BioRad).
Statistical methods
Linear and piecewise bilinear regressions were performed in SAS to assess correlation between relative transcript level and expanded GAA-TR allele length. Piecewise bilinear regression was used to identify a possible breakpoint, i.e. the estimated point at which the slope of the regression line changes. An estimate of the breakpoint and corresponding 95% confidence interval were obtained for short and long GAA-TR alleles and also the average of the two expanded GAA-TR alleles. The slope of the piecewise bilinear regression equation was tested for significant deviation from zero before and after the breakpoint. R-square values for the linear and piecewise bilinear regression were used to assess model improvement.
Results
Transcriptional deficiency in FRDA occurs upstream and downstream of the expanded GAA-TR mutation and is correlated with the length of the repeat
We measured FXN transcript levels both upstream and downstream of the expanded GAA-TR mutation, in order to differentiate between defects in transcriptional initiation and transcriptional elongation through the expanded repeat (Fig. 1A). For the upstream location, quantitative RT-PCR was performed in the immediate vicinity of the transcription start site of the FXN gene (Ex1), and the spliced product of exons 3 & 4 (Ex3-Ex4) was selected as the downstream location. Deficiency of transcript at both locations would suggest a defect in transcriptional initiation, but deficiency of only Ex3-Ex4 would suggest a defect in transcriptional elongation (Fig. 1A). In agreement with previous findings11, 12 we noted a severe deficiency of FXN transcript in FRDA cells at both the upstream and downstream locations, supporting the existence of a major defect in transcriptional initiation (Fig. 1B). As expected, heterozygous carriers of the expanded GAA-TR mutation showed levels of FXN transcript that were intermediate between FRDA patients and non-FRDA controls at both locations (Fig. 1B).
Figure 1. FXN transcriptional deficiency in FRDA extends upstream and downstream of the expanded GAA-TR mutation.

(A) Relevant portions of the FXN gene are depicted schematically, with the GAA-TR mutation in intron 1 and the FXN transcriptional start site (TSS) at −59 (position numbers are relative to the initiation codon, indicated as +1). FXN transcript was quantitatively measured both upstream (Ex1; in the vicinity of the TSS) and downstream (Ex3-Ex4) of the GAA-TR mutation. Solid lines depict the full-length FXN transcript and shorter predicted transcripts caused by defects in transcriptional elongation through the expanded GAA-TR mutation and by deficient transcriptional initiation due to FXN promoter silencing. Deficiency of transcript at both locations would suggest a defect in transcriptional initiation, and deficiency of only Ex3-Ex4 would suggest a defect in transcriptional elongation. (B) Quantitative RT-PCR showing significantly reduced amounts of FXN mRNA both upstream (Ex1) and downstream (Ex3-Ex4) of the expanded GAA-TR mutation in FRDA versus non-FRDA (CNTR) lymphoblastoid cells. Heterozygous carriers of the GAA-TR mutation (CARRIER) show transcript levels that are intermediate between FRDA and non-FRDA controls. Graphs represent the cumulative data from two complete experiments using three FRDA, three heterozygous carriers, and three non-FRDA (CNTR) lymphoblastoid cell lines, each assayed in triplicate. Error bars represent +/− SEM. ** = p<0.01, *** = p<0.001.
To test if transcriptional deficiency at both upstream and downstream locations was determined by the length of the expanded GAA-TR mutation, quantitative RT-PCR analysis was performed using cell lines from 16 FRDA patients who were homozygous for a wide range of expanded GAA-TR alleles (200 – 1122 triplets). A simple linear regression showed statistically significant, but moderate, correlations between FXN transcript levels and three separate measures of the length of the GAA-TR mutation; the lengths of the shorter, the longer and the average of the two expanded GAA-TR alleles (Table 1). However, the best fit was obtained with a piecewise bilinear regression model, which considerably enhanced the ability to account for the variation in transcript levels compared with the simple linear regression. However, this increased ability to account for repeat length-dependent transcript levels was seen only with the length of the shorter and the average of the two expanded GAA-TR alleles. An increase of ~35% in the R-square for the shorter allele and 13–22% for the average of the two expanded alleles was noted at both Ex1 and Ex3-Ex4 (Table 1). The bilinear regression model predicted a breakpoint, i.e. a point in the regression line at which the slope changed. This indicated that expanded GAA-TR alleles that were shorter than this threshold length showed very strong correlation with transcript levels that was statistically significant, but no correlation was seen with expanded alleles that were longer than this threshold (Fig. 2A & 2B; Table 1). This indicates that most of the variability in transcript levels is determined by variation in allele sizes up to ~400 triplets (423 triplets, 95%CI = 353,494 for Ex1 and 405 triplets, 95%CI = 346,464 for Ex3-Ex4) in the shorter of the two expanded alleles or up to ~700 triplets (706 triplets, 95%CI = 607,804 for Ex1 and 730 triplets, 95%CI = 564,896 for Ex3-Ex4) as the average of the two expanded alleles. Indeed, no further reduction in transcript levels was noted when expanded alleles contained >400 triplets in the shorter of the two expanded alleles or >700 triplets as the average of the two expanded alleles (Fig. 2A & 2B; Table 1).
Table 1.
Transcriptional deficiency in FRDA is modulated by the shorter of the two expanded GAA-TR alleles containing <400 triplets
| Linear
|
Piecewise Bilinear Regression
|
||||||
|---|---|---|---|---|---|---|---|
| Regression | |||||||
|
| |||||||
| GAA-TR allele* | Region | R2 | p | R2 | p, before breakpoint | Breakpoint (95% C.I.), triplets | p, after breakpoint |
|
| |||||||
| Short | Ex1 | 0.49 | 0.003 | 0.84 | 0.014 | 423 (353–494) | n.s. |
|
| |||||||
| Ex3-Ex4 | 0.56 | 0.001 | 0.89 | 0.002 | 405 (346–464) | n.s. | |
|
| |||||||
| Average | Ex1 | 0.57 | 0.001 | 0.79 | 0.021 | 706 (607–804) | n.s. |
|
| |||||||
| Ex3-Ex4 | 0.55 | 0.001 | 0.68 | 0.031 | 730 (564–896) | n.s. | |
|
| |||||||
| Long | Ex1 | 0.42 | 0.007 | 0.56 | 0.045 | 854 (649–1059) | n.s. |
|
| |||||||
| Ex3-Ex4 | 0.32 | 0.022 | 0.38 | n.s. | 853 (454–1251) | n.s. | |
Short / long / average refer to the shorter / longer / average of the two expanded GAA-TR alleles; C.I. = confidence interval; n.s. = not significant.
Figure 2. Repeat length-dependent FXN transcriptional deficiency in FRDA is modulated by the shorter of the two expanded GAA-TR alleles containing <400 triplets.
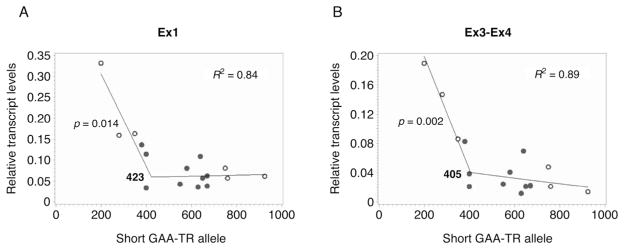
Relative FXN transcript levels (Y axis) of FRDA patients (n = 16), homozygous for a wide range of expanded GAA-TR alleles (200 – 1122 triplets), plotted against the length of the shorter of the two expanded alleles (X axis). (A) A piecewise bilinear regression model showed strong correlation (R2 = 0.84) between FXN transcript levels measured at Ex1 and the length of the shorter GAA-TR allele; highly significant correlation (p = 0.014) was seen with alleles shorter than the estimated breakpoint of 423 triplets (95% CI, 353 – 494 triplets). (B) A piecewise bilinear regression model showed strong correlation (R2 = 0.89) between FXN transcript levels measured at Ex3-Ex4 and the length of the shorter GAA-TR allele; highly significant correlation (p = 0.002) was seen with alleles shorter than the estimated breakpoint of 405 triplets (95% CI, 346 – 464 triplets). Note: open circles depict the six cell lines that were used for assessing repeat length-dependent FXN promoter silencing in Fig. 3B.
FXN promoter silencing in FRDA is dependent on the length of the expanded GAA-TR mutation
FRDA patients have a severe deficiency of transcriptional initiation due to epigenetic silencing of the FXN gene promoter.11, 12 Whereas FXN promoter silencing is caused by the upstream spread of repressive chromatin from the expanded GAA-TR mutation,12 it remains unknown if promoter function is modulated by the length of the repeat. We first tested FXN promoter activity by measuring FXN transcriptional initiation via metabolic labeling of nascent transcripts in three non-FRDA controls and three FRDA patient cell lines (homozygous for expanded GAA-TR alleles containing 750–1122 triplets). Quantitative RT-PCR was used to measure FXN transcript levels at Ex1, i.e., in the immediate vicinity of the transcription start site, following 2h and 4h of labeling of newly formed FXN transcript. We detected a severe deficiency of newly synthesized FXN transcript in FRDA at both the 2h (19-fold less; p<0.001) and 4h (15-fold less; p<0.001) time points (Fig. 3A). This deficiency of dynamic accumulation of FXN transcript levels noted in the immediate vicinity of the transcription start site indicates a severe deficiency of transcriptional initiation in FRDA.
Figure 3. FXN promoter silencing in FRDA is dependent on the length of the expanded GAA-TR mutation.
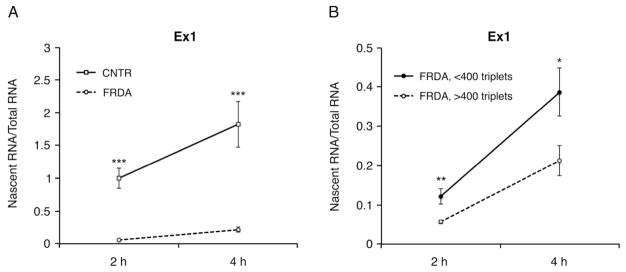
(A) Quantitative RT-PCR of metabolically labeled nascent FXN transcript for the indicated incubation times (2h & 4h) is shown at Ex1, i.e., in the vicinity of the transcription start site, for FRDA patients (n = 3) and non-FRDA controls (CNTR; n = 3). FRDA cells showed 15–19 fold less nascent FXN transcript compared with non-FRDA cells at both time points assayed. The graph represents cumulative data from two complete experiments with each cell line assayed in triplicate. Error bars represent +/− SEM. *** = P<0.001. (B) Quantitative RT-PCR of metabolically labeled FXN nascent transcript for the indicated incubation times (2h & 4h) is shown at Ex1, for FRDA patients with the shorter of the two expanded GAA-TR alleles containing <400 triplets (n = 3) and >400 triplets (n = 3). FRDA cells with shorter GAA-TR alleles showed 2-fold more nascent FXN transcript compared with FRDA cells with longer GAA-TR alleles at both time points assayed. The graph represents cumulative data from two complete experiments with each cell line assayed in triplicate. Error bars represent +/− SEM. * = p<0.05, ** = p<0.01.
In order to determine if FXN promoter silencing and the deficiency of transcriptional initiation are dependent on repeat length, we compared newly synthesized transcript levels in three FRDA cell lines in which the shorter of the two expanded alleles contained <400 triplets (200, 280 & 350 triplets) versus three FRDA cell lines in which the shorter of the two expanded alleles contained >400 triplets (750, 760 & 925 triplets; these six cell lines are depicted by open circles in Fig. 2A and 2B). The threshold length of 400 triplets was based on the breakpoint identified via bilinear regression, and was designed to test the hypothesis that the higher levels of FXN transcript noted with relatively short GAA-TR mutations could be explained by a correspondingly lesser severity of FXN promoter silencing. Indeed, FXN transcript levels at Ex1 revealed a significantly lower deficiency of newly synthesized FXN transcript in FRDA cell lines containing the shorter repeats at both the 2h (2-fold less; p<0.01) and 4h (1.8-fold less; p<0.05) time points (Fig. 3B). These data indicate that FXN promoter silencing and the resultant deficiency of transcriptional initiation are dependent on the length of the expanded GAA-TR mutation in FRDA.
Discussion
FXN transcriptional deficiency in FRDA is caused by at least two mechanisms. Impeded transcriptional elongation through the expanded GAA-TR contributes some of the deficiency,8–11 which is caused by a combination of repeat-proximal heterochromatin11, 14, 15 and abnormal DNA structures assumed by the expanded repeat.8–10, 16 However, the predominant mechanism is deficient transcriptional initiation caused by epigenetic silencing of the FXN gene promoter.11, 12 Until the discovery of the latter mechanism, deficient transcriptional elongation was considered to be the major cause for FXN transcriptional deficiency. Indeed, it is intuitive to think of long repeat tracts as posing a length-dependent barrier to transcriptional elongation, thus explaining the length-dependent deficiency of FXN transcript and correlation with phenotypic severity. However, epigenetic silencing of the FXN promoter produces a deficiency of the entire FXN transcript, i.e., both upstream and downstream of the expanded GAA-TR mutation – and not only downstream of the expanded GAA-TR, as would be expected with deficient transcriptional elongation. This led us to test the alternate hypothesis that FXN promoter silencing, and therefore transcriptional deficiency upstream (and downstream) of the expanded GAA-TR, would be dependent on the length of the expanded GAA-TR in FRDA.
To our knowledge, this is the first assessment of the severity of promoter silencing and the level of FXN transcriptional deficiency upstream of the expanded GAA-TR as a function of GAA-TR length in FRDA. We show that transcriptional deficiency upstream of the expanded GAA-TR is correlated significantly with repeat length. Furthermore, via direct measurement of promoter function we show that silencing of the FXN gene promoter is also related to the length of the expanded GAA-TR. These observations serve to further substantiate the causal relationship between the expanded GAA-TR mutation in intron 1 and silencing of the FXN gene promoter.
Our data support the notion that repeat-mediated variability in epigenetic silencing of FXN promoter function is an important determinant of phenotypic variability in FRDA. It is known that the length of the shorter of the two expanded alleles accounts for most of the genotype-phenotype correlation in FRDA.3–7 For instance, its length is estimated to account for as much as 50% of the variation in age of onset.4 Indeed, individuals with ages of onset >25 y and >40 y typically have at least one expanded allele that contains <500 and <300 triplets, respectively.17 Durr et al.3 found that the correlation between age of onset and the length of the shorter of the two expanded GAA-TR alleles was best explained by a quadratic regression model wherein alleles that contained <500 triplets had an inordinately greater contribution to phenotypic variability compared with alleles that contained >500 triplets. In a longitudinal natural history study over 2 years using a large heterogeneous cohort stratified by the size of the shorter of two GAA-TR alleles, Regner et al.18 showed that individuals with <300 triplets progressed slowly and with a high degree of variability compared to individuals with longer GAA-TR alleles. Similarly, albeit using another ataxia rating scale, Metz and collegues19 found that the rate of disease progression as a function of the length of the shorter of the two expanded GAA-TR alleles was most prominent with alleles containing <600 triplets. All of these observations are congruous with our finding that the strongest correlation between repeat length and FXN transcriptional deficiency is seen when the shorter of the two expanded GAA-TR alleles contains <400 triplets. Indeed, small changes in the length of the expanded GAA-TR allele have a striking effect on FXN transcript level as long as the shorter of the two expanded alleles contains <400 triplets. Furthermore, using a quantitative assay for DNA methylation, Evans-Galea et al.15 found that the severity of repeat–proximal heterochromatin was inversely correlated with FXN expression and the age of onset. Taken together, these data indicate that longer GAA-TR alleles result in more severe epigenetic silencing of the FXN gene, and the length of the shorter of the two expanded alleles serves as a quantitative modulator of epigenetic silencing, and thereby the phenotypic severity in FRDA.
A potential caveat of our study is that it relies on the measurement of expanded GAA-TR alleles and the function of the FXN gene promoter in (early passages of) patient-derived lymphoblastoid cell lines. Additional experiments would have to be conducted to confirm the validity of our data in other cell types and tissues. Another relevant question is how the absolute lengths of the expanded GAA-TR alleles in lymphoblastoid cell lines compare with those seen in cell types typically used for diagnosis of patients, e.g. peripheral blood leukocytes. Sizes of expanded GAA-TR alleles seen in early passages of lymphoblastoid cell lines when compared with the blood samples they were derived from are either not different in length (40%) or are shorter by ~100 triplets (60%).20 This suggests that our estimate of a threshold length of ~400 triplets in the shorter of the two expanded GAA-TR alleles will likely be comparable to that seen in peripheral blood leucocytes. It also remains to be seen how well the expanded GAA-TR allele lengths as measured in lymphoblastoid cell lines or peripheral blood leucocytes correlate with those seen in disease-relevant tissues such as dorsal root ganglia (DRG). Despite the clear age-dependent propensity for accumulation of larger expanded GAA-TR alleles in a subset of DRG cells, the constitutional allele lengths measured by conventional diagnostic assays showed relatively small length changes across various neuronal and non-neuronal tissues from autopsies.21 With the obvious caveat that the latter studies have only been conducted in few individuals, these data suggest that the measurement of expanded GAA-TR allele lengths in early-passage lymphoblastoid cell lines or peripheral blood leukocytes, at least using conventional assays, is likely a reasonable indicator of the constitutional GAA-TR alleles in FRDA.
Since the expanded GAA-TR mutation is located in an intron, the coding sequence of the FXN gene remains intact in individuals with FRDA. Reactivation of the transcriptionally silenced FXN gene in FRDA is therefore an attractive therapeutic strategy because it would be expected to produce a normally functional frataxin protein. Indeed, several candidate molecules designed to increase FXN transcriptional activity are currently in various stages of preclinical and clinical development. Among these molecules, 2-aminobenzamide derivatives14, 22, 23 and nicotinamide24, 25 serve as histone deacetylase inhibitors that specifically reverse FRDA-associated heterochromatin. However, it is presently unknown if these drugs, and those being developed by others, result in reversal of the transcriptional initiation and / or elongation defects in FRDA. Our data indicate that therapies designed to reverse the epigenetic defect in FRDA would be most effective if they reactivated the silenced FXN promoter, with the possibility of deriving further benefit from reversal of the transcriptional elongation defect.
Acknowledgments
This research was supported by a grant from the National Institutes of Health (R01 NS072418) to S.I.B.
References
- 1.Bidichandani SI, Delatycki MB. Friedreich Ataxia. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 2.Campuzano V, Montermini L, Molto MD, Pianese L, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 3.Durr A, Cossee M, Agid Y, Campuzano V, et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335:1169–1175. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- 4.Filla A, De Michele G, Cavalcanti F, Pianese L, et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet. 1996;59:554–560. [PMC free article] [PubMed] [Google Scholar]
- 5.Monros E, Molto MD, Martinez F, Canizares J, et al. Phenotype correlation and intergenerational dynamics of the Friedreich ataxia GAA trinucleotide repeat. Am J Hum Genet. 1997;61:101–110. doi: 10.1086/513887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montermini L, Richter A, Morgan K, Justice CM, et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997;41:675–682. doi: 10.1002/ana.410410518. [DOI] [PubMed] [Google Scholar]
- 7.Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, et al. Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet. 1999;87:168–174. doi: 10.1002/(sici)1096-8628(19991119)87:2<168::aid-ajmg8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62:111–121. doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohshima K, Montermini L, Wells RD, Pandolfo M. Inhibitory effects of expanded GAA. TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J Biol Chem. 1998;273:14588–14595. doi: 10.1074/jbc.273.23.14588. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J Biol Chem. 2001;276:27171–27177. doi: 10.1074/jbc.M101879200. [DOI] [PubMed] [Google Scholar]
- 11.Kumari D, Biacsi RE, Usdin K. Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J Biol Chem. 2011;286:4209–4215. doi: 10.1074/jbc.M110.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chutake YK, Costello WN, Lam C, Bidichandani SI. Altered Nucleosome Positioning at the Transcription Start Site and Deficient Transcriptional Initiation in Friedreich Ataxia. J Biol Chem. 2014;289:15194–15202. doi: 10.1074/jbc.M114.566414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pianese L, Turano M, Lo Casale MS, De Biase I, et al. Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J Neurol Neurosurg Psychiatry. 2004;75:1061–1063. doi: 10.1136/jnnp.2003.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 15.Evans-Galea MV, Carrodus N, Rowley SM, Corben LA, et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann Neurol. 2012;71:487–497. doi: 10.1002/ana.22671. [DOI] [PubMed] [Google Scholar]
- 16.Burnett R, Melander C, Puckett JW, Son LS, et al. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA. TTC repeats in Friedreich’s ataxia. Proc Natl Acad Sci U S A. 2006;103:11497–11502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bidichandani SI, Garcia CA, Patel PI, Dimachkie MM. Very late-onset Friedreich ataxia despite large GAA triplet repeat expansions. Arch Neurol. 2000;57:246–251. doi: 10.1001/archneur.57.2.246. [DOI] [PubMed] [Google Scholar]
- 18.Regner SR, Lagedrost SJ, Plappert T, Paulsen EK, et al. Analysis of echocardiograms in a large heterogeneous cohort of patients with friedreich ataxia. Am J Cardiol. 2012;109:401–405. doi: 10.1016/j.amjcard.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 19.Metz G, Coppard N, Cooper JM, Delatycki MB, et al. Rating disease progression of Friedreich’s ataxia by the International Cooperative Ataxia Rating Scale: analysis of a 603-patient database. Brain. 2013;136:259–268. doi: 10.1093/brain/aws309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bidichandani SI, Purandare SM, Taylor EE, Gumin G, et al. Somatic sequence variation at the Friedreich ataxia locus includes complete contraction of the expanded GAA triplet repeat, significant length variation in serially passaged lymphoblasts and enhanced mutagenesis in the flanking sequence. Hum Mol Genet. 1999;8:2425–2436. doi: 10.1093/hmg/8.13.2425. [DOI] [PubMed] [Google Scholar]
- 21.De Biase I, Rasmussen A, Endres D, Al-Mahdawi S, et al. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007;61:55–60. doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- 22.Gottesfeld JM, Rusche JR, Pandolfo M. Increasing frataxin gene expression with histone deacetylase inhibitors as a therapeutic approach for Friedreich’s ataxia. J Neurochem. 2013;126 (Suppl 1):147–154. doi: 10.1111/jnc.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plasterer HL, Deutsch EC, Belmonte M, Egan E, Lynch DR, Rusche JR. Development of frataxin gene expression measures for the evaluation of experimental treatments in Friedreich’s ataxia. PLoS One. 2013;8:e63958. doi: 10.1371/journal.pone.0063958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan PK, Torres R, Yandim C, Law PP, et al. Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich’s ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet. 2013;22:2662–2675. doi: 10.1093/hmg/ddt115. [DOI] [PubMed] [Google Scholar]
- 25.Libri V, Yandim C, Athanasopoulos S, Loyse N, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: an exploratory, open-label, dose-escalation study. Lancet. 2014 doi: 10.1016/S0140-6736(14)60382-2. [DOI] [PubMed] [Google Scholar]