

Abstract
Although chelator-based radiolabeling techniques have been used for decades, concerns about the complexity of coordination chemistry, possible altering of pharmacokinetics of carriers, and potential detachment of radioisotopes during imaging have driven the need for developing a simple yet better technique for future radiolabeling. Here, the emerging concept of intrinsically radiolabeled nanoparticles, which could be synthesized using methods such as hot-plus-cold precursors, specific trapping, cation exchange, and proton beam activation, is introduced. Representative examples of using these multifunctional nanoparticles for multimodality molecular imaging are highlighted together with current challenges and future research directions. Although still in the early stages, design and synthesis of intrinsically radiolabeled nanoparticles has shown attractive potential to offer easier, faster, and more specific radiolabeling possibilities for the next generation of molecular imaging.
1. Introduction
With the rapid growing interests in using radioisotopes for nanooncology, a broad spectrum of radiotracers has been generated for positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging in different diseases.[1,2] So far, different radioisotopes have been labeled to carriers, such as antibodies, peptides, nanoparticles, and so forth, for in vivo biomarker expression level imaging, early tumor detection, drug biodistribution pattern studies, and so on.[3–5] The most widely used radiolabeling strategy involves the use of exogenous chelators which could coordinate with certain radioisotopes to form stable complexes.[6,7] Well-established chelators, such as 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), p-isothiocyanatobenzyl-desferrioxamine (Df-Bz-NCS) and diethylene triamine pentaacetic acid (DTPA), etc., have been employed for radiolabeling of copper-64 (64Cu, t1/2 = 12.7 h), zirconium-89 (89Zr, t1/2= 78.4 h) and indium-111 (111In, t1/2= 2.8 d) for imaging in preclinical studies.[8,9] Different isotopes vary significantly in their coordination chemistry, making selection of the right chelator for a specific isotope vital; however, this could be tricky and even impossible to achieve in some cases. For example, so far, it is still a major challenge to radiolabel certain isotopes such as arsenic-72 (72As, t1/2 = 26 h) and germanium-69 (69Ge, t1/2 = 39.1 h). In addition, although most of the radiolabeling could be done under mild conditions, in some cases successful radiolabeling might require very harsh conditions (e.g. high reaction temperature with prolonged incubation time), causing the possible degradation of small biomolecules or antibodies.
The other concerns of using traditional radiolabeling strategies include the possible altering of pharmacokinetics of carriers and potential detachment of radioisotopes, which could lead to problems such as off-targeting and false positives. Studies performed with radiolabeled monoclonal antibodies demonstrated that the incorporation of the chelator may not always be site specific, and could adversely affect the biological behaviour of radio-immunoconjugates.[10,11] Since the carrier itself is not labeled and PET (or SPECT) only detects signals from the radioisotope, the integrity (or stability) of the radiolabeled system in the complicated physiological environment should always be well-addressed before in vivo imaging applications. Detachment of the radioisotopes from the carriers can also lead to potential transchelation to proteins, causing erroneous interpretation of the results.[12,13] Therefore, successful chelator-based radiolabeling requires in-depth knowledge of the coordination chemistry and selection of the best chelator for every radioisotope.
To address these concerns, recent research has been focusing on developing more reliable chelator-free radiolabeling techniques, which could fully take advantage of the unique physical and chemical properties of well-selected inorganic or organic nanoparticles for radiolabeling, and more importantly, offer an easier, faster, and more specific radiolabeling possibility. Herein, we introduce the emerging concept of intrinsically radiolabeled nanoparticles, which could be synthesized, so far, via four different methods without using any chelators. Mechanisms behind each synthetic method will be discussed and compared, with our focus on the simplicity as well as stability of the resulting nanoparticles. Representative examples of using these multifunctional nanoparticles for multimodality molecular imaging will also be highlighted. Finally, we will discuss the current challenges and future research directions in this field.
2. Synthetic Strategies and Multimodality Imaging
Although intrinsic radiolabeling of nanoparticles with PET/SPECT isotopes is a relatively new area of research, the first report of this kind possibly dates back to 1979, when cabon-14 (14C, t1/2 = 5700 y) labeled poly(methyl-2–14C-methacrylate) nanoparticles were synthesized and used for studying the in vivo distribution and elimination patterns in mice and rats.[14] In the following sections, we will discuss the latest synthetic strategies for various kinds of intrinsically radiolabeled nanoparticles, starting with the addition of radioactive precursors during the reaction.
2.1. Hot-plus-Cold Precursors
Adding trace levels of radioactive (or hot) precursors together with normal non-radioactive (or cold) precursors during the synthesis of nanoparticles is a simple, straightforward, and also the most widely used method to achieve intrinsically radioactive nanoparticles. Using this method, selected radioisotopes could be well embedded into the crystal lattice of final nanocrystals, resulting in intrinsically radioactive nanoparticles with high stability.
The 64Cu-labeled copper sulfide (CuS) nanoparticles are one such class of nanoparticles which could be easily prepared within 1 hour by adding 64CuCl2 (hot precursor) into the mixture of CuCl2 and Na2S (cold precursors) in a 95 °C water bath (Figure 1 a).[15] Semiconductor CuS nanoparticles are a new class of photo-thermal coupling agents which can absorb light in the near-infrared (NIR) region and convert it to thermal energy for thermal ablation of tumor cells.[15,16] [64Cu]CuS nanoparticles synthesized in this study showed not only the photo-thermal property from the CuS matrix, but also PET imaging capability thanks to the doping of 64Cu into the CuS crystal structure. After PEGylation, PEG-[64Cu]CuS nanoparticles allowed for clear visualization of U87MG glioblastoma xenografts 24 h post injection (p.i.) through enhanced permeability and retention (EPR) effect (Figure 1 a), showing great potential for synergetic cancer imaging and photo-thermal therapy.
Figure 1.
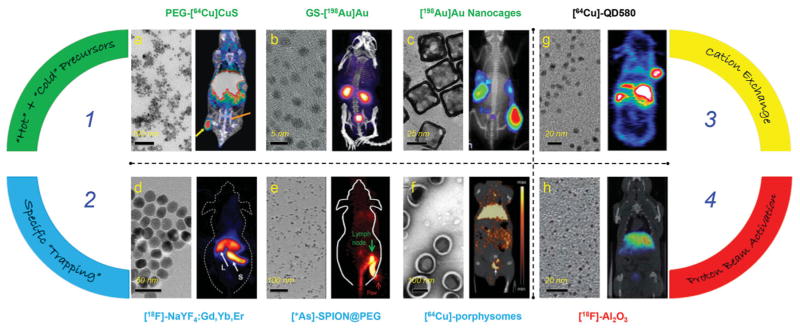
Representative transmission electron microscopy (TEM) (left panel) and in vivo PET/SPECT (right panel) images of different intrinsically radiolabeled nanoparticles prepared through four different methods. Route 1: Hot-plus-cold precursors. a) TEM image of PEG-[64Cu]CuS nanoparticles and PET/CT image of U87MG xenograft mouse at 24 h p.i. Tumor and bladder were marked with yellow and orange arrows, respectively. Reproduced with permission.[15] Copyright 2010, American Chemical Society. b) TEM image of GS-[198Au]Au nanoparticles and SPECT/CT image of mouse acquired 10 minutes after injection. Reproduced with permission.[19] Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. c) TEM image of [198Au]Au nanocages and 198Au induced Cerenkov luminescence image in EMT-6 tumor bearing mice at 24 h p.i. Reproduced with permission.[20] Copyright 2013, American Chemical Society. Route 2: Specific trapping. d) TEM image of [18F]-NaYF4:Gd3+/Yb3+/Er3+ and whole-body micro-PET image taken 15 minutes p.i. The arrows point at liver (L) and spleen (S). Reproduced with permission.[30] Copyright 2011, Elsevier. e) TEM image of water soluble SPIONs and lymph node PET imaging with *As-SPION@PEG (* = 71, 72, 74, 76) at 2.5 h p.i. Green and red arrows point to lymph node and paw, respectively. Reproduced with permission.[33] Copyright 2013, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. f) TEM image of porphysomes and micro PET/CT image in orthotopic PC3 tumor using [64Cu]-porphysomes at 24 h p.i. White arrow points to prostate tumor. Reproduced with permission.[36] Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Route 3: Cation exchange. g) TEM image of [64Cu]-QD580 and coronal PET image of [64Cu]-QD580 in U87MG bearing mouse at 17 h p.i. Reproduced with permission.[40] Copyright 2014, American Chemical Society. Route 4: Proton beam activation. h) TEM image of [18 F]-Al2O3 nanoparticles and PET/CT fused image at 60–80 min p.i. Reproduced with permission.[44] Copyright 2012, American Chemical Society.
Besides CuS nanoparticles, 64Cu has also been integrated into other platforms such as iron oxide (Fe3O4) and gold (Au) nanoparticles.[17,18] In one study, 64Cu was alloyed directly into the lattice of the Au nanostructure by the extra addition of 64CuCl2 into non-radioactive gold chloride and copper (II) acetylacetonate precursors prior to heating, resulting in 64Cu alloyed Au nanoparticles (or [64Cu]Au).[18] The strategy afforded stable radiolabeling and precise control of the specific activity by varying the initial activity of the 64CuCl2 precursor. As synthesized ≈10 nm sized [64Cu] Au showed significantly improved in vivo stability when compared with 64Cu-DOTA.
The same strategy could be used to create intrinsically radiolabeled Au nanoparticles with well-controlled size and morphology by replacing 64Cu with SPECT isotopes, such as gold-198 (198Au; t1/2 = 3.14 d). One of the most interesting examples is the synthesis of NIR-emitting ultrasmall (≈3 nm) radioactive glutathione (GS)-coated luminescent [198Au] Au nanoparticles (Figure 1 b) via a facile one step thermal reduction.[19] As synthesized GS-[198Au]Au exhibited renal clearance capability and desirable in vivo pharmacokinetics, which could be monitored both by using SPECT and NIR optical imaging (Figure 1 b). In another example, [198Au]Au nanoparticles with a cage-like morphology (Figure 1c) were also prepared and used for studying the in vivo Cerenkov luminescence (500–550 nm).[20] Cerenkov luminescence is an emerging concept in molecular imaging, referring to the emission of light (in visible and near-infrared regions) during the decay of radionuclides such as 18F, 131I, 64Cu, and so forth.[21] Since no excitation light is required, the issues of reduced penetration depth and autofluorescence from background are inherently resolved. Moreover, the phenomenon bridges the gap between nuclear and optical imaging modalities and combines the advantages of both.[20] [198Au]Au nanocages (edge length ≈33 nm) showed impressive passive targeting effect and high imaging contrast in EMT-6 tumors (Figure 1 c). In addition, mouse serum stability of [198Au]Au nanocages showed no obvious dissociated 198Au3+ over a week, indicating the high stability of integrated 198Au in the nanostructure. No in vivo SPECT imaging result was shown in this research.
Some other noteworthy examples of intrinsically radiolabeled nanoparticles synthesized via this route include cerium- 141 (141Ce, t1/2 = 32.5 d) doped cerium oxide nanoparticles,[22] cadmium-109 (109Cd, t1/2 = 461.4 d) doped core–shell quantum dots,[23]111In- and 64Cu-doped Fe3O4,[17,24] and samarium-153 (153Sm, t1/2 = 46.3 h) doped upconversion nanoparticles (Table 1).[25,26] Although adding hot precursors during the synthesis of nanoparticles is a generally applicable strategy, limitations of using the right radioactive precursors, possible crystal mismatch when using foreign radioactive ions, as well as the prolonged reaction time under harsh synthetic conditions (in some cases) still exist. Developing a strategy for a faster and more specific synthesis of intrinsically radiolabeled nanoparticles under milder conditions might become more attractive.
Table 1.
Representative examples of intrinsically radiolabeled nanoparticles synthesized via four major routes.
| Strategies | Radioisotopes (half-life) | Nanoparticles | Multimodality Imaging In Vivo | Refs. |
|---|---|---|---|---|
| Hot-plus-Cold Precursors | 64Cu (12.6 h) | PEG-CuS | PET | [15] |
| Dextran-Fe2O3 | PET/MRI | [17] | ||
| PEG-Au | PET/CT | [18] | ||
| 198Au (2.69 d) | Au | SPECT/NIR | [19] | |
| Au Nanocages | CLa) | [20] | ||
| 153Sm (46.3 h) | NaLuF4:Yb,Tm | SPECT/UCLb) | [25] | |
| NaLuF4:Yb,Tm@NaGdF4 | SPECT/CT/MRI/UCL | [26] | ||
| 141Ce (32.5 d) | CeO2 | N/Ac) | [22] | |
| 109Cd (461.4 d) | Core–Shell QDs | NIRFd) | [23] | |
| 111In (2.8 d) | Fe3O4 | N/A | [24] | |
| Specific Trapping | 18F (109.8 min) | NaYF4 | PET/UCL | [27] |
| NaYF4:Yb,Er | PET/UCL | [28] | ||
| NaYF4:Gd,Yb,Er | PET/MRI/UCL | [29] | ||
| 64Cu (12.6 h) | Ferritin Nanocages | PET | [32] | |
| Porphysomes | PET | [36] | ||
| 72As (26 h) | SPION | PET/MRI | [33] | |
| 69Ge (39.05 h) | SPION | PET/MRI | [35] | |
| Cation Exchange | 64Cu (12.6 h) | CdSe/ZnS | PET/CL | [40] |
| 153Sm (46.3 h) | NaLuF4:Yb,Gd,Tm | SPECT/UCL | [41] | |
| Proton Beam Activation | 18F (109.8 min) | Al2O3 | N/A | [43] |
| 13N (9.97 min) | Al2O3 | N/A | [44] |
Cerenkov Luminescence;
Upconversion Luminescence;
Not Applicable;
Near-infrared Fluorescence.
2.2. Specific Trapping
Specific trapping is an interesting synthetic method which takes advantage of the specific absorption (or reaction) of certain radioisotopes with appropriate nanoparticles. Usually, the labeling could be done in a fast and highly specific manner. Trapping fluorine-18 (18F, t1/2 = 109.8 min) to rare-earth nanoparticles is one of the representative examples. Research showed that 18F could be labeled to NaYF4 nanoparticles through a facile inorganic reaction between rare-earth cations (e.g., yttrium ions [Y3+]) and radioactive fluoride ions.[27,28] Due to the ultra low solubility constant (8.62 × 10−21) of yttrium trifluoride (YF3), the 18F-labeling to NaYF4 could be done at room temperature within 5 min.[27] The same trapping mechanism has recently been used for the synthesis of a triple modality imaging probe of [18F]-NaYF4:Gd,Yb,Er and similar others,[27,29,30] showing attractive potential in using these probes for cancer imaging and biodistribution studies (Figure 1 d). The high labeling specificity of 18F to NaYF4 as well as high in vivo stability of final [18F]-NaYF4:Gd,Yb,Er were also demonstrated by low 18F-labeling yield in non-rare-earth nanoparticles (e.g., silica, zinc oxide nanoparticles) and low uptake of 18F in bones, respectively.[27]
Ferritin is another interesting nanoplatform which has an ≈8 nm sized cavity for sequestering a wide range of divalent metals with high affinity.[31] For example, trapping 64Cu into ferritin nanocages could be achieved by simply adding 64Cu2+ into a mixture of heavy and light ferritin chains in acidic conditions, and then adjusting the pH back to 7.4 to reform [64Cu]ferritin nanocages.[32] The chains could also be pre-functionalized with Arg-Gly-Asp (RGD) peptides for targeting integrin αv β3. Stability tests with [64Cu]ferritin nanocages in fetal bovine serum (FBS) showed that less than 10% of the radioactivity was released out of ferritin cages after 24 h incubation, indicating the high stability of [64Cu] ferritin in vitro.[32]
Although the same mechanism could apply to other divalent radiometals, trapping non-metal radioisotopes, such as 72As, inside ferritin nanocages might not be easily achieved. By taking advantage of high affinity of AsIII (or AsV) for superparamagnetic iron oxide nanoparticles (SPION), we reported the first example of chelator-free labeling of 72As to SPION, forming a novel PET/magnetic resonance imaging (MRI) dual-modal imaging agent (Figure 1 e).[33] The underlying mechanism of arsenic trapping by SPION involves the formation of highly stable arsenic complexes, where AsIIIO3 trigonal pyramids or AsVO4 tetrahedra occupy vacant FeO4 tetrahedral sites on the octahedrally terminated (111) surface of the magnetite nanoparticles.[34] The labeling of *As (* = 71, 72, 74, 76) to SPION was later demonstrated to be fast, iron-concentration-dependent, and highly specific. Although the in vivo stability of *As-SPION still needs to be improved, the PEGylated *As-SPION showed improved serum stability and less bladder uptake in vivo. PET/MRI dual modal lymph node mapping using *As-SPION@PEG was also demonstrated in vivo (Figure 1 e). Germanium-69, (69Ge, t1/2 = 39.05 h) is another novel potential PET radioisotope, whose in vivo applications are hampered by its complex coordination chemistry in aqueous medium. Distribution of germanium species varies with concentration and pH under aqueous condition; thus, traditional radiolabeling techniques prove futile. To circumvent this challenge, we also exploited the high affinity of germanium for metal oxides to develop the first chelator-free 69Ge labeled SPION based agent for PET/MRI imaging and lymph node mapping.[35]
Porphysomes are liposome-like nanovesicles, which are composed of macrocyclic porphyrins, and possess the inherent propensity to chelate metal ions.[36] Incorporation of the radiometals into porphysomes could result in intrinsically radiolabeled porphysomes without compromising their inherent photo-thermal properties.[37] Using this strategy, researchers have developed [64Cu]-porphysomes, a novel and robust tumor imaging agent which could be easily synthesized through a fast (30 min), one-pot, high yielding (>95%) procedure (Figure 1 f).[36] It is also worthy to note that the snug fit of 64Cu into the cavity ensures that the in vivo pharmacokinetics and biodstribution of [64Cu]-porphysomes remain unaltered. As-synthesized [64Cu]-porphysomes were found to be highly stable with only less than 2% of 64Cu detachment after 48 h incubation in FBS. [64Cu]-porphysomes were then used for prostate cancer imaging and showed selective accumulation in orthotopic prostate tumors (up to 6.83 ± 1.08%ID/g at 24 h p.i.) with low nonspecific accumulation in surrounding healthy prostate (Figure 1 f). Considering the inherent photo-thermal properties and their high biocompatibility, [64Cu]-porphysomes could become a very attractive theranostic nanoplatform for future clinical translation.[37]
2.3. Cation Exchange
The third strategy for making intrinsically radiolabeled nanoparticles is called cation exchange, which is also a fast and specific, yet not fully explored method in this field. Cation exchange reactions have been utilized in a large number of ionic and semiconductor nanocrystals for complete and reversible exchange of cations for decades.[38] The process allows fast creation of novel nanosystems which could not possibly be synthesized using normal techniques at a relatively moderate temperature.[39] Although well-established in the field of chemistry, using cation exchange for developing intrinsically radiolabeled nanoparticles is still in early stages. So far, only 2 intrinsically radiolabeled nanoparticles of this kind have been reported.[40,41]
Inspired by the cation exchange among lanthanide ions in upconversion nanoparticles,[42] in one study, upconversion NaLuF4:Yb,Gd,Tm nanoparticles have been effectively post-labeled with 153 Sm for upconversion luminescence and SPECT dual-modality imaging.[41] Results showed that fast and highly efficient labeling of 153Sm to NaLuF4:Yb,Gd,Tm (<1 min, ≈99% labeling yield) could be achieved through exchanging 153Sm with lanthanide ions (e.g., Lu3+, Y3+, etc.) in the original host lattice. Both in vitro and in vivo stability investigations confirmed the high stability of [153Sm]-NaLuF4:Yb,Gd,Tm. In vivo real-time biodistribution of water soluble [153Sm]-NaLuF4:Yb,Gd,Tm was later investigated by SPECT imaging, highlighting the potential of these probes as multimodal molecular imaging agents in the future.
In another study, 64Cu and commercially available quantum dots (CdSe/ZnS, QD-580) were selected as a new combination for the synthesis of [64Cu]-QD580 via cation exchange between 64Cu and the original cations.[40] Results showed that 64Cu could replace Zn, and even diffuse inside the core to replace Cd under appropriate reaction conditions.[40] PEGylated [64Cu]-QD580 were then used for whole body PET imaging in U87 glioblastoma models, which showed as high as 12.5%ID/g tumor uptake at 17 h p.i. (Figure 1 g). Using Cerenkov luminescence as the internal light source, [64Cu]-QD580 also exhibited an interesting self-illuminating property due to the Cerenkov resonance energy transfer (CRET), which converts the blue Cerenkov luminescence to longer QDs emission wavelengths for in vivo optical imaging. The synergistic advantages of PET and QD-CRET make [64Cu]-QD580 a promising agent for future multimodal cancer imaging.
2.4. Proton Beam Activation
The last strategy to produce intrinsically radioactive nanoparticles involves direct irradiation of certain nanoparticles with protons.[43,44] In one study, oxygen-18 (18O) enriched aluminium oxide (Al2O3) nanoparticles were activated by 16 MeV proton bombardment in a cyclotron to produce nanoparticles containing 18F, via the 18O(p,n) 18F nuclear reaction without producing significant changes in the size or crystal structure.[43] Excellent activation yield up to 2.25 ± 0.16 MBq/mg of [18F]-Al2O3 could be obtained within a short time period (≈6 min). In vivo biodistribution studies in rodents showed fast accumulation of [18F]-Al2O3 in liver and other organs (Figure 1 h). TEM and X-Ray diffraction (XRD) confirmed no significant change in nanoparticle size, structure or composition after irradiation. In order to generalize the procedure to other synthetic or commercially available metal oxide nanoparticles, the same group adopted direct irradiation of Al2O3 nanoparticles with protons via the 16O(p,α) 13N nuclear reaction in a follow-up study.[44] So far only short-lived radioisotopes (e.g., 18F, t1/2 = 109.8 min; 13N, t1/2 = 9.97 min) have been embedded inside Al2O3 nanoparticles using this method. Also considering the difficulty in accessing the proton beams, compared with the other three strategies, chances to make this strategy widely used will be significantly lower. Direct irradiation may harm biological moieties on the surface of the nanoparticles, thus discouraging from the development of actively targeted nanoplatforms for future in vivo imaging purposes.
2.5. Challenges and Future Directions
Design, synthesis and biological applications of intrinsically radiolabeled nanoparticles have recently become growing interests in molecular imaging and cancer therapy. Challenges still exist for efficient synthesis of stable and multifunctional intrinsically radiolabeled nanoparticles. Firstly, although replacing cold elements from original nanoparticle with hot ones sounds like a generally applicable strategy, the size, morphology and crystal structure of final nanoparticle might change if wrong formulations of hot precursors are used. So far, most of the precursors used are radiometal chlorides. Caution needs to be taken when trying to dope nanoparticles with foreign radioactive ions to avoid undesired crystal mismatch and overcome the self-purification tendency of the nanoparticles.[45] Also, the half-life of the radioisotopes used must be considered since some of the reactions and post surface modifications might take longer time than one half-life of the radioisotopes. Equally important, radioactive wastes should be well-treated under strict safety protocols to avoid any unnecessary radio-exposure especially when certain long-lived isotopes (e.g., 109Cd, t1/2 = 461.4 d) are involved.
Secondly, for fast and specific synthesis of intrinsically radiolabeled nanoparticles, specific trapping and cation exchange are two of the best choices. So far, only limited combinations (72As/SPION,[33]69Ge/SPION,[35]18F/NaYF4,[27]64Cu/porphysome,[36] etc.) of radioisotopes and nanoparticles have been successfully investigated, and more new combinations are expected to be realized in the coming future. Considering the already well-established mechanisms of cation exchange, we also believe this method will embrace a faster growth in the next few years.
Thirdly, long-term potential toxicity is still one of the major roadblocks impeding the progress of intrinsically radioactive inorganic nanoparticles. Several studies have reported elaborate surface engineering of nanoparticles with various biocompatible macromolecules such as dextran, DT10, and so on, and subsequent cell viability assays to reduce any potential cytotoxicity.[22,25] No significant toxicity was observed which further encourages their in vivo applications. However, more intensive in vivo tests such as histology, hematology and serum biochemistry assays are warranted to determine their long-term influence on organ function, tissue damage and inflammation.[25] It is vital to thoroughly assess all aspects of nanoparticle toxicity beyond the perfunctory in vitro analysis for their successful translation to clinical settings.
Lastly, although most of the intrinsically radiolabeled nanoparticles discussed above showed high in vitro and in vivo stabilities with only limited percentages of radioisotope detachment, stability studies should always be well-investigated before any in vivo imaging applications to avoid any possible off-targeting and false positives.
3. Conclusion
In conclusion, an emerging concept of using intrinsically radiolabeled nanoparticles for multimodality molecular imaging was discussed in this concise article. Four major chelator-free radiolabeling methods, including hot-plus-cold precursors, specific trapping, cation exchange and proton beam activation, were summarized (Table 1) with our focus also on the nanoparticle stability. Representative examples of using these multifunctional nanoparticles for multimodality molecular imaging were also highlighted. Although still in the early stages, design and synthesis of intrinsically radiolabeled nanoparticles have shown an attractive potential in offering an easier, faster, more stable, and more specific radiolabeling technique for the next generation of molecular imaging.
Acknowledgments
This work is supported, in part, by the University of Wisconsin – Madison, the National Institutes of Health (NIBIB/NCI 1R01CA169365 and P30CA014520), the Department of Defense (W81XWH-11–1–0644), and the American Cancer Society (125246-RSG-13–099–01-CCE).
Contributor Information
Shreya Goel, Materials Science Program, University of Wisconsin-Madison, 1509 University Avenue Madison, WI 53706-15952, USA.
Dr. Feng Chen, Email: fchen@uwhealth.org, Department of Radiology, University of Wisconsin-Madison, School of Medicine and Public HealthE3/366 Clinical Science Center, 600 Highland Avenue Madison, WI 53792-32523, USA
Emily B. Ehlerding, Department of Medical Physics, Wisconsin Institutes for Medical Research, 1111 Highland Avenue, Room 1005 Madison, WI 53705-22754, USA
Prof. Weibo Cai, Email: wcai@uwhealth.org, Department of Radiology, University of Wisconsin-Madison, School of Medicine and Public HealthE3/366 Clinical Science Center, 600 Highland Avenue Madison, WI 53792-32523, USA. Department of Medical Physics, Wisconsin Institutes for Medical Research, 1111 Highland Avenue, Room 1005 Madison, WI 53705-22754, USA. University of Wisconsin, Carbone Cancer Center, UW Hospital and Clinics, 600 Highland Ave Madison, WI 53792, USA.
References
- 1.Cai W, Chen X. J Nucl Med. 2008;49(Suppl 2):113S. doi: 10.2967/jnumed.107.045922. [DOI] [PubMed] [Google Scholar]
- 2.Michalski MH, Chen X. Eur J Nucl Med Mol Imaging. 2011;38:358. doi: 10.1007/s00259-010-1569-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen F, Cai W. Small. 2014;10:1887. doi: 10.1002/smll.201303627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu D, Gambhir SS. C lin Breast Cancer. 2003;4(Suppl 1):S55. doi: 10.3816/cbc.2003.s.016. [DOI] [PubMed] [Google Scholar]
- 5.de Barros AB, Tsourkas A, Saboury B, Cardoso VN, Alavi A. EJNMMI Res. 2012;2:39. doi: 10.1186/2191-219X-2-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S. Adv Drug Delivery Rev. 2008;60:1347. doi: 10.1016/j.addr.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarko D, Eisenhut M, Haberkorn U, Mier W. Curr Med Chem. 2012;19:2667. doi: 10.2174/092986712800609751. [DOI] [PubMed] [Google Scholar]
- 8.Cutler CS, Hennkens HM, Sisay N, Huclier-Markai S, Jurisson SS. Chem Rev. 2013;113:858. doi: 10.1021/cr3003104. [DOI] [PubMed] [Google Scholar]
- 9.Cai W, Chen K, Mohamedali KA, Cao Q, Gambhir SS, Rosenblum MG, Chen X. J Nucl Med. 2006;47:2048. [PubMed] [Google Scholar]
- 10.Jeger S, Zimmermann K, Blanc A, Grunberg J, Honer M, Hunziker P, Struthers H, Schibli R. Angew Chem Int Ed. 2010;49:9995. doi: 10.1002/anie.201004243. [DOI] [PubMed] [Google Scholar]
- 11.Kukis DL, DeNardo GL, DeNardo SJ, Mirick GR, Miers LA, Greiner DP, Meares CF. Cancer Res. 1995;55:878. [PubMed] [Google Scholar]
- 12.Phillips WT, Goins BA, Bao A. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:69. doi: 10.1002/wnan.3. [DOI] [PubMed] [Google Scholar]
- 13.Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL, Anderson CJ. J Med Chem. 2004;47:1465. doi: 10.1021/jm030383m. [DOI] [PubMed] [Google Scholar]
- 14.Kreuter J, Tauber U, Illi V. J Pharm Sci. 1979;68:1443. doi: 10.1002/jps.2600681129. [DOI] [PubMed] [Google Scholar]
- 15.Zhou M, Zhang R, Huang M, Lu W, Song S, Melancon MP, Tian M, Liang D, Li C. J Am Chem Soc. 2010;132:15351. doi: 10.1021/ja106855m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goel S, Chen F, Cai W. Small. 2014;10:631. doi: 10.1002/smll.201301174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong RM, Gilbert DA, Liu K, Louie AY. ACS Nano. 2012;6:3461. doi: 10.1021/nn300494k. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Sultan D, Detering L, Cho S, Sun G, Pierce R, Wooley KL, Liu Y. Angew Chem Int Ed. 2014;53:156. doi: 10.1002/anie.201308494. [DOI] [PubMed] [Google Scholar]
- 19.Zhou C, Hao G, Thomas P, Liu J, Yu M, Sun S, Oz OK, Sun X, Zheng J. Angew Chem Int Ed. 2012;51:10118. doi: 10.1002/anie.201203031. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Liu Y, Luehmann H, Xia X, Wan D, Cutler C, Xia Y. Nano Lett. 2013;13:581. doi: 10.1021/nl304111v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y, Liu H, Cheng Z. J Nucl Med. 2011;52:2009. doi: 10.2967/jnumed.111.092965. [DOI] [PubMed] [Google Scholar]
- 22.Yang L, Sundaresan G, Sun M, Jose P, Hoffman D, McDonagh PR, Lamichhane N, Cutler CS, Perez JM, Zweit J. J Mater Chem B. 2013;1:1421. doi: 10.1039/c2tb00404f. [DOI] [PubMed] [Google Scholar]
- 23.Sun M, Hoffman D, Sundaresan G, Yang L, Lamichhane N, Zweit J. Am J Nucl Med Mol Imaging. 2012;2:122. [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng J, Jia B, Qiao R, Wang C, Jing L, Wang F, Gao M. Chem Commun. 2014;50:2170. doi: 10.1039/c3cc48948e. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Sun Y, Cao T, Peng J, Liu Y, Wu Y, Feng W, Zhang Y, Li F. Biomaterials. 2013;34:774. doi: 10.1016/j.biomaterials.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Zhu X, Peng J, Li F. ACS Nano. 2013;7:11290. doi: 10.1021/nn405082y. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Yu M, Liang S, Zhang Y, Li C, Mou T, Yang W, Zhang X, Li B, Huang C, Li F. Biomaterials. 2011;32:2999. doi: 10.1016/j.biomaterials.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Liu Q, Chen M, Sun Y, Chen G, Yang T, Gao Y, Zhang X, Li F. Biomaterials. 2011;32:8243. doi: 10.1016/j.biomaterials.2011.07.053. [DOI] [PubMed] [Google Scholar]
- 29.Liu Q, Sun Y, Li CG, Zhou J, Li CY, Yang TS, Zhang XZ, Yi T, Wu DM, Li FY. ACS Nano. 2011;5:3146. doi: 10.1021/nn200298y. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J, Yu M, Sun Y, Zhang X, Zhu X, Wu Z, Wu D, Li F. Biomaterials. 2011;32:1148. doi: 10.1016/j.biomaterials.2010.09.071. [DOI] [PubMed] [Google Scholar]
- 31.Uchida M, Flenniken ML, Allen M, Willits DA, Crowley BE, Brumfield S, Willis AF, Jackiw L, Jutila M, Young MJ, Douglas T. J Am Chem Soc. 2006;128:16626. doi: 10.1021/ja0655690. [DOI] [PubMed] [Google Scholar]
- 32.Lin X, Xie J, Niu G, Zhang F, Gao H, Yang M, Quan Q, Aronova MA, Zhang G, Lee S, Leapman R, Chen X. Nano Lett. 2011;11:814. doi: 10.1021/nl104141g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen F, Ellison PA, Lewis CM, Hong H, Zhang Y, Shi S, Hernandez R, Meyerand ME, Barnhart TE, Cai W. Angew Chem Int Ed. 2013;52:13319. doi: 10.1002/anie.201306306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morin G, Wang Y, Ona-Nguema G, Juillot F, Calas G, Menguy N, Aubry E, Bargar JR, Brown GE., Jr Langmuir. 2009;25:9119. doi: 10.1021/la900655v. [DOI] [PubMed] [Google Scholar]
- 35.Chakravarty R, Valdovinos HF, Chen F, Lewis CL, Ellison PA, Luo H, Meyerand ME, Nickles RJ, Cai W. Adv Mater. 2014 doi: 10.1002/adma.201401372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu TW, MacDonald TD, Shi J, Wilson BC, Zheng G. Angew Chem Int Ed. 2012;51:13128. doi: 10.1002/anie.201206939. [DOI] [PubMed] [Google Scholar]
- 37.Lovell JF, Jin CS, Huynh E, Jin H, Kim C, Rubinstein JL, Chan WC, Cao W, Wang LV, Zheng G. Nat Mater. 2011;10:324. doi: 10.1038/nmat2986. [DOI] [PubMed] [Google Scholar]
- 38.Son DH, Hughes SM, Yin Y, Paul Alivisatos A. Science. 2004;306:1009. doi: 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]
- 39.Sadtler B, Demchenko DO, Zheng H, Hughes SM, Merkle MG, Dahmen U, Wang LW, Alivisatos AP. J Am Chem Soc. 2009;131:5285. doi: 10.1021/ja809854q. [DOI] [PubMed] [Google Scholar]
- 40.Sun X, Huang X, Guo J, Zhu W, Ding Y, Niu G, Wang A, Kiesewetter DO, Wang ZL, Sun S, Chen X. J Am Chem Soc. 2014;136:1706. doi: 10.1021/ja410438n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y, Liu Q, Peng J, Feng W, Zhang Y, Yang P, Li F. Biomaterials. 2012;27:01306. [Google Scholar]
- 42.Dong C, van Veggel FC. ACS Nano. 2009;3:123. doi: 10.1021/nn8004747. [DOI] [PubMed] [Google Scholar]
- 43.Perez-Campana C, Gomez-Vallejo V, Martin A, San Sebastian E, Moya SE, Reese T, Ziolo RF, Llop J. Analyst. 2012;137:4902. doi: 10.1039/c2an35863h. [DOI] [PubMed] [Google Scholar]
- 44.Perez-Campana C, Gomez-Vallejo V, Puigivila M, Martin A, Calvo-Fernandez T, Moya SE, Ziolo RF, Reese T, Llop J. ACS Nano. 2013;7:3498. doi: 10.1021/nn400450p. [DOI] [PubMed] [Google Scholar]
- 45.Yang J, Chng LL, Yang X, Chen X, Ying JY. Chem Commun. 2014;50:1141. doi: 10.1039/c3cc47254j. [DOI] [PubMed] [Google Scholar]