

Abstract
Objective
Preeclampsia (PE) affects 2–8% of pregnancies worldwide and is a significant source of maternal and neonatal morbidity and mortality. However, the mechanisms underlying PE are poorly understood and major questions regarding etiology and risk factors remain to be addressed. Our objective was to examine whether abnormal expression of the cardiovascular developmental transcription factor, Nkx2-5, was associated with early onset and severe pre-eclampsia (EOSPE).
Methods
Using qPCR and immunohistochemical assay, we examined expression of Nkx2-5 and target gene expression in EOSPE and control placental tissue. We tested resulting mechanistic hypotheses in cultured cells using shRNA knockdown, qPCR and western blot.
Results
Nkx2-5 is highly expressed in racially disparate fashion (Caucasians > African Americans) in a subset of early EOSPE placentae. Nkx2-5 mRNA expression is highly correlated (Caucasians > African Americans) to mRNA expression of the preeclampsia marker sFlt-1, and of the Nkx2-5 target and RNA splicing factor, Sam68. Knockdown of Sam68 expression in cultured cells significantly impacts sFlt-1 mRNA isoform generation in vitro, supporting a mechanistic hypothesis that Nkx2-5 impacts EOSPE severity in a subset of patients via upregulation of Sam68 to increase sFlt-1 expression. Expression of additional Nkx2-5 targets potentially regulating metabolic stress response is also elevated in racially disparate fashion in EOSPE.
Conclusions
Expression of Nkx2-5 and its target genes may directly influence the genesis and racially disparate severity, and define a mechanistically distinct subclass of EOSPE.
Keywords: Preeclampsia, placenta, sFlt-1, Sam68, Nkx2-5, Xbp-1, Ccdc117, racial disparity
Introduction
Preeclampsia (PE) occurs in about 2–8% of pregnancies and is a leading cause of prematurity, and infant and maternal morbidity and mortality1, 2. PE, particularly early-onset or recurrent PE, has been associated with greater future risk for renal, metabolic and cardiovascular disease in both mothers and their children3–13. Despite the prevalence and serious consequences associated with PE, its initial etiology remains to be fully elucidated. Hypertension, proteinuria, and evidence of endothelial damage are major hallmarks of PE, and a central finding or component of PE is placental hypoxia, due to impaired remodeling of the spiral arteries leading to poor placental perfusion14.
Recent efforts to develop reliable diagnostic tests for PE have focused on the accurate assay of serum angiogenic factors such as placental growth factor or PlGF15, 16, the transforming growth factor beta (TGFβ) inhibiting factor endoglin17, and the anti-angiogenic factor sFlt-118, 19. sFlt-1 protein, a soluble truncated isoform of the Vascular Endothelial Growth Factor Receptor 1 (VEGFR1/Flt-1) in particular has emerged as a significant marker of PE in humans 20–23. mRNA encoding sFlt-1 is generated by an alternative splicing and premature polyadenylation of VEGFR1 transcripts: in the absence of splicing of VEGFR1 exon 13 to exon 14, transcripts are prematurely polyadenylated (pA) via cryptic sites in the intervening intron, and the resulting transcript encodes a truncated VEGFR1 receptor molecule lacking the transmembrane domain and c-terminal signaling domains. Soluble sFlt-1 thus acts as an antagonist for VEGFR signaling, particularly by PlGF24. Serum levels of sFlt-1 correlate directly with both increased severity, and with early-onset of PE20–23, 25. In addition, overexpression of sFlt-1 into pregnant animal models is sufficient to cause hypertension and proteinuria18, 26–29.
Current clinical studies are focused on determining the sensitivity and specificity of sFlt-1 levels to PE. In particular, sFlt-1/PlGF ratios may be particularly informative as to PE risk and severity30, 31. While the assay of these angiogenic factors has increasingly been shown to be informative and reliable for the general diagnosis of PE, few biomarkers currently provide insight as to how different PE etiologies lead to altered sFlt-1, PlGF or endoglin expression, or distinguish between potential subpopulations including racially disparate groups. This represents a critical gap of knowledge in the field complicating our ability to assess individual etiologic risk associations or long term prognoses, peculiar to PE subcategories.
The embryonic transcription factor Nkx2-5, a homolog of the Drosophila Tinman gene, is essential for normal heart development during embryogenesis, and its mutation is highly associated with the incidence of congenital heart disease in humans. While the majority of research on the function of Nkx2-5 is focused on its role and regulation in heart formation and in congenital heart disease32–35, studies of Nkx2-5 function in mice additionally detected a role in extraembryonic development. Homozygous knockout of Nkx2-5 in mice resulted in deficient amniotic sac blood vessel formation, and poor contact between PECAM positive endothelial cells and the apposing mesodermal and endodermal layers. Lineage tracing experiments assaying the expression of DNA recombinase under control of the Nkx2-5 gene locus showed evidence of at least transient Nkx2-5 expression in angiogenic amniotic sac populations including endothelial cells.36, 37. These observations suggested potential roles for Nkx2-5 in extraembryonic angiogenesis or vascular pathology in humans. We therefore examined the possibility that Nkx2-5 might be relevant to PE, a pregnancy-related disease characterized by vasospasm and abnormal vascular development, in which placental hypoxia and vascular insufficiency have been implicated as etiologic factors leading to hypertension, proteinuria and severe sequelae.
Materials and Methods
Patient Recruitment
The Institutional Review Board at the Medical University of South Carolina (MUSC) approved this investigation from a prospective study of early-onset severe PE (EOSPE) patients and control patients with normal term deliveries enrolled at MUSC from 2007 to 201138. All patients included in this investigation consented to collection of demographic and pregnancy outcome data. Subjects were recruited from the MUSC Labor and Delivery unit after confirmation of a diagnosis of EOSPE. EOSPE was defined according to the American College of Obstetrics and Gynecology criteria for severe preeclampsia in patients less than 34 weeks’ completed gestational age39. The demographics of the patient population described in this study are summarized in Supplementary Table 1. (Clinicaltrials.gov, #NCT00719654, HR # 17495)
Patients were excluded if they also had a diagnosis of chronic hypertension, pregestational diabetes, renal disease, lupus, or tobacco use. Subjects were followed until 6 weeks postpartum and excluded if they were noted to have chronic hypertension based on persistent hypertension. Demographic data were collected and included gestational age, maternal age, maternal parity, maternal prepregnancy body mass index (BMI), maternal systolic and diastolic blood pressure, and total urine protein excreted in 24 hours.
Placental Sampling, RNA extraction and cDNA synthesis
Approximately 1cm3 tissue samples were excised from newly delivered placentae at a distance half way between the base of the umbilical cord and the edge of the placental disc. Samples were rinsed thoroughly in ice cold phosphate buffered saline (PBS) then divided into aliquots and transferred to Qiagen RNAlater solution (Qiagen, Valencia, CA) for storage at −80C or fixed overnight in 4% paraformaldehyde/PBS for fixation and sectioning. RNA was purified from approximately 30 mg samples of placental tissue using RNeasy spin columns (Qiagen) according to manufacturer’s instructions. cDNA was reverse-transcribed from 500ng RNA samples using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA), diluted 5-fold with nuclease free water and used for quantitative PCR analysis.
Cell Culture and Lentiviral siRNA knockdown
HEK293 and HEK293T cells were maintained in DMEM (Gibco-Invitrogen, Grand Island, NY, #11960-51 +4.5g/L D-Glucose, - L-Glutamine, -sodium pyruvate) supplemented with 20mM l-glutamine (Gibco), 1kU/ml penicillin/streptomycin (HyClone, Logan UT), 1x non-essential amino acids (Gibco), and 10% ES-qualified FBS (Gibco). Doxycycline inducible lentiviruses (Thermo Fisher, Pittsburgh, PA) encoding shRNAs specific for different regions of Sam68 were produced in HEK293T cells by CaPO4 co-transfection of lentiviral backbone, packaging plasmid p8.74 and a plasmid encoding VSV-G protein40. Lentivirus were used to infect naïve HEK293 cells and stably transduced cell lines were selected by serial single cell density plating and puromycin selection according to manufacturers recommendations, and confirmed by visual detection of RFP co-expression upon doxycycline induction. For knockdown experiments, control and knockdown samples were plated in parallel, and lentiviral shRNA expression was induced for 24 hrs by addition of 100 ng/ml doxycycline to culture medium of knockdown samples. Protein and RNA samples were simultaneously extracted using RNeasy columns (Qiagen), and proteins further purified and desalted by acetone precipitation. cDNA synthesis and qPCR for siRNA experiments were as described as above.
Protein Analysis
Protein samples were resuspended in NUPAGE LDS sample buffer and protein concentration determined by BCA assay (Pierce Biologicals-Thermo Fisher). 20ug protein samples were separated on 10% Bis-Tris NuPAGE LDS PAGE gels (Novex-Invitrogen). Separated samples were transferred to nitrocellulose, blocked in Odyssey blocking buffer overnight (LI-COR Biosciences, Lincoln, NE), prior to probing with mouse anti-human Sam68 (Millipore, Billerica, MA) and rabbit-anti-human GAPDH (Fitzgerald, Acton, MA) antibodies. Protein bands were visualized using goat-anti mouse 700 and goat-anti rabbit 800 secondary antibodies (LI-COR). Signals were digitally visualized and quantified using an Odyssey CLx Infrared scanner (LI-COR) and ImageJ freeware available through NIH 41.
Quantitative RT-PCR
cDNA was prepared from total RNA from placental RNA samples using the iScript cDNA synthesis kit. Quantitative RT-PCR (qRT-PCR) for Nkx2-5, Nkx2-5 target genes and β-actin performed using the iQ SYBR green/iCycler amplification system (Bio-Rad, Hercules, CA) and gene-specific oligonucleotide primers (Supplementary Table 2) (Integrated DNA Technologies, Coralville, IA). qPCR assays were performed in triplicate. Results were corrected for PCR efficiency of individual assays as determined by PCR Miner and expressed as ratios of relative target and control β-actin mRNA based on C(t) values42.
Placental Immunohistochemistry
Formalin fixed, paraffin-embedded placental specimens were microtome-sectioned to 8μ thickness and subject to immunohistochemistry using the HistoMouse-Plus Broad Spectrum (AEC) kit (Invitrogen) and manufacturer protocols, and primary antibody to Nkx2-543, Sam68 (Millipore), Xbp1 (Abcam, Cambridge, MA) and Ccdc117 (Sigma, St Louis, MO). Immunostained sections were counterstained with H+E according to standard protocols prior to mounting and digital photography.
Statistical Analysis
Associations between gene expression levels were assessed using Spearman rank correlations, which are ideal for non-Gaussian data. Comparisons in expression levels between patient subgroups (e.g. intrauterine growth restriction [IUGR] vs. no IUGR) were assessed using non-parametric Wilcoxon rank sum tests. All statistical analyses were conducted using SAS v9.3 (Cary, NC), and p-values <0.05 were considered statistically significant.
Results
Nkx2-5 mRNA expression levels in placental samples obtained from a total of n=33 subjects with EOSPE were measured. As an indicator of the potential association of Nkx2-5 expression levels to PE severity, we examined in parallel the mRNA expression of sFlt-1, a significant marker of PE20–23. We detected significantly elevated expression of Nkx2-5 in the placentae of a subset of patients with EOSPE, independent of the presence (n=19) or absence (n=14) of IUGR. These elevations had the appearance of being correlated with elevations of sFlt-1 mRNA expression, particularly in Caucasian American (CA) (n=14) vs. African American (AA) (n=19) patients (Fig. 1A, B).
Figure 1. Expression of Nkx2-5 and sFlt-1 mRNA in EOSPE Placentae.
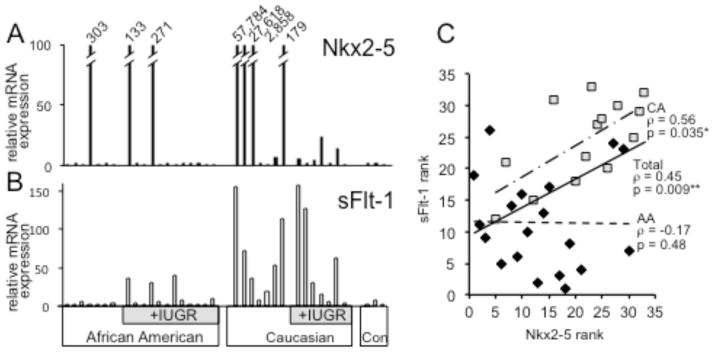
Histogram shows relative fold expression of mRNA for: A. Nkx2-5 and B. sFlt-1 as determined by qPCR in 33 EOSPE placentae and 3 control term placentae. Expression values are normalized to average expression in control term placentae and divided by ethnicity and absence or presence of IUGR. C. Scatter diagram showing Nkx2-5 expression rank (x-axis) vs. sFlt-1 expression rank (y-axis). Rank order values are diagrammed separately for CA (squares) vs. AA (diamonds). *significant (p ≤ 0.05); **highly significant (p ≤ 0.02)
Within the overall study population, a statistically significant and strong positive correlation was noted between elevated Nkx2-5 and elevated sFlt-1 mRNA expression (Spearman’s rank correlation rho= +0.45; p=0.009, n=33). When this analysis was stratified by race, it was noted that the correlation was quite strong in CAs (rho= +0.56; p=0.035; n=14) but not in AAs (rho= −0.17; p=0.48; n=19) (Fig. 1C). No such elevation was observed in a limited number of control samples from uncomplicated term pregnancies. We observed no significant correlation of expression profile with maternal age, maternal weight, or weight gain, body mass index, degree of hypertension, parity, gravidity, birth weight or placental mass. Perhaps due to small sample size, we observed no apparent significant correlation or differences in correlation with respect to IUGR status.
We previously identified the STAR RNA splicing family member Khdrbs1/Sam68 as a downstream target gene of Nkx2-5 in developing heart populations44. Sam68 is a signal-responsive RNA-binding protein that recognizes consensus U(U/A)AA sequences on single-stranded RNA via to affect both alternative splicing and alternative polyadenylation events of target molecules45, 46. These RNA processing events are similar to those controlling the generation of sFlt-1 isoforms from VEGFR1 transcripts47, and several A/U rich Intron 13 sequences critical for alternative splicing regulation are highly similar to consensus Sam68 RNA binding sites48. We therefore investigated the hypothesis that elevated Nkx2-5 expression could result in increased sFlt-1 production through up-regulation of Sam68 in our study population.
Consistent with this possibility, qPCR analysis detected higher levels of expression of Sam68 in PE placental samples that also show high levels of Nkx2-5 and sFlt1 expression (Fig. 2B). In the overall population we found statistically significant and positive correlations between both Nkx2-5 and Sam68 expression (rho= +0.47; p=0.006) and Sam68 and sFlt-1 expression (rho= +0.79; p<0.00001) (Fig. 2C). When stratified by race, these associations remain positive in CA (Nkx2-5-Sam68: rho= +0.40; p=0.16 (ns), and Sam68-sFlt1: rho= +0.56; p=0.04) though with less statistical significance in the smaller subpopulation. Associations between Nkx2-5 and Sam68 or Nkx2-5 and sFlt1 remained weak and not statistically significant in the AA population (Nkx2-5-Sam68; rho= −0.08; p=0.74, and Nkx2-5-sFlt1: rho= −0.17; p=0.48). Interestingly, Sam68 and sFlt1 levels remain significantly and positively correlated in the AA population (rho= +0.59; p=0.008). (Summarized in Supplementary Table 3).
Figure 2. Nkx2-5 and Sam68 co-expression in EOSPE placenta.
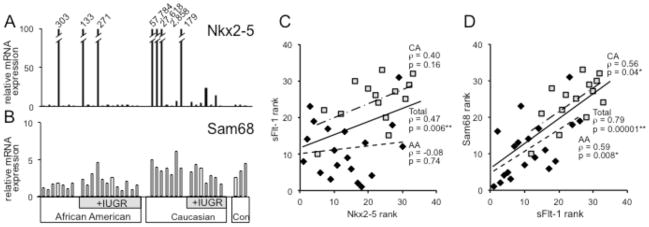
Relative fold expression of mRNA for A. Nkx2-5 and B. Sam68 as determined by qPCR in EOSPE and 3 control term placentae. Expression values are normalized and displayed as in Figure 1. Scatter diagrams showing ranked expression levels of Sam68 mRNA (x-axis) vs.: C. Nkx2-5 expression rank or D. sFlt-1 expression rank,(y-axis). Rank order values are diagrammed separately for CA (squares) vs. AA (diamonds). *significant (p ≤ 0.05); **highly significant (p ≤ 0.02)
In order to test whether Sam68 is functionally involved in the alternative splicing of sFlt-1 isoforms, we performed inducible, lentiviral-mediated RNA knockdown of Sam68 in HEK 293 cells, which natively express VEGFR1 mRNA (Fig. 3A). Reduction of Sam68 protein expression by siRNA interference (approx. 40%; Fig. 3B) resulted in a significant decrease in the amount of sFlt-1 mRNA isoforms relative to both control β-actin mRNA (approx. 25%; Fig. 3C) and to Exon13-Exon14 spliced isoforms (approx. 20%; Fig. 3D), supporting the hypothesis that Sam68 may be a functional mediator of VEGFR1 alternative splicing whose expression level influences the relative amount of sFlt-1 mRNA isoform production.
Figure 3. Effect of Sam68 expression levels on sFlt-1 VEGFR1 isoform splicing.

A. Sam68 and control GAPDH protein levels in triplicate control (left) and anti-Sam68 siRNA-expressing HEK293 cells. B. Quantification of of Sam68 protein in samples shown in A. shRNA induction results in approximately 40% relative reduction of Sam68 protein. C–D: Reduction of Sam68 protein by shRNA correlates with 25% loss of sFlt-1 VEGFR1 mRNA isoform relative to control β-actin (C) and 20% loss of sFlt-1 isoform relative to Exon12-13 spliced VEGFR1 isoforms (D). Statistical significance of expression changes are shown above as p values calculated by Students two-tailed t-test. **highly significant (p ≤ 0.02)
Our previous study of Nkx2-5 target genes during mouse heart development additionally identified a gene of yet unknown function, Ccdc117, as being directly regulated by Nkx2-5 44. While the function Ccdc117 is yet unknown, the Ccdc117 locus is physically proximal to that of the endoplasmic reticulum stress response mediator gene, Xbp1, in both mice and humans49. Xbp1 mRNA expression has previously been found to be elevated in placentae of patients with PE in the absence of fetal growth restriction as a putative response to oxidative stress 50.
As shown in Fig. 4A, Ccdc117 transcript levels were elevated in patients that have high Nkx2-5 expression, and we detected a positive and statistically significant correlation between Nkx2-5 and Ccdc117 expression levels in our EOSPE placenta samples (rho= +0.40; p=0.02; n=33). Again stratifying by race, a positive and highly significant Nkx2-5-Ccdc117 correlation was again observed in CAs (rho= +0.72; p=0.004; n=14), but not in AAs, which instead showed a non-significant negative correlation (rho= −0.23; p=0.34; n=19) (Fig. 4C).
Figure 4. Expression of Ccdc117 and Xbp1 mRNA in EOSPE Placentae.
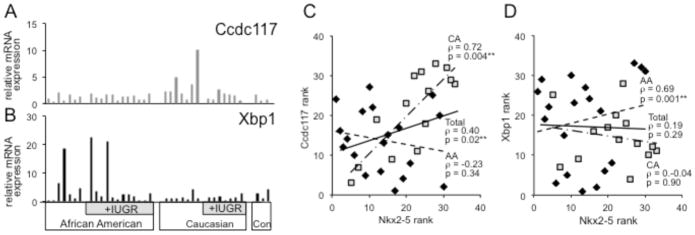
Histograms show relative fold expression of mRNA for A. Ccdc1171 and B. Xbp as determined by qPCR in EOSPE and control term placentae. Expression values are normalized to average expression in control term placentae and divided by ethnicity and absence or presence of IUGR. C. Scatter diagrams showing ranked expression levels of Nkx2-5 mRNA (y-axis) vs.: C. Ccdc117 expression rank; or D. Xbp1 expression rank (x-axis). Rank order values are diagrammed separately for CA (squares) vs. AA (diamonds). *significant (p ≤ 0.05); **highly significant (p ≤ 0.02)
Additional qPCR assay of placental mRNA samples from our EOSPE cohort detected elevation of Xbp1 transcript levels in some patients with elevated Nkx2-5 expression (Fig. 4B). Overall, there was no significant correlation between Xbp1 and Nkx2-5 expression in the overall population (rho= +0.19; p=0.29). However, when stratified by race, and in opposition to trends observed with Ccdc117, Xbp-1 expression correlated significantly and positively with Nkx2-5 elevation in AAs (rho= +0.69; p=0.001; n=19) but not significantly with Nkx2-5 elevation in CAs (rho= −0.04; p=0.90; n=14) (Fig. 4D) (Summarized in Supplementary Table 4).
Immunohistochemical analysis of trophoblastic villi from an EOSPE human placental sample detected co-expression of Nkx2-5 and Sam68 expression in syncytiotrophoblast cells, the principal site of sFlt-1 synthesis (Fig. 5A, D) consistent with both cell-autonomous Nkx2-5 direct regulation of Sam68, and with a potential role for Sam68 in regulating sFlt-1-producing splicing of VEGFR1 transcripts in trophoblast cells. Ccdc117 and Xbp1 are similarly co-expressed in syncytiotrophoblast cells of EOSPE placenta (Fig. 5C, D), consistent with their additional and cell-autonomous regulation by Nkx2-5. Collectively, these results suggest key roles for elevated Nkx2-5 and Nkx2-5 target gene expression in the genesis and racially-disparate modification of EOSPE in a select subpopulation.
Figure 5. Syncytiotrophoblast expression of Nkx2-5 and target genes in EOSPE placenta.

Immunohistochemistry detects protein expression of: A. Nkx2-5, B. Sam68, C. Ccdc117, and D. Xbp1 protein in syncytrophoblast layer of an EOSPE placenta. 50uM scale is shown in lower right of D.
Discussion
PE remains a significant cause of neonatal and maternal morbidity and mortality. To date, the cause of PE is not known, although many agree that PE is a two-stage disease as described by Roberts. The first stage involves poor placental perfusion, usually as a result of impaired vascular remodeling in early pregnancy. This leads to the second stage, which is the maternal syndrome of PE and involves endothelial and leukocyte activation. Determining the link between abnormal placentation and clinical disease has been a prime focus of PE research.
One of the most compelling putative links between poor placental perfusion and the clinical sequelae recognized in PE is sFlt-1. Maynard et al demonstrated that sFlt1, a soluble receptor for VEGF and PlGF, is increased in PE. Soluble Flt1 binds and inactivates VEGF and PlGF. The increase in sFlt1 and the corresponding decrease in PlGF correlate with the severity of PE. Additionally, Levine et al have shown that elevated sFlt1 and decreased PlGF, as well as decreased urinary PlGF, are predictive of the development of PE. While it is known that placental tissue is the primary source of sFlt-1, and that sFlt-1 alone is capable of inducing symptoms of PE in animals, the mechanism regulating generation of sFlt-1 isoforms and thus responsible for high levels of its expression in PE is yet unknown18, 51.
Additionally, other diseases such as diabetes, obesity, chronic hypertension, and systemic lupus erythematosus are associated with altered risk for PE,16, 52–55 and racial differences exist for PE, with AAs having higher rates and more severe disease as compared to CAs and Hispanics.56–58. These observations have led to the hypothesis that several disease entities of variable maternal, fetal and/or environmental etiology may incite or contribute to PE, but little is known about the cause of these differences.59.
We have found an unexpected and previously unreported elevation of expression of the developmental transcription factor Nkx2-5 in the placentae of a subset of predominantly CA patients with EOSPE that exhibited statistically significant correlation with the expression of sFlt-1. Related increases were observed in the expression of an RNA splicing factor downstream of Nkx2-5, Sam68. Immunolocalization studies and in vitro RNA interference experiments in cultured cells supported a mechanistic model whereby abnormally high Nkx2-5 expression levels potentiate high placental sFlt-1 expression and EOSPE through cell autonomous activation of Sam68 and preferential production of sFlt-1 encoding splice isoforms in these patients. Of note, the correlation of Sam68-sFlt-1 levels remains statistically strong in AAs as well as CAs, suggesting that while a strong functional link exists between Sam68 and sFlt-1 in both populations, Sam68 regulation and/or other PE modifiers may be driven by other parallel pathways not involving Nkx2-5 regulation in AAs.
As an example of such race-specific modifers, we detected parallel elevations in the expression of other Nkx2-5 target genes potentially related to metabolic stress responses in placental tissue, Ccdc117 and Xbp1. Their elevated expression in EOSPE were found to occur in racially disparate fashion, with elevated Nkx2-5 expression and Ccdc117 co-expression occurring with greater frequency in CAs as compared to AAs, but Xbp1 elevation occurring with greater frequency in AAs. These observations point to additional potential for differences in genetic or ethnicity-related epigenetic factors to modify disease risk with regard to Nkx2-5-related mechanisms.
By identifying novel differences in Nkx2-5-related gene expression in EOSPE potentially linked to mechanism via modulation of sFlt-1, and by further identifying subpopulations including racially disparate subgroups showing distinct disease-related gene expression profiles, the findings of this study are quite significant for the ability both to diagnose and treat PE related to elevated sFlt-1 expression. The detection of expression changes of other Nkx2-5 regulated genes, including one previously associated with PE, and known to participate in oxidative stress reactions, points to additional implications for modifiers of either PE etiology or severity. Defining studies such as this will play an important role in the ability to successfully treat PE both through interception of “final common pathway” pathophysiology and through prevention of initial inciting mechanisms.
Supplementary Material
Maternal age (MA), gestational age (GA), parity and gravidity are shown as median (range). Birthweight (BW), body mass index (BMI), blood pressure (BP) and proteinuria are expressed as median (interquartile range). *Data available for 16 of 19 patients. **Data available for 9 of 14 patients.
Statistical associations of Nkx2-5, Sam68 and sFlt-1 mRNA expression in EOSPE placentae in the overall population, and stratified by AA or CA race. *significant (p ≤ 0.05); **highly significant (p ≤ 0.02)
Statistical associations of Nkx2-5, Ccdc117 and Xbp1 expression in overall EOSPE population, and stratified by AA and CA race. **highly significant (p ≤ 0.02)
Acknowledgments
We thank Meaghan E. Flessa for technical assistance, Benjamin Parrott, Satomi Kohno and Louis J. Guillette for expertise for placental RNA purification, and Roger Newman, Carol L. Wagner and Ananth Karumanchi for constructive feedback on this manuscript.
Sources of Funding: KHL received funding from the American Heart Association (BGIA 12060120), pilot funding and consultation support from the South Carolina Translational Research Institute (NIH/NCATS TL1TR000061), and facilities and consultation support from the SC COBRE for Developmentally-based Cardiovascular Disease (P20 016434). PJN received funding from the South Carolina Translational Research Institute (NIH/NCATS TR000062). AJH received funding from the SURP program at the Medical University of South Carolina. This research was enabled by facilities and support of the Darby Children’s Research Institute (DCRI), Children’s Hospital at MUSC.
Footnotes
Declaration of Interest: Dr. Chang was a paid adjudicator for Alere 2013 (PETRA study), and received research support from Hologic, Inc. The other authors report no conflicts.
References
- 1.Duley L. The global impact of preeclampsia and eclampsia. Semin Perinatol. 2009;33:130–137. doi: 10.1053/j.semperi.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Preeclampsia. Lancet. 2010;376:631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- 3.Smith GC, Pell JP, Walsh D. Pregnancy complications and maternal risk of ischaemic heart disease: A retrospective cohort study of 129,290 births. Lancet. 2001;357:2002–2006. doi: 10.1016/S0140-6736(00)05112-6. [DOI] [PubMed] [Google Scholar]
- 4.Skjaerven R, Wilcox AJ, Klungsoyr K, Irgens LM, Vikse BE, Vatten LJ, Lie RT. Cardiovascular mortality after preeclampsia in one child mothers: Prospective, population based cohort study. BMJ. 2012;345:e7677. doi: 10.1136/bmj.e7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonsdottir LS, Arngrimsson R, Geirsson RT, Sigvaldason H, Sigfusson N. Death rates from ischemic heart disease in women with a history of hypertension in pregnancy. Acta Obstet Gynecol Scand. 1995;74:772–776. doi: 10.3109/00016349509021195. [DOI] [PubMed] [Google Scholar]
- 6.Irgens HU, Reisaeter L, Irgens LM, Lie RT. Long term mortality of mothers and fathers after preeclampsia: Population based cohort study. BMJ. 2001;323:1213–1217. doi: 10.1136/bmj.323.7323.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funai EF, Friedlander Y, Paltiel O, Tiram E, Xue X, Deutsch L, Harlap S. Long-term mortality after preeclampsia. Epidemiology. 2005;16:206–215. doi: 10.1097/01.ede.0000152912.02042.cd. [DOI] [PubMed] [Google Scholar]
- 8.Wikstrom AK, Haglund B, Olovsson M, Lindeberg SN. The risk of maternal ischaemic heart disease after gestational hypertensive disease. BJOG. 2005;112:1486–1491. doi: 10.1111/j.1471-0528.2005.00733.x. [DOI] [PubMed] [Google Scholar]
- 9.Lykke JA, Paidas MJ, Damm P, Triche EW, Kuczynski E, Langhoff-Roos J. Preterm delivery and risk of subsequent cardiovascular morbidity and type-ii diabetes in the mother. BJOG. 2010;117:274–281. doi: 10.1111/j.1471-0528.2009.02448.x. [DOI] [PubMed] [Google Scholar]
- 10.Lykke JA, Langhoff-Roos J, Sibai BM, Funai EF, Triche EW, Paidas MJ. Hypertensive pregnancy disorders and subsequent cardiovascular morbidity and type 2 diabetes mellitus in the mother. Hypertension. 2009;53:944–951. doi: 10.1161/HYPERTENSIONAHA.109.130765. [DOI] [PubMed] [Google Scholar]
- 11.Davis EF, Lazdam M, Lewandowski AJ, Worton SA, Kelly B, Kenworthy Y, Adwani S, Wilkinson AR, McCormick K, Sargent I, Redman C, Leeson P. Cardiovascular risk factors in children and young adults born to preeclamptic pregnancies: A systematic review. Pediatrics. 2012;129:e1552–1561. doi: 10.1542/peds.2011-3093. [DOI] [PubMed] [Google Scholar]
- 12.Carr DB, Newton KM, Utzschneider KM, Faulenbach MV, Kahn SE, Easterling TR, Heckbert SR. Gestational diabetes or lesser degrees of glucose intolerance and risk of preeclampsia. Hypertens Pregnancy. 2011;30:153–163. doi: 10.3109/10641950903115012. [DOI] [PubMed] [Google Scholar]
- 13.Vikse BE, Irgens LM, Karumanchi SA, Thadhani R, Reisaeter AV, Skjaerven R. Familial factors in the association between preeclampsia and later esrd. Clin J Am Soc Nephrol. 2012;7:1819–1826. doi: 10.2215/CJN.01820212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts JM, Gammill HS. Preeclampsia: Recent insights. Hypertension. 2005;46:1243–1249. doi: 10.1161/01.HYP.0000188408.49896.c5. [DOI] [PubMed] [Google Scholar]
- 15.Thadhani R, Mutter WP, Wolf M, Levine RJ, Taylor RN, Sukhatme VP, Ecker J, Karumanchi SA. First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab. 2004;89:770–775. doi: 10.1210/jc.2003-031244. [DOI] [PubMed] [Google Scholar]
- 16.Reuvekamp A, Velsing-Aarts FV, Poulina IE, Capello JJ, Duits AJ. Selective deficit of angiogenic growth factors characterises pregnancies complicated by preeclampsia. Br J Obstet Gynaecol. 1999;106:1019–1022. doi: 10.1111/j.1471-0528.1999.tb08107.x. [DOI] [PubMed] [Google Scholar]
- 17.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D’Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 18.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sflt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rana S, Karumanchi SA, Lindheimer MD. Angiogenic factors in diagnosis, management, and research in preeclampsia. Hypertension. 2014;63:198–202. doi: 10.1161/HYPERTENSIONAHA.113.02293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson CJ, Johnson DD, Chang EY, Armstrong DM, Wang W. Evaluation of placenta growth factor and soluble fms-like tyrosine kinase 1 receptor levels in mild and severe preeclampsia. Am J Obstet Gynecol. 2006;195:255–259. doi: 10.1016/j.ajog.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 21.Moore Simas TA, Crawford SL, Solitro MJ, Frost SC, Meyer BA, Maynard SE. Angiogenic factors for the prediction of preeclampsia in high-risk women. Am J Obstet Gynecol. 2007;197:244 e241–248. doi: 10.1016/j.ajog.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 22.Stepan H, Unversucht A, Wessel N, Faber R. Predictive value of maternal angiogenic factors in second trimester pregnancies with abnormal uterine perfusion. Hypertension. 2007;49:818–824. doi: 10.1161/01.HYP.0000258404.21552.a3. [DOI] [PubMed] [Google Scholar]
- 23.Karumanchi SA, Lindheimer MD. Advances in the understanding of eclampsia. Curr Hypertens Rep. 2008;10:305–312. doi: 10.1007/s11906-008-0057-3. [DOI] [PubMed] [Google Scholar]
- 24.He Y, Smith SK, Day KA, Clark DE, Licence DR, Charnock-Jones DS. Alternative splicing of vascular endothelial growth factor (vegf)-r1 (flt-1) pre-mrna is important for the regulation of vegf activity. Molecular Endocrinology. 1999;13:537–545. doi: 10.1210/mend.13.4.0265. [DOI] [PubMed] [Google Scholar]
- 25.Rana S, Hacker MR, Modest AM, Salahuddin S, Lim KH, Verlohren S, Perschel FH, Karumanchi SA. Circulating angiogenic factors and risk of adverse maternal and perinatal outcomes in twin pregnancies with suspected preeclampsia. Hypertension. 2012;60:451–458. doi: 10.1161/HYPERTENSIONAHA.112.195065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carr DB, Tran LT, Brateng DA, Kawamura C, Shofer JB, Karumanchi SA, Easterling TR. Hemodynamically-directed atenolol therapy is associated with a blunted rise in maternal sflt-1 levels during pregnancy. Hypertens Pregnancy. 2009;28:42–55. doi: 10.1080/10641950802132803. [DOI] [PubMed] [Google Scholar]
- 27.Lu F, Longo M, Tamayo E, Maner W, Al-Hendy A, Anderson GD, Hankins GD, Saade GR. The effect of over-expression of sflt-1 on blood pressure and the occurrence of other manifestations of preeclampsia in unrestrained conscious pregnant mice. Am J Obstet Gynecol. 2007;196:396 e391–397. doi: 10.1016/j.ajog.2006.12.024. discussion 396 e397. [DOI] [PubMed] [Google Scholar]
- 28.Costantine MM, Tamayo E, Lu F, Bytautiene E, Longo M, Hankins GD, Saade GR. Using pravastatin to improve the vascular reactivity in a mouse model of soluble fms-like tyrosine kinase-1-induced preeclampsia. Obstet Gynecol. 2010;116:114–120. doi: 10.1097/AOG.0b013e3181e10ebd. [DOI] [PubMed] [Google Scholar]
- 29.Mateus J, Bytautiene E, Lu F, Tamayo EH, Betancourt A, Hankins GD, Longo M, Saade GR. Endothelial growth factor therapy improves preeclampsia-like manifestations in a murine model induced by overexpression of sVEGF-1. American Journal of Physiology. Heart and Circulatory Physiology. 2011;301:H1781–1787. doi: 10.1152/ajpheart.00373.2011. [DOI] [PubMed] [Google Scholar]
- 30.Verlohren S, Herraiz I, Lapaire O, Schlembach D, Zeisler H, Calda P, Sabria J, Markfeld-Erol F, Galindo A, Schoofs K, Denk B, Stepan H. New gestational phase-specific cutoff values for the use of the soluble fms-like tyrosine kinase-1/placental growth factor ratio as a diagnostic test for preeclampsia. Hypertension. 2014;63:346–352. doi: 10.1161/HYPERTENSIONAHA.113.01787. [DOI] [PubMed] [Google Scholar]
- 31.Engels T, Pape J, Schoofs K, Henrich W, Verlohren S. Automated measurement of sflt1, plgf and sflt1/plgf ratio in differential diagnosis of hypertensive pregnancy disorders. Hypertens Pregnancy. 2013;32:459–473. doi: 10.3109/10641955.2013.827205. [DOI] [PubMed] [Google Scholar]
- 32.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor nkx2–5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 33.Benson DW. Genetic origins of pediatric heart disease. Pediatr Cardiol. 2010;31:422–429. doi: 10.1007/s00246-009-9607-y. [DOI] [PubMed] [Google Scholar]
- 34.Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. Nkx-2.5: A novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development (Cambridge, England) 1993;119:969. doi: 10.1242/dev.119.3.969. [DOI] [PubMed] [Google Scholar]
- 35.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. Nkx2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene csx/nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 37.Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. Efficient cre-mediated deletion in cardiac progenitor cells conferred by a 3′utr-ires-cre allele of the homeobox gene nkx2–5. Int J Dev Biol. 2002;46:431–439. [PubMed] [Google Scholar]
- 38.Robinson CJ, Wagner CL, Hollis BW, Baatz JE, Johnson DD. Maternal vitamin d and fetal growth in early-onset severe preeclampsia. Am J Obstet Gynecol. 2011;204:556 e551–554. doi: 10.1016/j.ajog.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, january 2002. Obstet Gynecol. 2002;99:159–167. doi: 10.1016/s0029-7844(01)01747-1. [DOI] [PubMed] [Google Scholar]
- 40.FLJ, Wesselschmidt RL, Schwartz PH. Human stem cell manual: A laboratory guide. 2007:461. [Google Scholar]
- 41.NIH. ImageJ. http://rsb.info.nih.gov/ij/
- 42.Zhao S, Fernald RD. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J Comput Biol. 2005;12:1047–1064. doi: 10.1089/cmb.2005.12.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlson CD, Warren CL, Hauschild KE, Ozers MS, Qadir N, Bhimsaria D, Lee Y, Cerrina F, Ansari AZ. Specificity landscapes of DNA binding molecules elucidate biological function. Proc Natl Acad Sci U S A. 2010;107:4544–4549. doi: 10.1073/pnas.0914023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barth JL, Clark CD, Fresco VM, Knoll EP, Lee B, Argraves WS, Lee KH. Jarid2 is among a set of genes differentially regulated by nkx2.5 during outflow tract morphogenesis. Dev Dyn. 2010;239:2024–2033. doi: 10.1002/dvdy.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of sam68. Nature. 2002;420:691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 46.McLaren M, Asai K, Cochrane A. A novel function for sam68: Enhancement of hiv-1 rna 3′ end processing. RNA. 2004;10:1119–1129. doi: 10.1261/rna.5263904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas CP, Andrews JI, Liu KZ. Intronic polyadenylation signal sequences and alternate splicing generate human soluble flt1 variants and regulate the abundance of soluble flt1 in the placenta. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2007;21:3885–3895. doi: 10.1096/fj.07-8809com. [DOI] [PubMed] [Google Scholar]
- 48.Huckle WR, Roche RI. Post-transcriptional control of expression of sflt-1, an endogenous inhibitor of vascular endothelial growth factor. J Cell Biochem. 2004;93:120–132. doi: 10.1002/jcb.20142. [DOI] [PubMed] [Google Scholar]
- 49.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at ucsc. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lian IA, Loset M, Mundal SB, Fenstad MH, Johnson MP, Eide IP, Bjorge L, Freed KA, Moses EK, Austgulen R. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without preeclampsia. Placenta. 2011;32:823–829. doi: 10.1016/j.placenta.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmad S, Ahmed A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ Res. 2004;95:884–891. doi: 10.1161/01.RES.0000147365.86159.f5. [DOI] [PubMed] [Google Scholar]
- 52.Catalano PM. Management of obesity in pregnancy. Obstet Gynecol. 2007;109:419–433. doi: 10.1097/01.AOG.0000253311.44696.85. [DOI] [PubMed] [Google Scholar]
- 53.Germain S, Nelson-Piercy C. Lupus nephritis and renal disease in pregnancy. Lupus. 2006;15:148–155. doi: 10.1191/0961203306lu2281rr. [DOI] [PubMed] [Google Scholar]
- 54.Powe CE, Thadhani R. Diabetes and the kidney in pregnancy. Semin Nephrol. 2011;31:59–69. doi: 10.1016/j.semnephrol.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 55.Roberts JM, Pearson GD, Cutler JA, Lindheimer MD. Summary of the nhlbi working group on research on hypertension during pregnancy. Hypertens Pregnancy. 2003;22:109–127. doi: 10.1081/PRG-120016792. [DOI] [PubMed] [Google Scholar]
- 56.Bigelow CA, Pereira GA, Warmsley A, Cohen J, Getrajdman C, Moshier E, Paris J, Bianco A, Factor SH, Stone J. Risk factors for new-onset late postpartum preeclampsia in women without a history of preeclampsia. Am J Obstet Gynecol. 2013 doi: 10.1016/j.ajog.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 57.Shahabi A, Wilson ML, Lewinger JP, Goodwin TM, Stern MC, Ingles SA. Genetic admixture and risk of hypertensive disorders of pregnancy among latinas in los angeles county. Epidemiology. 2013;24:285–294. doi: 10.1097/EDE.0b013e31828174cb. [DOI] [PubMed] [Google Scholar]
- 58.Knuist M, Bonsel GJ, Zondervan HA, Treffers PE. Risk factors for preeclampsia in nulliparous women in distinct ethnic groups: A prospective cohort study. Obstet Gynecol. 1998;92:174–178. doi: 10.1016/s0029-7844(98)00143-4. [DOI] [PubMed] [Google Scholar]
- 59.Jebbink J, Wolters A, Fernando F, Afink G, van der Post J, Ris-Stalpers C. Molecular genetics of preeclampsia and hellp syndrome - a review. Biochim Biophys Acta. 2012;1822:1960–1969. doi: 10.1016/j.bbadis.2012.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Maternal age (MA), gestational age (GA), parity and gravidity are shown as median (range). Birthweight (BW), body mass index (BMI), blood pressure (BP) and proteinuria are expressed as median (interquartile range). *Data available for 16 of 19 patients. **Data available for 9 of 14 patients.
Statistical associations of Nkx2-5, Sam68 and sFlt-1 mRNA expression in EOSPE placentae in the overall population, and stratified by AA or CA race. *significant (p ≤ 0.05); **highly significant (p ≤ 0.02)
Statistical associations of Nkx2-5, Ccdc117 and Xbp1 expression in overall EOSPE population, and stratified by AA and CA race. **highly significant (p ≤ 0.02)