

Abstract
Estrogen receptors alpha (ERα) and beta (ERβ) are nuclear transcription factors that are involved in the regulation of many complex physiological processes in humans. Modulation of these receptors by prospective therapeutic agents is currently being considered for prevention and treatment of a wide variety of pathological conditions, such as, cancer, metabolic and cardiovascular diseases, neurodegeneration, inflammation, and osteoporosis. This review provides an overview and update of compounds that have been recently reported as modulators of ERs, with a particular focus on their potential clinical applications.
Keywords: Estrogen receptors, Estrogen receptor subtype α, Estrogen receptor subtype β, modulators, agonists, antagonists, therapeutic applications
1. Estrogen receptors subtypes alpha (ERα) and beta (ERβ)
For half a century, menopausal symptoms, such as osteoporosis, hot flashes, and urogenital atrophy, have been treated with hormone therapy (HT), comprising estrogens alone or estrogens and progestins. In fact, because of their beneficial actions in non-reproductive tissues, such as bone, brain, and urogenital tract, estrogens would be ideal drugs if they did not have serious adverse effects, consisting mainly of increasing the risk of breast and endometrial cancer, as well as of thromboembolisms and strokes [1].
The physiological functions of estrogenic compounds are modulated largely by the estrogen receptors subtypes alpha (ERα) and beta (ERβ). These proteins have actions in the cell nucleus, regulating transcription of specific target genes by binding to associated DNA regulatory sequences. In humans, both receptor subtypes are expressed in many cells and tissues, and they control key physiological functions in various organ systems, such as reproductive, skeletal, cardiovascular and central nervous systems, as well as in specific tissues (such as breast and sub-compartments of prostate and ovary). ERα is present mainly in mammary gland, uterus, ovary (thecal cells), bone, male reproductive organs (testes and epididymis), prostate (stroma), liver, and adipose tissue. By contrast, ERβ is found mainly in the prostate (epithelium), bladder, ovary (granulosa cells), colon, adipose tissue, and immune system. Both subtypes are markedly expressed in the cardiovascular and central nervous systems. There are some common physiological roles for the two ERs, such as in the development and function of the ovaries, and in the protection of the cardiovascular system. The alpha subtype has a more prominent role on the mammary gland and uterus, as well as on the preservation of skeletal homeostasis and the regulation of metabolism. The beta subtype seems to have a more profound effect on the central nervous and immune systems, and it generally counteracts the ERα-promoted cell hyperproliferation in tissues such as breast and uterus [2–3].
Estradiol and hormone replacement therapies target both the ERs, but this often leads to an increased risk of breast and endometrial cancers, and thromboembolism. Also, selective estrogen receptor modulators (SERMs) target both receptor subtypes, although they display tissue-selective agonist/antagonist activities through mechanisms distinct from those involving ER subtype-selective binding or activation. An ideal SERMs would possess antagonist activity in the mammary gland and uterus, and agonist activity in other target tissues that benefit from an estrogen-like action, such as the cardiovascular, skeletal, and central nervous systems [4]. Alternatively, subtype-selective ligands could be used to elicit beneficial estrogen-like activities and reduced side effects, based on the different distributions and relative levels of the two ER subtypes in different estrogen target tissues noted above. In this regard, there appears to be particular promise for the use of ERβ-selective agonists.
The development of compounds possessing ER subtype specificity still constitutes a major challenge. The amino acid sequence of ERα and ERβ displays 59% sequence identity in their respective ligand binding domains (LBD), which represents a significant difference. However, the differences within the ligand binding cavities are at only two amino acid positions: Leu384 and Met421 in ERα are replaced by Met336 and Ile373, respectively, in ERβ; not only are these conservative changes between hydrophobic residues, they are also reciprocal, with each ER subtype having one methionine residue and then either a leucine or isoleucine residue. Perhaps more significant is the fact that in most crystal structures, the ERβ binding pocket is smaller and narrower than that of ERα, which is more likely due to the greater sequence diversity beyond the ligand binding pocket than to the small differences at the two pocket residue positions. The overall effect of these very subtle differences makes it quite difficult to design subtype-selective ligands. Nevertheless, such efforts have been greatly aided by major advances in the structural biology of estrogen receptors over the past two decades that have shed light on the molecular basis for the binding modes and the regulation of agonist vs. antagonist activities of ER-ligands [5–6]. Consequently, nowadays investigators have a good context in which to consider ligand interactions with the ERs that are likely to be crucial for good binding affinity, and these, together with accumulating information on structure-binding and structure-activity relationships, have led to the formulation of reliable ER-pharmacophore models [7].
Generally, a good ER-ligand should possess two OH groups that are linked by a lipophilic central scaffold, which places them at a distance of about 11 Å. At least one of these hydroxyls should be a phenol or a phenol-bioisostere. One of these OH groups forms a strong attractive interaction with an H-bond network comprising residues (ERα/β numbering) Glu353/305, Arg394/346 and a water molecule, where the phenolic OH group of the A-ring of estradiol binds. The antipodal hydroxyl group, instead, mimics the 17β-OH group of estradiol and forms an additional H-bond with residue His524/475 in the estradiol D-ring pocket. Beyond these two key, energetically important interactions, which appear to be commensurate between the two ERs, subtype-selectivity needs to arise from the different shape and hydrophobicity of the central scaffold, which can only count on weakly differential attraction/repulsion balances with the non-conserved residues Leu384(ERα)/Met 336(ERβ), and Met421(ERα)/Ile373(ERβ), as well as differences in the propensity of the two ERs to form pockets of different sizes. In any case, the selective activation of ERα or ERβ may depend not only on a selective receptor binding affinity, but also on selective activation of each receptor subtype [8].
2. Potential clinical applications of ERα and ERβ modulators
2.1. Cancer
In many breast cancers, ERα activation by estrogens is generally considered responsible for enhanced proliferation, whereas this is counteracted by the presence of ERβ, which exerts an antiproliferative effect [9]. Therefore, theoretically, breast cancer patients with estrogen-responsive disease should respond positively to treatment with ERα-antagonists and/or ERβ-agonists. However, the situation is a bit more complicated because responses will depend on the expression level of these two receptors in the cancer tissue and also on the stage of the neoplasia. For example, it was recently demonstrated that ductal breast cancer that is highly proliferative shows a high expression of ERα and a low expression of ERβ when it is early stage. High-grade invasive ductal cancer then loses the expression of both receptors. On the other hand, early-stage lobular breast cancer shows abundant levels of both ERα and ERβ, whereas advanced stages of this disease present only ERα expression, ERβ having been lost [10]. These examples would support the use of ERβ-selective agonists only in the case of early-stage ductal cancer, since it may prevent or delay development of more invasive disease. Correspondingly, an ERα-antagonist may be helpful for the treatment of advanced lobular cancer (but not early stage disease), or early-stage ERα-positive ductal cancer.
Activation of ERβ in early prostate cancer, as well as in benign prostatic hyperplasia, leads to an antiproliferative and pro-apoptotic effect [11]. Furthermore, ERβ seems to inhibit the epithelial-mesenchymal transition (EMT) in prostate cancer cells by destabilizing hypoxia-inducible factor 1α (HIF-1α), thus reducing their invasive potential [12]. Therefore, there is hope that an ERβ-selective agonist might be used to treat these pathologies.
In human malignant pleural mesothelioma, ERβ was found to modulate cell proliferation by effectively inactivating the epidermal growth factor receptor (EGFR), thus exerting a tumor repressive effect [13], and a similar function was also observed in renal cell carcinoma cell lines [14].
Loss of ERβ expression is generally found in colorectal cancer, and the degree to which it is reduced correlates with increasingly poor prognosis [15]. ERβ was demonstrated to have a protective effect against the development and progression of colon cancer by inhibiting cell proliferation in vitro and tumor formation in vivo [16–17]. Therefore, selective stimulation of ERβ might be useful as a preventive and/or therapeutic strategy against this type of neoplasia.
The role of ERs in lung cancer has been extensively studied, but it still remains quite controversial. In fact, in some studies ERβ was found to be the main receptor mediating the proliferative effects of estrogens in lung cancer cells [18]. Furthermore, ERβ appears to be involved with insulin-like growth factor-1 receptor (IGF-1R) in a synergistic promotion of lung adenocarcinoma development in murine models [19]. On the other hand, other studies indicate that ERβ may have prognostic value, since its overexpression appears to be a positive prognostic marker in stage II and III non-small cell lung cancer (NSCLC) patients [20], whereas its absence, together with the presence of ERα, was associated with a poorer prognosis in patients affected by the same type of cancer [21]. This effect seems to be gender-dependent, since women with ERβ-positive tumors did not show any significant difference in mortality from those with ERβ-negative tumors, whereas men with ERβ-positive tumors displayed a significant reduction in mortality [22]. In any case, ERβ-activation in human non-small cell lung cancer cells in vitro produced a stimulatory effect on cell proliferation [23]. So, the real potential for the use of ERβ-agonists in lung cancer is still rather puzzling. Other types of tumors that might potentially be treated with ER ligands will be mentioned in the specific sections below.
2.2. Neuropathies
Much evidence underlies the important role of estradiol in the central nervous system (CNS) for different kind of diseases, including pathologies associated with depression and anxiety. A recent study reports that aged female mice respond favorably to E2 administration and exhibit greater anti-anxiety and antidepressant behavior [24]. In fact, estrogens were found to modulate the synthesis and degradation of serotonin, as well as the expression of serotonin receptors [25]. Activation of both ERα and ERβ caused a neuroprotective effect, in particular against toxicity associated with Aβ accumulation, therefore suggesting a potential role in the prevention and treatment of Alzheimer’s disease [26]. In animal models, ERα proved to play a key role in the prevention and treatment of multiple sclerosis, which was superior to that of ERβ [27]. ERβ ligands have some capacity to reverse symptoms of established disease, and the use of selective ERα agonists is controversial, because of the increased risk of breast and endometrial cancers and thromboembolic events. Finally, administration of soy isoflavones, which are known ERβ-selective agonists, to healthy male individuals caused a general enhancement of their cognitive abilities [28], thus further supporting the beneficial role of ERβ in supporting memory and learning functions.
2.3. Cardiovascular disease
The regular production of estrogens in pre-menopausal women places them at a lower risk for cardiovascular disease, but this cardioprotective effects vanishes after menopause [29]. The mechanisms involved in this cardioprotective role of estrogens are currently under investigation, and both ERs seem to be involved at least to some extent [30]. In particular, ERα is highly expressed in endothelial cells and plays a role in mediating the effects of estrogens in the vascular endothelium, whereas ERβ stimulates the production of nitric oxide. Therefore, the activation of both receptors has a beneficial hypotensive effect caused by vascular wall dilation [31–32]. A recent study [33] demonstrated that the non-nuclear fraction of ERα, which is linked to plasma membrane, is selectively responsible for the vascular actions elicited by estrogens, such as rapid dilatation, acceleration of endothelial repair, and endothelial NO production. This observation is particularly useful since a selective cytoplasmic stimulation of ERα would promote a beneficial cardioprotective effect, without eliciting all those side effects associated to nuclear activation of this receptor.
2.4. Osteoporosis
Osteoporosis is a major health problem associated with estrogen deficiency in postmenopausal women due to the marked increase of bone resorption that is not compensated by bone formation. Many studies have demonstrated that estrogens play a crucial role in bone homeostasis, not only in women but also in men [34]. Estrogen replacement therapy (ERT) is clinically used to prevent osteoporosis [35]; however, long-term ERT may stimulate proliferation of breast and uterus through ERα, increasing the risk of cancer in these organs. Bone cells express both estrogen receptor, ERα and ERβ, although ERα seems to play the major role [36]. The use of a SERM, such as raloxifene, seems to reduce the risk caused by ERT and, at the same time, to counteract osteoporosis [37].
2.5. Obesity and metabolic disorders
ERβ may play an important role in regulating metabolic pathway and adipose tissue function [38]. Obesity and related metabolic diseases represent major problems in modern society, and the few drugs approved for treatment have side effects. Recent studies highlight the beneficial effect of E2 on hyperglycemia, oxidative stress and on the liver dysfunction in diabetic rats [39]. In fact, estrogen deficiency causes metabolic dysfunction, obesity, metabolic syndrome, and type 2 diabetes. Hence, there is a great potential for the treatment of these pathologies with appropriate ER-activating agents, in particular, those behaving as SERMs or acting through ERβ [40].
2.6. Menopause
There are several ongoing investigations aimed at establishing the utility of estrogen-based treatments in reversing menopausal changes in women, such as, loss of bone density, hot flushes and cardiovascular problems. Nowadays, a great deal of attention is being devoted to the administration of selective ERβ-activators, since they are considered likely to be safer than non-specific estrogens, due to the lack of stimulation of ERα. One of these studies concerns the administration of an herbal extract, MF101 [41], which proved to reduce the frequency of hot flushes in a phase II clinical trial [42].
3. Update on chemical classes comprising ERα and ERβ modulators
Over the past few years, many comprehensive reviews have exhaustively covered progress in the development of ER ligands [43–47]. We herein describe an update of the most recently reported molecules that are able to interact with the ERs, with particular focus on those displaying likely potential as perspective therapeutic agents.
3.1. Steroid derivatives
Several synthetic estradiol analogues were tested in animal models to understand the role of both ER subtypes in pathophysiology. Among the steroid derivatives tested, two molecules have been used in several in vivo studies as ERs selective ligands: 8β-VE2 (1, Fig. 1), a 46-fold ERβ-selective compound obtained by the addition of a vinyl group at 8β position of E2 scaffold [48], and 16α-LE2 (2, Fig. 1), an ERα-selective compound obtained with the fusion of a lactone ring across the 16α and 17α positions of E2 [49].
Fig. 1.

Steroids derivatives.
Both ERs are thought to play important roles in cardiovascular diseases, having beneficial short and long-term cardiovascular effects [50], although the respective roles of ERα and ERβ have not yet been clarified [51]. In a recent study, ovariectomized atherosclerosis-prone apoE-/- mice were fed a high-cholesterol diet and received daily subcutaneous injections of 8β-VE2 for 5 weeks. Compared with controls, treatment with 8β-VE2 reduced aortic arch atherosclerotic lesion areas by 34% of total and 75% of dense lesions, without altering the serum lipid profile, while the treatment with fulvestrant, a nonselective ERs antagonist devoid of any agonistic activity, completely abrogated the beneficial vascular effects of 8β-VE. Furthermore, blood samples collected at regular intervals reveal that 8β-VE2 reduces serum levels of proinflammatory cytokines. These results demonstrate that ERβ may play a crucial role in maintaining vascular homeostasis [52]. On the other hand, ERα seems to be important in maintaining the physiological cardiac glucose uptake in normotensive and nondiabetic female mice. In fact, administration of 16α-LE2 to ovariectomized mice completely restored cardiac glucose uptake, whereas this effect was strongly reduced in ERα knockout mice, although the role of ERβ in this process still needs to be investigated [53].
ERs, and ERβ in particular, also play important roles in skeletal muscle regeneration processes after injuries. Ovariectomized female Wistar rats with noxetin-induced damage have been used to investigate the role of both ERs in skeletal muscle regeneration using E2, 8β-VE2 and 16α-LE2. First, serum levels of creatine kinase (CK) were analyzed to investigate the extent of injury, and although all the compounds decrease CK levels, 8β-VE2 proved to be the most potent agent in protecting the cell membrane. Furthermore, βERKO mice experienced increased muscle damage [54], thus highlighting a predominant role for ERβ in this repair process.
There is considerable evidence that endogenous estrogens play a significant role as modulators of immune responses and autoimmune diseases. Therapeutic effects of 8β-VE2 and 16α-LE2 have been evaluated in ArKO (aromatase-knockout) male mice, in which estrogen deficiency is associated with a significant increase in lipopolysaccharide (LPS)-induced serum interleukin-6 (IL-6), tumor necrosis factor, monocyte chemotactic protein 1 and interferon-γ levels, all of which are significantly abrogated by administration of 16α-LE2, but not 8β-VE2. Furthermore, only 16α-LE2 is effective in reducing arthritis severity in ArKO mice with collagen-induced arthritis (CIA), suggesting a dominant role of ERα in this process, although both ligands are effective in LPS-induced cytokine expression and antigen-induced arthritis (AIA) [55].
5-Androstenediol (3, Fig. 1) and 3β-androstanediol (4, Fig. 1) are two steroid hormone derivatives with an interesting ERβ-selective profile. 5-Androstenediol has a higher ERβ affinity (ERβ-RBA = 17%) with a 3-fold selectivity for ERβ, while 3β-androstanediol has a lower affinity (ERβ-RBA = 7%) and a comparable selectivity [56]; both compounds were tested in animal models.
5-Androstenediol was found to act as an ERβ-selective modulator by suppressing the inflammatory responses of microglia and astrocytes, which are responsible for the maintenance of homeostasis in CNS. Microglia seem to play an important role in experimental autoimmune encephalomyelitis (EAE). Furthermore, anti-inflammatory effects of ER-selective ligands within the CNS have been extensively evaluated in animal models of EAE [57]. After the observation that some ERβ selective with an indazole scaffold are effective in inhibiting inflammatory responses in microglia while E2 and other ERβ ligands are not, a panel of ERβ natural steroids was screened for their ability to suppress induction of IL-6 in microglia upon LPS stimulation. Although most of the compounds have no activity, 5-androstenediol represses the expression of IL-6 in the substantia nigra, indicating that it can suppress microglia-mediated inflammation in vivo. Furthermore, this study demonstrates that the binding of 5-androstenediol to ERβ promotes the interaction of the receptor with the CtBP repressor, resulting in transcriptional repression that regulates microglia-mediated inflammation [58].
3β-Androstanediol modulates the LPS-induced inflammatory response in the arterial vascular wall in vivo in castrated mice. Interestingly, as observed above for 5-androstenediol, E2 is ineffective on the same parameters modified by 3β-androstanediol, suggesting the existence of different ligand-selective pathways for the activity of ERβ [59]. Another study reveals this different behavior between 3β-androstanediol and E2 in prostate cancer models in vivo, by using a bioluminescence imaging (BLI) assay. In this study, PC3-Luc cells were subcutaneously injected in the shoulder of BALB/c nu/nu nude mice, and after three weeks, a silastic pellet continuously releasing 3β-androstanediol was implanted into one group. Interestingly, after two weeks of treatment, tumor proliferation and metastatic spreading was significantly inhibited. It was hypothesized that the different effect of E2 vs. 3β-androstanediol was due to a different conformation of Helix 12 imposed by the two ligands could lead to a differential co-regulator complex recruitment to the regulatory regions of the target genes [60]. Other evidence supports the important role of ERβ in prostate cancer: its apoptotic function seems to be mediated by the upregulation of FOXO3a, a member of the FOXO family transcription factors, which upregulates PUMA (p53-upregulated modulator of apoptosis), a potent pro-apoptotic factor. Athymic mice were injected with PC3-control or PC3-ERβ cells, and after 40 days visible tumors developed. Apoptotic bodies were found in ERβ-expressing tumors but not in control tumors. Treatment of PC3-cells with 3β-androstanediol led to an increase in the level of pro-apoptotic factor p53-upregulated modulator of apoptosis (PUMA). It is important to note that PC3 cells are also non-responsive to E2, as described above for the other two studies. Furthermore, this study shows that when paired with the ERα-selective agonist propyl pyrazole triol (PPT), 3β-androstanediol no longer increases PUMA expression. The fact that PC3 cells fail to respond to E2 may be explained by considering that simultaneous activation of the two ER subtypes could lead to opposing effects that neatly cancel out one another [61].
The positive results of these first in vivo experiments raise the interesting question concerning the stability of the 3-OH group present in these steroid derivatives, because it could be converted to the corresponding ketone by 3-hydroxysteroid dehydrogenases, thus producing metabolites with a lower affinity for the ERs but increased affinity for androgen receptor (AR) [62]. Further studies will be needed to verify the extent to which these compounds undergo metabolism in vivo.
In recent years, one of the main strategies used to selectively targeted estradiol derivatives has been to conjugate them to other specific molecules to try to avoid proliferative side effects due to ERα stimulation in sensitive reproductive tissues, such as breast and uterus. Among these E2 conjugates are several considered as antiosteoporosis agents. A recent study reports the generation of a new female osteoblast-specific ERαKO mouse (pOC-ERαKO), which was used to clarify the estrogen signaling pathway in bone cells in vivo. pOC-ERαKO mice show decreased cancellous bone mass in the proximal tibia, vertebra, and distal femur, and decreased cortical bone mass in the tibial midshaft at 12 and 18 weeks of age, indicating that ERα in osteoblasts is required for appropriate bone mass and strength in female mice [63]. To overcome the ERα-related side effects and to selectively target estrogen effects in bones, different estradiol-conjugates were prepared. BTE2-A1 (5) is a novel bone-targeted estrogen, which was obtained by conjugation of estradiol to a mimic of the tricarbonylmethane system of the tetracycline ring A, a portion that binds very well to the bone mineral hydroxyapatite. BTE2-A1 shows a slight preference for ERα, with a RBA = 16% and, interestingly, shows a higher binding affinity to hydroxyapatite than that of tetracycline in vitro. After subcutaneous administration of this compound in vivo, a partial dissociation of the effects of estradiol in bone vs. uterus was noted, indicating that this bone-targeting strategy has some merit, although further studies will be required to clarify the mechanism of action [64].
Novel estrogen conjugates with integrin-binding RGD peptides, notably 3,17-estradiol-RGD tetrapeptides (6–8, Fig. 1) and 3-estrone-RGD octapeptides (9–11, Fig. 1), show high oral anti-osteoporosis activity in an ovariectomized mouse model. Serum calcium content and alkaline phosphatase (ALP) levels in mice given 110.3 nmol/kg of these compounds orally were significantly lower than those of mice receiving control estrogen, and both femur weight and femur ash and mineral levels of mice receiving control estrogen were lower than those receiving conjugates. Finally, these compounds showed a lower risk of endometrial hyperplasia and thromboembolic events [65]. In addition to these conjugates, 17-estradiol-RGD octapeptides (such as 12, Fig. 1) and 3-estradiol-RGD octapeptides (such as 13, Fig. 1) were evaluated and assayed as anti-osteoporosis agents in ovariectomized mice. These conjugates caused a reduction of serum calcium and ALP levels when compared to estrogen control, showed femur weights and femur ash, and higher mineral values. The anti-osteoporosis activity of these octapeptide derivatives was significantly higher than that of estradiol and tetrapeptide analogues, while the risk of thrombogenesis and endometrial hyperplasia was lower, suggesting that the derivatization of estradiol with RGD peptides may be a good strategy to further increase the potency of these conjugates [66]. Unfortunately, the affinities and selectivities of compounds 6–13 for ERs have not been reported.
Another anti-osteoporosis compound was obtained by conjugation of E2 with iminodiacetic acid (14, SE2, Fig. 1), a well-known, high-affinity calcium chelator, in order to obtain a preferential distribution to bone. SE2 shows a greater affinity for bone and a lower affinity for uterus, when compared to E2, and in osteoblasts, it maintains high affinity for ERα, similar to that of E2. Furthermore, in this study, SE2 was effective in preventing OVX-induced bone loss in rats, decreasing bone turnover and improving bone mass and trabecular architecture [67].
Some interesting E2-conjugates consist in poly(amide)polyamine (PAMAM) dendrimers (EDCs), in particular those linked by shorter tethers to PAMAM through a 17α-phenylethynyl unit, show good affinity for the ERs, which are often comparable with those of the parent ligands alone, with a slight preference for ERα (for example, compound 15: ERα-RBA = 3.8 %, ERβ-RBA = 1.9 %, Fig. 1) [68]. Recently, EDC 15 was shown to promote cardiovascular protection in mice. In particular, this EDC interacts only with the membrane ERα receptor, thus showing exclusive non-nuclear action. Interestingly, in mice the EDC 15 does not stimulate proliferative effects due to ERα activation in sensitive reproductive tissues and, therefore, does not induce uterine or breast cancer growth. This selective effect is due to the highly preferential activation of the ERs present in extra-nuclear sites [69].
3.2. Benzopyrans
Many efforts to find new SERMs have focused on the design of benzopyran derivatives. In particular, McKie et al. [70] screened commercially available phenolic compounds using a cellular assay based on inhibition of the release of the cytokine interleukin-6 (IL-6) from an estrogen receptor-negative osteosarcoma cell line (U2OS), which had been transfected with ERα. The results of this first screening inspired the design and synthesis of a series of new potent benzopyran ERα ligands, with good receptor binding affinities, but devoid of undesirable proliferative effects on breast cancer MCF-7 cells. This study led to 16 (Fig. 2) as a potential lead compound for the prevention and treatment of osteoporosis and breast cancer. Compound 16 preferentially binds to ERα (RBA = 85%), and it showed an IC50 value of 0.38 nM in the IL-6-based cellular assay, thus demonstrating an improvement over reference compounds such as tamoxifen and raloxifene, whereas the anti-proliferative activity on MCF-7 cell line was quite comparable to that of raloxifene (IC50 = 7.7 nM). Moreover, when compound 16 was tested in immature or ovariectomized rat uterine bioassay, it did not increase significantly uterine weight, confirming the lack of unwanted uterine stimulatory activity [71]. In 2004, benzopyranone analogue CC-8490 of undisclosed structure (with a reported molecular weight of 586.05) belonging to this class of compounds, proved able to induce apoptosis in glioma cells in a time- and dose-dependent manner, and also to suppress tumor growth in vivo and prolong survival in malignant glioma xenograft models [72]. Interestingly, the pro-apoptotic effect of CC-8490 seemed to be non ER-related, because the tested cell lines did not express estrogen receptors α or β, suggesting a mechanism which excludes the ER signaling pathway. Otherwise, a positive correlation between nuclear-factor (NF)-κB activation and resistance to SERM-induced apoptosis was found, suggesting that the NF-kB pathway acts as a pro-survival mechanism in glioma cells and its inhibition should accentuate the antiglioma activity of CC-8490.
Fig. 2.

Benzopyran derivatives 16–22.
These findings led Celgene Corporation, in partnership with the US National Cancer Institute, to start in 2003 a phase I clinical trial of CC-8490 in patients affected by recurrent or refractory high-grade gliomas [73] in the search for agents to treat brain tumors; this compound was found to be safe and well-tolerated. This trial is currently listed on clinicaltrials.gov as being completed, and CC-8490 is passed to a phase 2 trial, but no additional information was found on the Celgene Corporate website.
Compound 17 (LY3201, Fig. 2) [74] belongs to the cyclopentane benzopyran series, and it showed a high selectivity for ERβ (Ki = 0.44 nM, β/α = 19). Recent studies at Eli Lilly investigated the effects of LY3201 in brain and identified this derivative as a possible therapeutic agent for CNS diseases. Benzopyran 17 was administered to mice as subcutaneous pellets (0.04 mg/d), and after 3 days it was found to provoke a shift in the balance between excitatory and inhibitory signaling in favor of inhibition. This effect was mainly attributed to an increased production of γ-aminobutyric acid (GABA), which is one of the main inhibitory transmitters in brain. It also increased the rate of removal of glutamate, the primary excitatory neurotransmitter in central nervous system, which occurs by glutamine synthase in astrocytes that take up glutamate and convert it into glutamine [75]. Considering this potential anxiolytic action of compound 17 and the well-known involvement of estrogens on the serotonin system, further in vivo tests were performed to investigate possible effects on serotoninergic neurons of the dorsal raphe, which are involved in anxiety and depression. In ovariectomized rats, where the enzyme responsible of serotonin synthesis, tryptophan hydroxylase (TPH), is down-regulated due to the absence of estrogens, treatment with LY3201 in a continuous-release fashion for 3 days caused in increase in the number of TPH-positive neurons; however, if the benzopyran 17 was administered 10 weeks after estrogen deprivation, it was no longer able to restore TPH-positive neurons. These findings suggest a potential use of this ERβ-selective agonist in combination with selective serotonin re-uptake inhibitors to shorten the onset of their anti-depressant effect in postmenopause [76].
A further beneficial effect of ER-ligands against neurodegenerative diseases was demonstrated in an animal model of multiple sclerosis, consisting of an experimental autoimmune encephalomyelitis affected mice, where compound 17 caused a down-regulation of inflammatory pathways and led to a decreased mortality and a reduction of severity of symptoms in survived mice. Activated microglia, which express only ERβ, are the cells responsible of immunological defense in the CNS. Upon treatment with 17, microglial activation was markedly reduced, and the production of inflammatory molecules produced by microglia was also reduced. Moreover, T-cell reactivity was compromised by this ERβ-selective activator, due to the induction of the enzyme indole-amine-2,3-dioxygenase, which causes T-cell death through depletion of tryptophan [77].
Piperidinoethoxyphenyl-substituted benzopyran 18 (Fig. 2) named CDRI-85/287, an ER ligand which was previously discovered and evaluated for its potent antiestrogenic activity in the uterus and anti-implantation properties in animal models [78–81], recently has attracted great interest for its growth inhibitory activity on breast cancers [82]. CDRI-85/287 in its racemic form displayed a slight preference for ERα over ERβ, with low relative binding affinities of 0.25% for ERα and 0.15% for ERβ, although previous studies assigned a higher ER binding affinity to the L-enantiomer [83]. Despite the low binding affinity for ERs in vitro, compound 18 inhibited the growth of the ER-positive breast cancer cell lines MCF-7 and T47D with IC50 values of about 5 μM, without exerting toxic effects on healthy cells, and the beneficial activity on breast cancer is reflected in vivo where at dose of 20 mg/kg the benzopyran prevented growth of MCF-7 xenografts in nude mice after 8 days of administration. The central role of ERα and ERβ in the anti-cancer activity of compound 18 was confirmed by the loss of cytotoxicity when 18 was added to cells in which the expression of the two receptors was knocked down by using siRNA. Moreover, it has been demonstrated that CDRI-85/287 reduced the expression of ERα while it increased that of ERβ, suggesting that the influence on ER expression may mediate, at least in part, its anti-proliferative activity on ER-positive breast cancer. In any case, the considerable difference between the potent activity in vivo and the modest binding to ERs may be attributed to benzopyran metabolites, which, unlike the parent compound, could bind with high affinity to the ERs and have potent biological activity. This compound exerted some anti-proliferative action by interfering with the EGFR/Akt pathway in the ER-negative human breast cancer cell line MDA-MB-231 and inducing apoptosis in cultured cells, while it arrested tumor growth in mice. This additional activity contributes to the ER-mediated mechanism, confirming the anti-tumorigenic potential of CDRI-85/287 in breast cancer [84].
Benzopyranobenzoxapanes 19–22 (Fig. 2) [85] were designed and synthesized as therapeutic agents to treat postmenopausal symptoms, and they are derived from a previous series of benzopyran derivatives discovered in 2006 [86], which were further developed by Johnson & Johnson to improve their stability, pharmacokinetic and pharmacological profiles. Compound 19, the representative derivative of this class of chromenes, is an ERα-selective ligand with an IC50 of 1.1 nM (ERβ-IC50 = 21 nM). This compound displayed excellent activity as an ER antagonist when tested in two cell-based functional assays, Ishikawa endometrial cell alkaline phosphatase secretion and MCF-7 breast cancer proliferation assay, with IC50 values in the low nanomolar range. In the immature rat uterotrophic model, it was administered once daily (1 mg/kg) for 3 days, and it showed antagonistic behavior, exerting a 135% inhibition of uterus weight compared to control animals. Its di-methoxy analogue 22 is about 60-fold less active on ERα and does not bind ERβ, with the result that it is inactive in in vitro assays, even though it retained strong uterotrophic inhibition. The two mono-methoxy analogues, 20 (R2 = Me, Fig. 2) and 21 (R1 = Me, Fig. 2), have modest binding affinities to ERs and weak potencies in cellular assays, which are definitely less than that of 19. Interestingly, compound 21 has a better binding affinity for ERβ (ERβ-IC50 = 7 nM) than the parent compound 19, but it loses beta-selectivity, because it also binds ERα with a similar affinity (IC50 = 9 nM). However, both the mono-methoxy compounds 20 and 21 showed strong inhibitory activities on uterus. Any other replacement of the piperidine ring on the amino side chain inverted activity in the uterotrophic assay, so this basic moiety appears essential for its in vivo antagonism. Furthermore, at least one hydroxyl group (R1 or R2) is required for the binding to ERs, as demonstrated by the reduction in binding to ER when hydroxyl substituents R1 and/or R2 are methylated. Rather different effects were obtained when these four lead compounds were evaluated in pharmacokinetic studies, in which they were administered intravenously or orally to rats. The bisphenolic compound 19 had poor bioavailability, probably because of rapid metabolism in vivo. The di-methoxy analogue 22 by contrast, showed good bioavailability with improved absorption in tissues, and it is metabolized in vivo to mono-methoxy derivatives 20 and 21, which in turn were tested and displayed excellent bioavailabilities, even superior to that of compound 22.
After these encouraging results, compound 22 and its metabolites 20 and 21 were further evaluated in different animal models to test their tissue selectivity as potential SERMs. In the adult ovariectomized rat model, they were dosed orally once daily for 2 or 6 weeks, and the responses were typical of an ideal SERM: prevention of bone loss, decrease of plasma total cholesterol level and increase in vaginal fluidity without significant stimulation of the uterus. Moreover, in the ovariectomized rat hot flush model, compound 22 was able to reduce the rise in tail temperature, revealing it to be a suitable candidate to treat hot flushes and, generally, an optimal SERM candidate.
3.3. Cyclohexylphenols, cycloalkylene and diphenylmethane derivatives
Fluorinated cyclohexylphenol AC-186 (23, Fig. 3) was discovered by Acadia Pharmaceutical as a selective ERβ agonist [87–88] and proved to be active in vivo against neurodegenerative diseases in mouse models of Alzheimer’s disease and Parkinson’s disease. In combination with a selective androgen receptor agonist ACP-105, in male gonadectomized triple transgenic Alzheimer’s disease mice (3xTg-AD mice), AC-186 decreased amyloid-β levels in the brain by increasing the amyloid-β degrading enzymes neprilysin and insulin-degrading enzyme and improved cognition after 7 months of treatment [89]. Moreover, chronic treatment with the same combination led to increased levels of the androgen receptor in the brain, which is known to be beneficial for Alzheimer’s disease, an disorder that is often characterized by a pathological reduction of testosterone and estrogen. The combination of the two androgen and estrogen receptor modulating agents modified spatial memory and decreased anxiety-like behavior in 3xTg-AD mice. In a rat model of Parkinson’s disease, AC-186 23 prevented motor, cognitive and sensorimotor gating deficits when it was administered 1 day prior to induction of substantia nigra lesions. In the same animal model, it exerted a gender-specific effect, protecting the viability and functionality of dopamine neurons in the substantia nigra only in male rats: this selective neuroprotection is under hormonal influence but the exact mechanism needs further investigation [90].
Fig. 3.
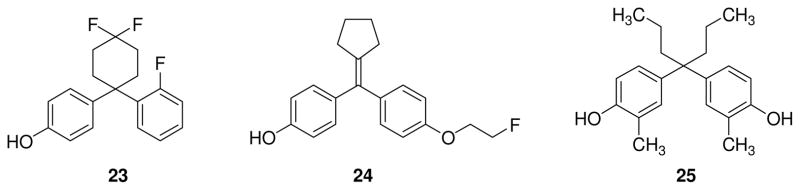
Cyclohexylphenol 23, cyclopentylene 24, and diphenylmethane derivative 25.
Fluorinated diaryl-substituted cyclic alkylidene derivatives such as compound 24 (Fig. 3) were developed as candidate ligands for positron emission tomography (PET) imaging of ERs [91]. Cyclopentylene 24 showed a good affinity for ERβ, similar to that of estradiol, and also a good ER subtype selectivity (RBA = 88.6 %, β/α = 6.4) thanks to the lower affinity to ERα (RBA = 13.8%). Biodistribution studies of compound 24 labeled with 18F demonstrated a selective uptake in target ER-positive tissues, such as uterus, ovaries, kidneys and spleen, when compared to non-target tissues such as blood, muscle, but it had a certain degree of accumulation in the brain, probably because it is a small lipophilic molecule that can easily pass the blood-brain barrier; in addition, key controls to demonstrate that uptake was due to ER binding were lacking. Micro-PET imaging of [18F]24 in immature female rats confirmed uptake in liver, kidneys and urinary bladder, suggestive of selective elimination by a renal route.
On the other hand, among the diphenylmethane derivatives, a new ERα-selective antagonist was recently discovered, compound 25 (Fig. 3), showing an IC50 value of 4.9 nM on ERα and of 140 nM on ERβ [92].
3.4. Benzoxazole, indole and indazole derivatives
Researchers at Wyeth identified benzoxazole ERB-041(26, Fig. 4) as one of the most potent and selective ERβ agonists, with a 200-fold selectivity for ERβ and a low IC50 value (5.4 nM) [93]. Due to its excellent binding affinity and selectivity, ERB-041 represented one of the most promising ERβ ligands for therapeutic uses, and it was tested in two inflammation animal models with excellent results that encouraged its clinical development as a drug for the treatment of inflammatory pathologies [51]. In 2010, the first results of a phase II clinical trial were published [94]. The aim of this study was to verify the efficacy and the safety of ERB-041 in patients with rheumatoid arthritis. Although this compound was found to be safe and well-tolerated, the trial failed to show efficacy after oral administration of ERB-041 for 12 weeks, despite the very promising results previously obtained in animal models [95]. It is interesting to note that the ERα selective ligand ORG37663 (undisclosed structure) also gave disappointing results in a phase IIa clinical trial in postmenopausal female rheumatoid arthritis patients [96].
Fig. 4.
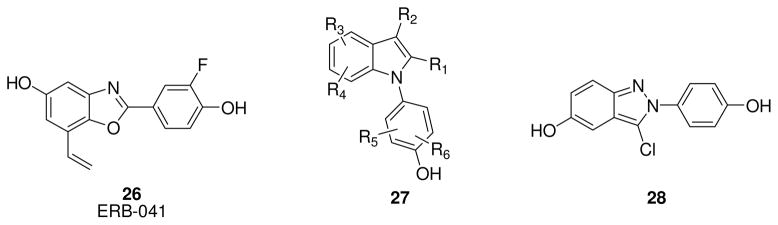
Structure of ERB-041 26, KB9520 27 and chloroindazole 28.
Another potential application of ERB-041 was for the treatment of endometriosis, which is considered an inflammatory disease characterized by ectopic growth of endometrial-like tissue throughout the peritoneum. ERB-041 showed interesting results in in vivo animal models of endometriosis, reducing lesion size in nude mice implanted with human endometrial tissue [97], and it was used in a phase II, double-blind, randomized, placebo-controlled trial in reproductive aged women to evaluate its safety and efficacy in reducing symptoms associated with endometriosis, at 75 mg and 150 mg doses [98]; no results have yet been published, however [99].
Recently, a new study indicates the efficacy of ERB-041 as potent skin cancer chemopreventive. In fact, topical application of ERB-041 substantially decreased UVB-induced skin tumor development in SKH-1 hairless mice. Remarkably, ERB-041 not only reduces cutaneous hyperplasia but also cytokine production, including IL6 and IL10. Probably, the response is not mediated by only estrogen response elements, but other transcription factors could play a significant role in this process. The observation that ERB-041 treatment decreases both WNT3a and WNT7b expression in immortalized human squamous cell carcinoma leads to the presumption that it may act by modulating the WNT signaling pathway [100]. While these results are intriguing, it is unclear whether some of the protective activities seen in this study were not due to a “sunscreen” effect of the topically applied drug.
The indole scaffold has been exhaustively explored to produce new ERs ligands. Among them worth mentioning is KB9520, a compound patented by Karo Bio [101]. It has an undisclosed structure that belongs to the indole class (whose general structure 27 is depicted in Fig. 4), and it displays a 700-fold ERβ selectivity. KB9520 has so far proved to be a promising antineoplastic agent in several in vitro and in vivo assays. The first published results of KB9520 in in vivo studies revealed that this compound showed a strong antiproliferative effect, suppressing lymphoma growth in male C57BL/6J mice subcutaneously engrafted with EG/cells, whereas the ERα-agonist PPT had no effect. Furthermore, KB9520 did not affect growth of the uterus, thus confirming its excellent ERβ-selective profile also in vivo [102]. Later, KB9520 was found to be active in vivo for the treatment of cholangiocarcinoma (CCA), a relatively rare neoplasm expressing both ERs that has poor prognosis in most cases. In fact, daily treatment with KB9520 in a rodent model of CCA resulted in reduction of tumor liver infiltration and tumor-associated collagen deposition, without any significant toxic side effects [103]. Interestingly, in both studies the effect of KB9520 is abolished when it is co-administered with fulvestrant, suggesting an ERβ-mediated antiproliferative effect of KB9520. Finally, KB9520 has recently found application in the treatment of malignant pleural mesothelioma (MMe), a rare aggressive and treatment-resistant cancer with very poor prognosis. Mice injected with cancer cells expressing endogenous ERβ and treated with KB9520 experienced a decrease in tumor size and again KB9520 treatment was not toxic. Interestingly, ERβ seems to interfere with the mitochondrial respiratory chain complex and mitochondrial ATP production in tumor cells [104].
Finally, chloroindazole 28 (Fig. 4), an ERβ agonist with remarkably high selectivity [105], was recently found to reduce inflammation in microglia and, therefore, may be potentially useful for the therapy of neurodegenerative diseases driven by inflammatory pathways, such as Parkinson’s, Alzheimer’s, Amyotrophic Lateral Sclerosis (ALS), and Multiple Sclerosis (MS) [58].
3.5. Diarylethane derivatives
One of the most widely utilized ERβ-selective agonist is represented by a diarylethane derivative, diarylproprionitrile (DPN, 29, Fig. 1). This compound possesses a chiral center, but it has generally been studied as a racemic mixture, which demonstrates a 170-fold greater relative potency for ERβ [106–107]. Recently, the single enantiomers of DPN (S-, and R-DPN, Fig. 5) were obtained by enantioselective synthesis, so that they could be separately assayed for binding affinity and transcriptional activity. The R-enantiomer (30, Fig. 5) displayed higher affinity (ERβ-RBA = 32.6%) and selectivity (β/α ratio = 332) for ERβ than its counterpart of S-configuration (31, Fig. 5, ERβ-RBA = 9.7%; β/α ratio = 147) [108]. Therefore, R-DPN should now be considered as the preferred enantiomer for biological studies of ERβ function.
Fig. 5.
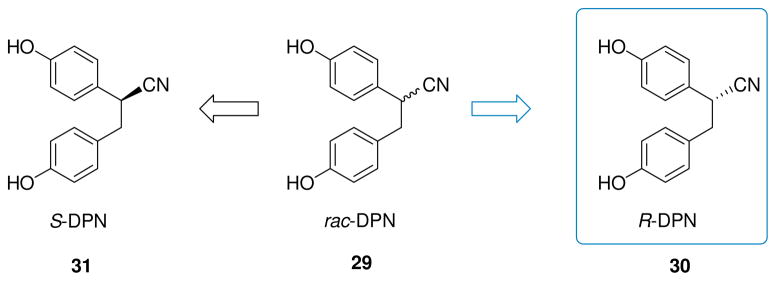
Separate enantiomers of DPN.
3.6. Tetrahydrofluorenones
Among the tetrahydrofluorenone derivatives developed by researchers at Merck as ERβ-selective ligands [109], two were tested in in vivo models for their antidepressant activity: SERM-beta1 (32, Fig. 6, chirality not specified, however it is likely to be the S-enantiomer) [109], a 129-fold ERβ-selective ligand, and SERM-beta2 (33, Fig. 6), displaying a 176-fold preference for ERβ. Due to their considerable lipophilicity, both compounds proved to be potent, selective and brain penetrant ERβ-agonists, exhibiting antidepressant-like effects in the Porsolt forced swim test, by modulating tryptophan hydroxylase 1 (TPH1) expression in raphe nuclei, and stimulating neurogenesis in the dentate gyrus. These data suggest that ERβ plays an important role in the regulation of mood disorders, since no increase in TPH1 mRNA expression is found in β-ERKO mice. Interestingly, in contrast to common antidepressant drugs such as selective serotonin reuptake inhibitors (SSRIs), which need several days of treatment to begin to exert their effects, SERM-beta1 and SERM-beta2 induce hippocampal neurogenesis after only 4 hours of exposure. Furthermore, these compounds do not have proliferative activity in MCF cells and on the immature rat uterus [110]. A better metabolic profile of SERM-beta2 over SERM-beta1 may be hypothesized, since the phenolic OH group, which generally undergoes an extensive phase II metabolic transformation (glucuronation, sulfation), is made less nucleophilic and less accessible by the Cl and F atoms occupying both ortho positions.
Fig. 6.
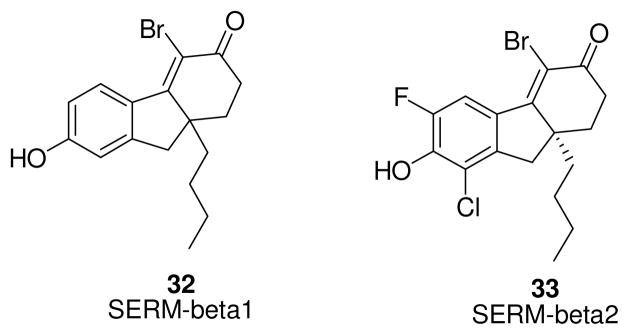
Tetrahydrofluorenone derivatives.
3.7. Isoquinolinones
Two ERβ-selective isoquinolinone derivatives, β-LGND1 (34, Fig. 7) and β-LNGD2 (35, Fig. 7), developed by GTx Inc., when tested in animal models of induced obesity, were found to be potentially useful for the treatment and prevention of obesity. Both compounds show an ERβ selectivity greater than 100-fold, without significant stimulation of any of the other receptors belonging to the nuclear receptor superfamily. In fact, β-LGND1 and β-LNGD2 induce proliferation in Ishikawa endometrial cells only at the highest concentration tested (1 μM), whereas they fail to stimulate uterine growth in vivo. The cyano-substituted derivative (β-LGND1) repressed body weight gain induced by high-fat diet in mice, prevented loss in bone mineral content, and reduced the increase in white adipose tissue weight. Furthermore, it decreased serum cholesterol and leptin levels and, at the same time, prevented accumulation of fat in the liver. Moreover, it altered the expression of genes involved in energy homeostasis and adipogenesis. The bromo-substituted analogue (β-LNGD2), when submitted to the same assays, gave similarly good results. In fact, this derivative also showed promising results for the treatment of obesity in mice, being effective not only in the prevention of obesity in high fat diet-fed animals but also in its treatment in obese animals. In vitro and in vivo mechanistic studies indicated that the anti-obesity effects were due to inhibition of PARR-γ transactivation operated by ERβ activation [111].
Fig. 7.
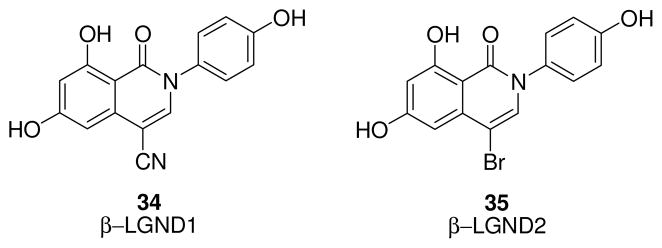
β-LGND1 and β-LNGD2 compounds.
In addition, β-LNGD2 also showed interesting cardioprotective properties, suggesting that estrogens inhibit cardiac hypertrophy and progression in animal models and humans [58]. In particular, β-LNGD2 substantially prevented AngII-induced hypertension, cardiac hypertrophy and cardiac fibrosis in ovariectomized female mice. Interestingly, this compound was active in wild type (WT) mice but inactive in β-ERKO mice, thus confirming the key role of ERβ in mediating this effect. β-LGND2 action prevents phosphorylation of serine 632 of histone deacetylase 4, which is implicated in the modulation of cardiac disease [112]. More recently, β-LNGD2 was found to promote ERβ-mediated in vitro and in vivo anti-angiogenic effects, highlighting application of this compound for the treatment of diabetic retinopathy and other similar ocular diseases arising from dysregulated angiogenesis [113].
3.8. Schiff bases: oximes and imines
The possibility of modulating ERβ/ERα selectivity in a class of oxime derivatives was conducted by analyzing the effects of variously substituted aryl substituents bound to the salicylaldoxime central scaffold [114]. The evolution of these compounds led to the production of two ERβ-selective ligands (oxime 36: ERβ-RBA = 4.21 %, β/α= 65; oxime 37: ERβ-RBA = 7.01 %, β/α= 64, Fig. 8) [115], displaying agonist properties on ERβ. A further increase in ERβ-selectivity was later obtained by a spatial inversion of the relative positions of the phenol and oxime groups of the central salicylaldoxime core of compounds 36 and 37, generating new oxime derivatives (compounds 38 and 39, Fig. 8). These two oxime derivatives display a very interesting profile of both ERβ affinity and selectivity (38: ERβ-RBA = 130 %, β/α= 29, 39: ERβ-RBA = 87.1 %, β/α= 46), and profiled as full agonist on ERβ, with a remarkable beta-selectivity in the functional assays [116]. In particular, ERβ-EC50 values were found to be 0.23 nM for 38 and 1.3 nM for 39. Furthermore, 39 shows a remarkable functional subtype selectivity, with a β/α transcription potency ratio 50-fold higher than that of estradiol.
Fig. 8.
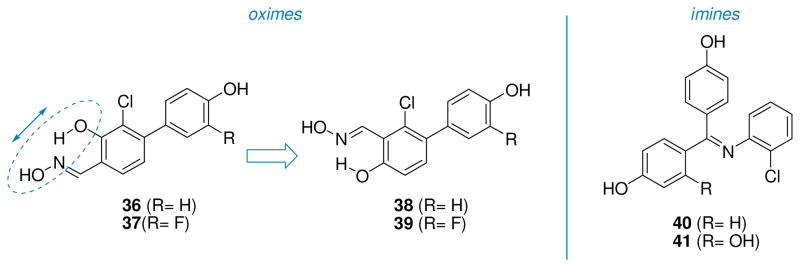
Schiff base derivatives: oximes (36–39) and imines (40–41).
A series of triaryl-imines produced compounds with similar binding affinities for ERα and ERβ; however, they generally show a very interesting ERα-agonist and ERβ-partial agonist/antagonist profile. In particular, representative imines containing a 2-chlorophenyl substituent on the nitrogen atom (40 and 41, Fig. 8) displayed ERα-EC50 values of 1 nM and 0.1 nM, and ERβ-IC50 values (in the presence of 10 nM E2) of 19 nM and 6 nM, respectively. X-ray analysis of these triaryl-imines in the ERα ligand-binding domain revealed that they cause a distortion of this pocket, enlarging the separation of helices 3 and 11, which seems to affect the stability of the C-terminal AF2 portion of ER that is involved in the recruitment of coactivators [117].
3.9. Carboranes
Among carboranes containing phenol substituents were several derivatives having good binding affinities for the ERs, and a recent study showed that o-carboranes, but not m-carboranes, are ERβ-selective ligands. In particular, n-propyl-substituted o-carborane 42 (Fig. 9) shows an interesting 7.4-fold beta selectivity in ER-binding assays, with RBA values of 11.8% on ERα and of 87% on ERβ [118]. Most importantly, this compound displayed only moderate estrogenic activity in MCF-7 cells and, therefore, may constitute a promising starting point for the development of ERβ-selective carborane-containing ligands. Furthermore, C,C′-difluoro-p-carboranyl phenol 43 (Fig. 9) was reported to have a higher beta selectivity (10-fold), though a lower binding affinity (ERβ-RBA = 50%) than 42 [119].
Fig. 9.
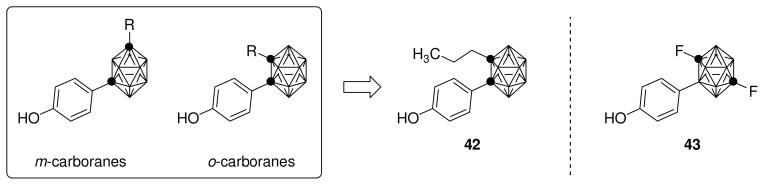
Carborane derivatives.
3.10. Phytoestrogens
Genistein (44, Fig. 10) is an isoflavone usually found as the glucoside genistin in soybeans that is cleaved to aglycone by bacterial β-glucosidases in the gut. This benzopyran-based natural compound is widely reported as an ERβ selective ligand (ERβ-RBA = 36%, ERα-RBA = 5%) [120], and it has been investigated as a therapeutic agent in different animal models. Some studies were designed to assess the possible beneficial effects of genistein on skeletal muscle structural composition in a model of the decline in skeletal muscle mass and muscle strength that affects postmenopausal women due to decreased estrogen levels. In ovariectomized mice, genistein normalized the expression of isoform I of myosin heavy chain molecule (MHC-I), characterized by slow contracting velocity and low force production, in absence of high intensity exercise, whereas the fast type isoform 2 (MHC-II) expression was increased after treatment with genistein in a group of mice exposed to high intensity exercise. These findings suggested that genistein increased the muscle composition in fast fibers in the presence of a high intensity exercise and that this effect is mainly mediated through ERβ [121].
Fig. 10.

Phytoestrogen compounds 44–53.
ERβ influence on skeletal muscle was further evaluated in an animal model of muscle injury induced by injection of notexin, a myotoxin that provokes skeletal muscle damage and impairment of neuromuscular transmission in ovariectomized rats. A high serum level of creatine kinase activity was used as an indicator of muscle injury, and daily treatment with genistein significantly contributed to its reduction. On the other hand, the same injury provoked in ERβ-knockout mice led to an evident increase of creatine kinase activity. When embryonic myosin heavy chain (MHC) was analyzed as a marker for the formation of new muscle fibers and for muscle repair, genistein-treated ovariectomized animals showed a marked increase of MHC expression, whereas it was reduced in ERβ-knockout mice. These data collectively underlined the beneficial involvement of ERβ activation for skeletal muscle growth and regeneration [54].
There are many reports in the literature about neuroprotection exerted by genistein in vitro and in vivo, and in a rat model of Alzheimer’s disease (AD), where Aβ-lesioned mice were pre-treated with genistein, the deficits in learning, memory and recognition were evidently reduced. Treatment with the ER antagonist fulvestrant abolished the beneficial effects of genistein, confirming that the tested phytoestrogen acts through an estrogenic pathway [122]. A recent study confirmed the preventive role of genistein in the establishment of AD pathology: genistein reduced the brain deposition of Aβ plaques by increasing Aβ clearance in two different transgenic mouse models of AD, APP23 mice with genetic deficiency of aromatase in which the brain contains non-detectable levels of estrogens, and in ovariectomized APP23 mice, which still contain a certain level of estrogens in their brains. The low estrogen concentration in APP23 mice with genetic deficiency of aromatase is characterized by a more severe formation of Aβ plaques and an extensive AD neuropathology than that found in ovaricetomized mice, suggesting that the deficiency of endogenous estrogens in the brain could increase the risk of Alzheimer’s disease. This estrogen-deficient mouse model was even more sensitive to the reduction of plaque formation by genistein than by endogenous estrogens, thus supporting the beneficial effect of this exogenous phytoestrogen [123]. An intriguing study of genistein on brains of ovariectomized rats showed that genistein mimicked the estradiol-induced effects by enhancing place learning, but it also impaired response learning compared to untreated animals. Hence, the estrogenic action of genistein is probably site-specific, according to the brain regions that are critical for each cognitive task [124]. Genistein proved to be effective in protection from neuronal death after ischemia, if given few minutes after injury in an animal model of global cerebral ischemia, and this neuroprotective action involves ERβ, as demonstrated by the total loss of the genistein-mediated effect when a selective ERβ antagonist was administered before ischemia induction [125].
Certain anti-cancer effects of genistein have been reported in many studies. For example, it showed antiproliferative and antiapoptotic activities via ERβ in colorectal cancer in vivo [126]. Moreover, genistein administration in mice prevented the formation of well-differentiated prostate carcinoma [127]. Some rather different effects of genistein in cancer have also been found, such as the recent report that genistein increased metastatic spread in two different xenograft models of prostate cancer [128]. These contrasting effects of genistein in prostate cancer models may be understood by considering that in vitro genistein at physiological doses has some agonist activity on certain androgen receptor mutants, which are responsible for growth and progression of advanced prostate cancer, whereas at higher doses it inhibits proliferation of the same cells [129]. The inhibitory effects of high genistein concentrations may not be ER dependent, as this compound is known to be a receptor tyrosine kinase inhibitor, though a relatively weak one [130].
A variety of many other biological effects of genistein in animals are well documented in literature, exemplified by its ERβ-mediated detrimental effects on mandibular condylar cartilage, which is made thinner after oral administration of genistein to female rats [131]. A protective role of genistein on dental pulp has been observed in vivo; so, it might prove useful as a capping agent to decrease the inflammation at the site of pulp exposure [132]. In an animal model, genistein had effects on sexual behavior, which was impaired when male rats were exposed to genistein in the neonatal period [133].
Genistein proved to be effective in preventing and treating diabetes, by maintaining the function and stimulating the proliferation of pancreatic islet β-cells in animal models. However, considering the multiple mechanisms of action of this versatile isoflavone, it is plausible that different non ER-mediated mechanisms may contribute to the anti-diabetic effect [134]. A study of the regulation of energy homeostasis in obese female ovariectomized Wistar rats fed a high-fat diet showed that genistein improved the utilization of glucose and lipids. In fact, triglyceride accumulation was reduced in liver and muscle, and hepatic glucose uptake was increased, without any significant effect in adipose tissue, probably due to the low concentration of genistein in this tissue. Thus, lipid and glucose metabolism is generally affected positively when ER is activated by genistein [135]. Many clinical studies involving genistein are presently ongoing or completed, some involving, for example, the treatment of neurodegenerative diseases, breast, prostate, bladder and colorectal cancer, metabolic syndrome, etc., to further explore the multifaceted pharmacological profile of this phytoestrogen [73].
Many examples of combinations of different phytoestrogens have been explored for the treatment of several pathologies [136]. For example, genistein when administered together with two other ERβ-selective ligands, daidzein (45, Fig. 10) and equol (46, Fig. 10), resulted in a synergistic neuroprotective effect in vivo, greater than the effects of the single agents, yet without affecting the reproductive system. Daidzein 45 is a second isoflavone abundant in soybeans, whereas equol 46 is produced from daidzein by bacteria in the gut of certain individuals (in particular the S-enantiomer is formed in humans). Many studies were conducted to determine whether the positive health effects of soybeans could be attributed to the metabolic product equol, but the results were often variable, and they did not take into account the variability in equol production by different individuals.
The combination of genistein with daidzein and equol increased mitochondrial bioenergetic function and led to the overexpression of mitochondrial antiapoptotic proteins and of Aβ-degrading enzymes in the brain of adult female ovariectomized rats. Fortunately, the combination of phytoestrogens had no relevant effect on the uterus. In vitro, the ERβ binding of isolated genistein is higher than that of the other two phytoestrogens, whereas the ERβ selectivity of the equal molar mixture of genistein/daidzein/equol is increased (β/α 82.6) when compared to genistein alone (β/α 60.0), suggesting that the mechanism by which they exert their positive combination effects against neurodegeneration is more complex than a simple sum of individual effects, perhaps revealing some complex competitive and synergistic interactions among them [137].
This phytoestrogenic combination, referred as phyto-β-SERM formulation, was extensively studied and tested in vivo. For example, in 2011 the research group of Zhao et al. reported that the phyto-β-SERM formulation alleviated the typical menopause symptoms in an ovariectomized mouse model of human menopause. The formulation prevented the rise in skin temperature, hair loss and deficit in spatial memory function, and reversed the decline in neurological functionality caused by hormone loss. Notably, by comparison, a simple diet enriched with soy extract had no effect on these ovariectomy-induced changes [138]. The therapeutic potential of this formulation was confirmed in a transgenic mouse model of AD: when phyto-β-SERM administration was started before the appearance of pathology, the formulation alleviated the typical neurological symptoms associated with the disease and attenuated the formation of Aβ plaques [139]. All these studies on phyto-β-SERM formulation included racemic equol, but recent findings have shown that no significant differences if only the enantiomer S-equol is used in its place [140].
Coumestrol 47 (Fig. 10) is a phytoestrogen derived from sprouting plants like alfalfa, clover and beans, and it belongs to the coumestane family. It is a potent ERβ and ERα ligand, with binding affinities comparable to those of 17β-estradiol [120]. Coumestrol displayed a neuroprotective effect when administered before and after global cerebral ischemia in a mouse model; however, this effect was only partially reversed by an ER-antagonist, suggesting that other non ER-mediated mechanisms may be influencing the neuroprotection [141–142].
ERβ-selective flavonone liquiritigenin 48 (Fig. 10), isolated from the root of Glycyrrhiza uralensis, is an active component of MF101 (Menerba™), a plant extract of 22 herbs traditionally used in Chinese medicine to treat vasomotor symptoms (hot flushes) associated with menopause. Despite the mixed composition, Cvoro et al. demonstrated that MF101 is ERβ-selective, because it specifically activated an ERβ estrogen response element leading to specific gene activation, although no binding preference for this receptor subtype was observed with this extract [143]. MF101 at the dose of 10 g/day for 12 weeks induced a significant reduction of frequency of hot flushes in postmenopausal women and appeared to be safe and well tolerated, and without any effect on the uterus level in a phase 2 clinical trial [42]. Presently, MF101 is under evaluation in a phase 3 clinical trial in collaboration with Bionovo Inc. [144], which previously patented this herbal extract [145]. Moreover, MF101 has been clinically evaluated for improving sleep quality in postmenopausal women [146]. Liquiritigenin also has a tumor-suppressor function, demonstrated by the reduction of the tumor size in a xenograft model of glioma, with a concomitant increase of nuclear ERβ expression [147].
Caffeic acid phenylethyl ester 49 (Fig. 10), a component of beehive propolis, is a natural compound known for its several pharmacological and biochemical properties [148]. Estrogen receptor binding affinity assays of this ester derivative of caffeic acid revealed an IC50 of 0.12 μM on ERα and of 0.049 μM on ERβ, thus showing good selectivity for ERβ. Moreover, it decreased ERα expression in breast cancer cell lines without promoting cell proliferation in MCF-7 cells, and in vivo it caused no increase in uterine weight relative to control [149].
An estrogen-like effect was observed with medicarpin (50, Fig. 10), a member of the pterocarpan subgroup of isoflavones, which are infection-induced phytoalexins usually present in many dietary legumes. Medicarpin demonstrated positive effects in skeletal growth in vivo, by stimulating osteoblast differentiation and mineralization, an action selectively mediated by ERβ, as demonstrated by siRNA knockdown of ERβ in osteoblasts. This estrogenic action was devoid of any stimulatory effects on the uterus or stimulation breast cancer cell proliferation. Interestingly, like 17β-estradiol, medicarpin exerted an osteoprotective effect against bone loss after ovariectomy, preventing premature T cell senescence and enhancing CD28 expression, a membrane glycoprotein considered the phenotypic marker of cellular senescence [150–151].
Zearalenone 51(Fig. 10) is a mycotoxin produced by numerous Fusarium species, and it is found as a contaminant in mold-infected maize, wheat and oats. This structurally intriguing compound is a resorcylic acid lactone, consisting of a phenolic ring fused to a 14-membered macrocylic lactone, and it has binding affinities for ERα and ERβ with IC50 values in the nanomolar range (ERα-RBA = 4.3%, ERβ-RBA = 6.0%) [152]. When given to ovariectomized mice, zearalenone increased uterine weight. Orally administered zearalenone is rapidly adsorbed and biotransformed, producing reduced metabolites that likely contribute its estrogenic action, and an interaction between zearalenone and a mixture of soybean isoflavones was reported in vivo in pre-pubertal female pigs [153].
Silymarin is an extract of the milk thistle (Silybum marianum) composed of a mixture of flavonoids and polyphenols, like silibinin, isosilibinin, silichristin and silidianin. Among them [154], silibinin 52 (Fig. 10), an ERβ-selective ligand, is the major bioactive component of this natural extract, and it is endowed of anticancer, antioxidant and antinflammatory properties. Male mice fed with a silymarin-enriched diet experienced a reduction in the progression of colorectal cancer and showed increase ERβ expression in the intestinal mucosa, suggesting the protective role that this receptor plays in this aggressive tumor. Moreover, the combination of silymarin with lignin showed a more potent, synergistic effect in suppressing intestinal tumorigenesis [155]. Interestingly, other ER-related effects of silymarin in vivo are improved endothelial dysfunction, which could prevent cardiovascular diseases in menopause [156], and antiosteoporotic activity in ovariectomized rats, but these are associated with a mild increase in uterine weight [157–158]. In a rat model of Parkinson’s disease, silymarin exerted a neuroprotective effect, which appears to be ER mediated because it was partially reversed by fulvestrant administration [159]. However, the complex composition of silymarin makes it difficult to determine which biological effects can be ascribed with certainty to silibinin or to other components of this extract.
Diarylheptanoid 53 (Fig. 10) was isolated from Curcuma comosa Roxb., a plant of the Zingiberaceae family [160], and it has been traditionally used by menopausal women in Thailand for its estrogenic activity. The activity of 53 on ERβ gene expression in HeLa cells is of 75% relative to estradiol (which is set at 100%), and is similar to that of genistein. This compound was later found to stimulate estrogen-regulated genes in MCF-7 breast cancer cells, predominantly by agonist action on ERα, as determined by several cellular studies and supported by molecular modeling [161–163]. Unfortunately, compound 53 caused marked uterine stimulation in mice, although less so than that of 17β-estradiol. This action was ERα-mediated, because it was blocked by an antiestrogen and not observed in ERα-knockout mice. ERα is highly expressed in endothelial cells, where it mediates the effects of estrogens in the vascular endothelium. Long-term treatment of ovariectomized rats with diarylheptanoid 53 increased ERα levels and endothelial nitric oxide synthase (eNOS), leading to relaxation of the rat aorta, effects of potential benefit to menopausal women [164].
3.11. ER ligands used in positron emission tomography (PET)
For use in PET studies, particularly imaging ER in breast cancers, some ERs ligands have been purposely modified by introducing radionuclides, such as fluorine-18 or bromine-76 [165–169].
Recent studies, focused on the development of ERβ-selective ligands for PET, involved the synthesis of 8βFEE2 (54, Fig. 11), derived from the ERβ selective ligands 8βVE2 (1, Fig. 1), and its evaluation in in vivo model of immature female rodents, and of Br-041 (55, Fig. 11), belonging to the class of benzoxazole derivatives with the same scaffold of ERB-041 (26, Fig. 4). 8βFEE2 maintains good selectivity for ERβ (β/α=170), but its affinity is 10-fold lower than that of 8βVE2, whereas Br-041 shows about the same beta-selectivity of ERB-041 (β/α ~500), with a 16-fold higher ERβ affinity (ERβ-RBA of Br-041 is 50.3% vs. 3.22% of ERB-041) [170]. Efforts PET agents for the in vivo imaging of ERα and ERβ are motivated by the prognostic value of the expression level of this receptor in cancer patients [165]. It is known, for example, that levels of the two ER subtypes change during the progression of breast cancer, so the independent measurement of both ERs might be useful in predicting disease outcome and response to endocrine therapies [171–172]. Unfortunately, despite efforts so far to obtain 18F-labeled compound 54 and 76Br-labeled compound 55, little evidence of ERβ-mediated uptake was observed in model systems, underlining the need for improved in vivo models to develop ERβ-selective radiopharmaceuticals for use as PET imaging agents to measure ERβ levels in breast tumors.
Fig. 11.
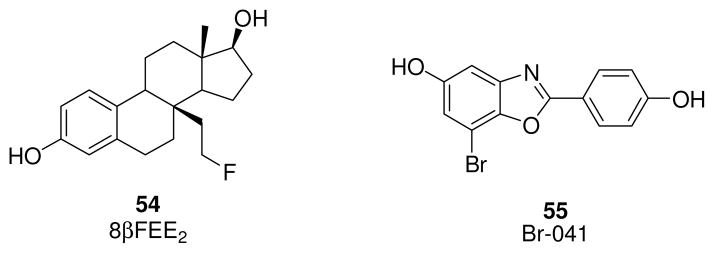
ERβ selective ligands useful for PET studies.
4. Conclusions
From a clinical perspective, ERβ-selective agonists might be therapeutically useful for some types of cancer, hot flashes and several other inflammatory diseases. Since most of the undesirable proliferative effects of estrogens involve activation of ERα, fewer efforts have been devoted to the development of pharmaceuticals that ERα-selective agonists than for ERβ-selective agonists. Nevertheless, ERα undoubtedly has a significant role in preventing osteoporosis, obesity, cardiovascular diseases and diabetes. Therefore, tissue-selective or non-nuclear-selective ERα agonists may prove to be safer, provided they stimulate ERα in tissues such as bone, fat, and vascular endothelium, but not in breast or endometrium. On the other hand, ERα antagonists have already proved to be effective agents for inhibiting cell proliferation in hormone-sensitive breast cancer. Overall, a deeper knowledge of the diverse biological roles of ERα or ERβ in different tissues, developed through future studies, will help in the decision of which receptor subtype should best be targeted for the management or cure of various individual pathological disorders.
As efforts to discover and develop new and more specific subtype-selective ligands continue to be made, whether they are agonists or antagonists, several longstanding challenges should be recognized. One relates to the fundamental role played by the phenolic hydroxyl groups that are nearly ubiquitous in ER-binding compounds, since their presence constitutes a major metabolic liability. As a class, phenolic compounds are often characterized by poor pharmacokinetic properties (low bioavailability, short half-life, etc.), because their OH functional groups generally undergo a rapid phase II sulfation and glucuronidation. Adequate consideration should thus be given to bioisosteric replacements for these groups or other strategies, such as the insertion of electron-withdrawing or sterically-hindered groups close to the phenol OH portion, to moderate the phase II metabolic conjugation process, provided these modifications do not compromise the receptor binding. Alternatively, many effective ER-targeted drugs, such as tamoxifen used in breast cancer therapy or mestranol used in oral contraceptives, are prodrugs in which the phenolic function is either missing or masked by derivatization, respectively, but then is revealed by specific hydroxylation or metabolic cleavage. These prodrugs typically have better pharmacokinetic properties than their active metabolites. General guidance from the literature suggests that the development of effective ERβ-selective agonists will be most successful by designing non-flat non-symmetric molecules, which are able to fully exploit the opportunities for weak hydrophobic attractive interactions offered by the ERβ binding pocket while, at the same time, avoiding promiscuous interactions with other steroid receptors or steroid-modifying enzymes [173].
HIGHLIGHTS.
Estrogens exert beneficial actions in bone, brain, and urogenital tract, but they can have serious adverse effects in breast and uterus.
The actions of estrogens are mediated by two receptor subtypes (ERα and ERβ).
The development of subtype-selective ligands is challenging due to the similarity of the ligand binding pockets in ERα and ERβ.
The most promising ER-ligands for clinical use are those eliciting an ERβ-selective activation.
So far, in contrast to SERMs, no ERβ-selective agonists have been successfully introduced into the clinic.
Acknowledgments
Intramural funding support from the University of Pisa (to F. M.) is gratefully acknowledged. Support from the National Institutes of Health (PHS R01CA025836 and R01DK015556, to J. A. K.) is gratefully acknowledged.
ABBREVIATIONS
- ER
estrogen receptor
- ERα
estrogen receptor subtype alpha
- ERβ
estrogen receptor subtype beta
- LBD
ligand binding domain
- RBA
relative binding affinity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM. Writing Group for the Women’s Health Initiative: Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 2.Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JÅ International Union of Pharmacology. LXIV. Estrogen Receptors. Pharmacol Rev. 2006;58:773–781. doi: 10.1124/pr.58.4.8. [DOI] [PubMed] [Google Scholar]
- 3.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JÅ. Estrogen receptors: how do they signal and what are their targets. Physiological Reviews. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 4.Jordan VC. Selective estrogen receptor modulation: a personal perspective. Cancer Res. 2001;61:5683–5687. [PubMed] [Google Scholar]
- 5.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, Öhman L, Greene GL, Gustafsson JÅ, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 6.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nature Struct Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 7.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 8.Leitman DC, Paruthiyil S, Vivar OI, Saunier EF, Herber CB, Cohen I, Tagliaferri M, Speed TP. Regulation of specific target genes and biological responses by estrogen receptor subtype agonists. Curr Opin Pharmacol. 2010;10:629–636. doi: 10.1016/j.coph.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 10.Huang B, Omoto Y, Iwase H, Yamashita H, Toyama T, Coombes RC, Filipovic A, Warner M, Gustafsson JÅ. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc Natl Acad Sci USA. 2014;111:1933–1938. doi: 10.1073/pnas.1323719111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McPherson SJ, Hussain S, Balanathan P, Hedwards SL, Niranjan B, Grant M, Chandrasir UP, Toivanen R, Wang Y, Taylor RA, Risbridger GP. Estrogen receptor-β activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFα mediated. Proc Natl Acad Sci USA. 2010;107:3123–3128. doi: 10.1073/pnas.0905524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM. ERβ Impedes Prostate Cancer EMT by Destabilizing HIF-1α and Inhibiting VEGF-Mediated Snail Nuclear Localization: Implications for Gleason Grading. Cancer Cell. 2010;17:319–332. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinton G, Thomas W, Bellini P, Manente AG, Favoni RE, Harvey BJ, Mutti L, Moro L. Estrogen receptor β exerts tumor repressive functions in human malignant pleural mesothelioma via EGFR inactivation and affects response to gefitinib. PLoS ONE. 2010;5:e14110. doi: 10.1371/journal.pone.0014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu CP, Ho JY, Huang YT, Cha TL, Sun GH, Yu DS, Chang FW, Chen SP, Hsu RJ. Estrogen Inhibits Renal Cell Carcinoma Cell Progression through Estrogen Receptor-β Activation. PLoS ONE. 2013;8:e56667. doi: 10.1371/journal.pone.0056667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudolph A, Toth C, Hoffmeister M, Roth W, Herpel E, Jansen L, Marx A, Brenner H, Chang-Claude J. Expression of oestrogen receptor β and prognosis of colorectal cancer. Br J Cancer. 2012;107:831–839. doi: 10.1038/bjc.2012.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Ström A, Gustafsson JÅ. Tumor repressive functions of estrogen receptor β in SW480 colon cancer cells. Cancer Res. 2009;69:6100–6106. doi: 10.1158/0008-5472.CAN-09-0506. [DOI] [PubMed] [Google Scholar]
- 17.Weige CC, Allred KF, Allred CD. Estradiol alters cell growth in nonmalignant colonocytes and reduces the formation of preneoplastic lesions in the colon. Cancer Res. 2009;69:9118–9124. doi: 10.1158/0008-5472.CAN-09-2348. [DOI] [PubMed] [Google Scholar]
- 18.Chakraborty S, Ganti AK, Marr A, Batra SK. Lung cancer in women: role of estrogens. Expert Rev Respir Med. 2010;4:509–518. doi: 10.1586/ers.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang H, Liao Y, Xu L, Zhang C, Liu Z, Deng Y, Jiang Z, Fu S, Chen Z, Zhou S. Estrogen and insulin-like growth factor 1 synergistically promote the development of lung adenocarcinoma in mice. Int J Cancer. 2013;133:2473–2482. doi: 10.1002/ijc.28262. [DOI] [PubMed] [Google Scholar]
- 20.Verma MK, Miki Y, Sasano H. Aromatase in human lung carcinoma. Steroids. 2011;76:759–764. doi: 10.1016/j.steroids.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Kawai H, Ishii A, Washiya K, Konno T, Kon H, Yamaya C, Ono I, Minamiya Y, Ogawa J. Estrogen Receptor α and β are Prognostic Factors in Non–Small Cell Lung Cancer. Clin Cancer Res. 2005;11:5084–5089. doi: 10.1158/1078-0432.CCR-05-0200. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz AG, Prysak GM, Murphy V, Lonardo F, Pass H, Schwartz J, Brooks S. Nuclear estrogen receptor beta in lung cancer: expression and survival differences by sex. Clin Cancer Res. 2005;11:7280–7287. doi: 10.1158/1078-0432.CCR-05-0498. [DOI] [PubMed] [Google Scholar]
- 23.Hershberger PA, Stabile LP, Kanterewicz B, Rothstein ME, Gubish CT, Land S, Shuai Y, Siegfried JM, Nichols M. Estrogen receptor beta (ERbeta) subtype-specific ligands increase transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human non-small cell lung cancer cells. J Steroid Biochem Mol Biol. 2009;116:102–109. doi: 10.1016/j.jsbmb.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walf AA, Frye CA. Estradiol reduces anxiety- and depression-like behavior of aged female mice. Physiology & Behavior. 2010;99:169–174. doi: 10.1016/j.physbeh.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osterlund MK. Underlying mechanisms mediating the antidepressant effects of estrogens. Biochim Biophys Acta. 2010;1800:1136–1144. doi: 10.1016/j.bbagen.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Pike CJ, Carroll JC, Rosario ER, Barron AM. Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol. 2009;30:239–258. doi: 10.1016/j.yfrne.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)α and ERβ ligand treatment. Proc Natl Acad Sci USA. 2007;104:14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorp AA, Sinn N, Buckley JD, Coates AM, Howe PR. Soya isoflavone supplementation enhances spatial working memory in men. Br J Nutr. 2009;102:1348–1354. doi: 10.1017/S0007114509990201. [DOI] [PubMed] [Google Scholar]
- 29.Vitale C, Mendelsohn ME, Rosano GM. Gender differences in the cardiovascular effect of sex hormones. Nature Rev Cardiol. 2009;6:532–542. doi: 10.1038/nrcardio.2009.105. [DOI] [PubMed] [Google Scholar]
- 30.Murphy E. Estrogen signaling and cardiovascular disease. Circ Res. 2011;109:687–696. doi: 10.1161/CIRCRESAHA.110.236687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors α and β. J Biol Chem. 2005;280:19704–19710. doi: 10.1074/jbc.M501244200. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JÅ, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor β. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]
- 33.Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, Boudou F, Sautier L, Vessière E, Kim SH, Lière P, Fontaine C, Krust A, Chambon P, Katzenellenbogen JA, Gourdy P, Shaul PW, Henrion D, Arnal JF, Lenfant F. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111:E283–E290. doi: 10.1073/pnas.1322057111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nilsson S, Gustafsson JÅ. Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clin Pharmacol Ther. 2011;89:44–55. doi: 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- 35.Michael JS. Selective estrogen receptor modulators: the ideal estrogen replacement. Primary care Update OB/Gyns. 2001;8:25–30. doi: 10.1016/s1068-607x(00)00066-4. [DOI] [PubMed] [Google Scholar]
- 36.Harris HA, Katzenellenbogen JA, Katzenellenbogen BS. Characterization of the Biological Roles of the Estrogen Receptors, ERα and ERβ, in Estrogen Target Tissues in Vivo through the Use of an ERα-Selective Ligand. Endocrinology. 2002;143:4172–4177. doi: 10.1210/en.2002-220403. [DOI] [PubMed] [Google Scholar]
- 37.Recker RR, Mitlak BH, Ni X, Krege JH. Long-term raloxifene for postmenopausal osteoporosis. Curr Med Res Opin. 2011;27:1755–1761. doi: 10.1185/03007995.2011.606312. [DOI] [PubMed] [Google Scholar]
- 38.Ropero AB, Alonso-Magdalena P, Quesada I, Nadal A. The role of estrogen receptors in the control of energy and glucose homeostasis. Steroids. 2008;73:874–879. doi: 10.1016/j.steroids.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 39.Ahmed MA, Hassanein KMA. Effects of estrogen on hyperglycemia and liver dysfunction in diabetic male rats. Int J Physiol Pathophysiol Pharmacol. 2012;4:156–166. [PMC free article] [PubMed] [Google Scholar]
- 40.Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34:309–338. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stovall DW, Pinkerton JV. MF-101, an estrogen receptor beta agonist for the treatment of vasomotor symptoms in peri- and postmenopausal women. Curr Opin Investig Drugs. 2009;10:365–371. [PubMed] [Google Scholar]
- 42.Grady D, Sawaya GF, Johnson KC, Koltun W, Hess R, Vittinghoff E, Kristof M, Tagliaferri M, Cohen I, Ensrud KE. MF101, a selective estrogen receptor beta modulator for the treatment of menopausal hot flushes: a phase II clinical trial. Menopause. 2009;16:458–465. doi: 10.1097/gme.0b013e31818e64dd. [DOI] [PubMed] [Google Scholar]
- 43.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multi-functional medicines 1. Receptor interactions. J Med Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 44.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multi-functional medicines 2. Clinical considerations and new agents. J Med Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 45.Minutolo F, Macchia M, Katzenellenbogen JA, Katzenellenbogen BS. Med Res Rev. 2011;31:364–442. doi: 10.1002/med.20186. [DOI] [PubMed] [Google Scholar]
- 46.Paterni I, Bertini S, Granchi C, Macchia M, Minutolo F. Estrogen receptor ligands: a patent review update. Expert Opin Ther Pat. 2013;23:1247–1271. doi: 10.1517/13543776.2013.805206. [DOI] [PubMed] [Google Scholar]
- 47.Nilsson S, Koehler KF, Gustafsson JÅ. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Disc. 2011;10:778–792. doi: 10.1038/nrd3551. [DOI] [PubMed] [Google Scholar]
- 48.Schering AG. Preparation of 8β-hydroxycarbyl-substituted estratrienes for use as selective estrogens. 2001077139. WO. 2001
- 49.Jenapharm G.m.b. H & Co. Preparation of 19-nor-17α-pregna-1,3,5(10)-trien-17β-ols containing a 21,16α lactone ring with a preference for estrogen receptor alpha. 2002026763. WO. 2002
- 50.Babiker FA, de Windt LJ, van Eickels M, Grohe C, Meyer R, Doevendans PA. Estrogenic hormone actions in the heart: regulatory network and function. Cardiovascular Research. 2002;53:709–719. doi: 10.1016/s0008-6363(01)00526-0. [DOI] [PubMed] [Google Scholar]
- 51.Harris HA. Recent lessons from in vivo studies. Mol Endocrinol. 2007;21:1–13. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 52.Sun J, Ma X, Chen YX, Rayner K, Hibbert B, McNulty M, Dhaliwal B, Simard T, Ramirez D, O’Brien E. Attenuation of atherogenesis via the anti-inflammatory effects of the selective estrogen receptor beta modulator 8β-VE2. J Cardiovasc Pharmacol. 2011;58:399–405. doi: 10.1097/FJC.0b013e318226bd16. [DOI] [PubMed] [Google Scholar]
- 53.Arias-Loza PA, Kreissl MC, Kneitz S, Kaiser FR, Israel I, Hu K, Frantz S, Bayer B, Fritzemeier KH, Korach KS, Pelzer T. The Estrogen Receptor-α Is Required and Sufficient to Maintain Physiological Glucose Uptake in the Mouse Heart. Hypertension. 2012;60:1070–1077. doi: 10.1161/HYPERTENSIONAHA.111.190389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Velders M, Schleipen B, Fritzemeier KH, Zierau O, Diel P. Selective estrogen receptor-β activation stimulates skeletal muscle growth and regeneration. FASEB J. 2012;26:1909–1920. doi: 10.1096/fj.11-194779. [DOI] [PubMed] [Google Scholar]
- 55.Yang YH, Ngo D, Jones M, Simposon E, Fritzemeier KH, Morand FE. Endogenous Estrogen Regulation of Inflammatory Arthritis and Cytokine Expression in Male Mice, Predominantly via Estrogen Receptor α. Arthritis & Reumatism. 2010;62:1017–1025. doi: 10.1002/art.27330. [DOI] [PubMed] [Google Scholar]
- 56.Kuiper GGJM, Shughrue PJ, Merchenthaler I, Gustafsson JÅ. The estrogen receptor β subtype: A novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrin. 1998;19:253–286. doi: 10.1006/frne.1998.0170. [DOI] [PubMed] [Google Scholar]
- 57.Gold SM, Voskuhl RR. Estrogen treatment in multiple sclerosis. J Neurol Sci. 2009;286:99–103. doi: 10.1016/j.jns.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP Transrepression Pathway Negatively Regulates Microglia-Mediated Inflammation. Cell. 2011;145:584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Norata GD, Cattaneo P, Poletti A, Catapano AL. The androgenderivative5α-androstane-3β,17β-diol inhibit stumor necrosis factor α and lipopolysaccharide induced inflammatory response in human endothelial cells and in mice aorta. Atherosclerosis. 2010;212:100–106. doi: 10.1016/j.atherosclerosis.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 60.Dondi D, Piccolella M, Biserni A, Della Torre S, Ramachandra B, Locatelli A, Rusmini P, Sau D, Caruso D, Maggi A, Ciana P, Poletti A. Estrogen receptor β and the progression of prostate cancer: role of 5α-androstane-3β,17β-diol. Endocrine Related Cancer. 2010;17:731–742. doi: 10.1677/ERC-10-0032. [DOI] [PubMed] [Google Scholar]
- 61.Dey P, Ström A, Gustafsson JÅ. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer. Oncogene. 2014 doi: 10.1038/onc.2013.384. published online 30 Sep 2013; [DOI] [PubMed] [Google Scholar]
- 62.Gibb W, Hagerman DD. The specificity of the 3β-hydroxysteroid dehydrogenase activity of bovine ovaries toward dehydroepiandrosterone and pregnenolone: Evidence for multiple enzymes. Steroids. 1976;28:31–41. doi: 10.1016/0039-128x(76)90123-9. [DOI] [PubMed] [Google Scholar]
- 63.Melville KM, Kelly NH, Khan SA, Schimenti JC, Ross FP, Main RP, van der Meulen MC. Female mice lacking estrogen receptor-alpha in osteoblasts have compromised bone mass and strength. J Bone Miner Res. 2014;29:370–379. doi: 10.1002/jbmr.2082. [DOI] [PubMed] [Google Scholar]
- 64.Neale JR, Richter NB, Merten KE, Grant KT, Singh S, Waite LC, Emery NK, Smith NB, Cai J, Pierce WM. Bone selective effect of an estradiol conjugate with a novel tetracycline-derived bone-targeting agent. Bioorg Med Chem Lett. 2009;19:680–683. doi: 10.1016/j.bmcl.2008.12.051. [DOI] [PubMed] [Google Scholar]
- 65.Zhao M, Liu J, Zhang X, Peng L, Li C, Peng S. 3D QSAR of novel estrogen–RGD peptide conjugates: Getting insight into structural dependence of anti-osteoporosis activity and side effect of estrogen in ERT. Bioorg Med Chem. 2009;17:3680–3689. doi: 10.1016/j.bmc.2009.03.057. [DOI] [PubMed] [Google Scholar]
- 66.Zhao M, Liu J, Zhang X, Peng S. Synthesis, evaluation and 3D QSAR analysis of novel estrogen–RGD octapeptide conjugates with oral anti-osteoporosis activity. Eur J Med Chem. 2009;44:1689–1704. doi: 10.1016/j.ejmech.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 67.Zhao Q, Liu X, Zhang L, Shen X, Qi J, Wang J, Qian N, Deng L. Bone Selective Protective Effect of a Novel Bone-seeking Estrogen on Trabecular Bone in Ovariectomized Rats. Calcif Tissue Int. 20130;93:172–183. doi: 10.1007/s00223-013-9739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim SH, Katzenellenbogen JA. Hormone-PAMAM Dendrimer Conjugates: Polymer Dynamics and Tether Structure Affect Ligand Access to Receptor. Angew Chem Int. 2006;45:7243–7248. doi: 10.1002/anie.200601923. [DOI] [PubMed] [Google Scholar]
- 69.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. Non-nuclear estrogen receptor α signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2320–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKie JA, Bhagwat SS, Brady H, Doubleday M, Gayo L, Hickman M, Jalluri RK, Khammungkhune S, Kois A, Mortensen D, Richard N, Sapienza J, Shevlin G, Stein B, Sutherland M. Lead identification of a potent benzopyranone selective estrogen receptor modulator. Bioorg Med Chem Lett. 2004;14:3407–3410. doi: 10.1016/j.bmcl.2004.04.081. [DOI] [PubMed] [Google Scholar]
- 71.Signal Pharmaceuticals, Inc. Compounds and methods for modulation of estrogen receptors. 6291456B1. US. 2000
- 72.Hui AM, Zhang W, Chen W, Xi D, Purow B, Friedman GC, Fine HA. Agents with selective estrogen receptor (ER) modulator activity induce apoptosis in vitro and in vivo in ER-negative glioma cells. Cancer Res. 2004;64:9115–9123. doi: 10.1158/0008-5472.CAN-04-2740. [DOI] [PubMed] [Google Scholar]
- 73. [Accessed 25 February 2014]; http://www.clinicaltrials.gov.
- 74.Richardson TI, Dodge JA, Durst GL, Pfeifer LA, Shah J, Wang Y, Durbin JD, Krishnan V, Norman BH. Benzopyrans as selective estrogen receptor β agonists (SERBAs). Part 3: Synthesis of cyclopentanone and cyclohexanone intermediates for C-ring modification. Bioorg Med Chem Lett. 2007;17:4824–4828. doi: 10.1016/j.bmcl.2007.06.052. [DOI] [PubMed] [Google Scholar]
- 75.Tan XJ, Dai YB, Wu WF, Kim HJ, Barros RP, Richardson TI, Yaden BC, Warner M, McKinzie DL, Krishnan V, Gustafsson JÅ. Reduction of dendritic spines and elevation of GABAergic signaling in the brains of mice treated with an estrogen receptor β ligand. Proc Natl Acad Sci USA. 2012;109:1708–1712. doi: 10.1073/pnas.1121162109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suzuki H, Barros RP, Sugiyama N, Krishnan V, Yaden BC, Kim HJ, Warner M, Gustafsson JÅ. Involvement of estrogen receptor β in maintenance of serotonergic neurons of the dorsal raphe. Mol Psychiatry. 2013;18:674–680. doi: 10.1038/mp.2012.62. [DOI] [PubMed] [Google Scholar]
- 77.Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JÅ. Targeting estrogen receptor β in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2013;110:3543–3548. doi: 10.1073/pnas.1300313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saeed A, Sharma AP, Durani N, Jain R, Durani S, Kapil RS. Structure-activity relationship of antiestrogens. Studies on 2,3-diaryl-1-benzopyrans. J Med Chem. 1990;33:3210–3216. doi: 10.1021/jm00174a018. [DOI] [PubMed] [Google Scholar]
- 79.Sharma AP, Saeed A, Durani S, Kapil RS. Structure-activity relationship of antiestrogens. Effect of the side chain and its position on the activity of 2,3-diaryl-2H-1-benzopyrans. J Med Chem. 1990;33:3216–3222. doi: 10.1021/jm00174a019. [DOI] [PubMed] [Google Scholar]
- 80.Sreenivasulu S, Singh MM, Setty BS, Kamboj VP. Effect of a pure nonsteroidal antiestrogen, CDRI-85/287, on implantation-associated histological and biochemical changes in the rat uterus. Contraception. 1993;48:597–609. doi: 10.1016/0010-7824(93)90121-m. [DOI] [PubMed] [Google Scholar]
- 81.Dwivedi A, Bansode FW, Setty BS, Dhar JD. Endometrial steroid receptors during decidualization in rhesus monkey (Macaca mulatta); their modulation by anti-oestrogen CDRI-85/287. Human Reproduction. 1999;14:1090–1095. doi: 10.1093/humrep/14.4.1090. [DOI] [PubMed] [Google Scholar]
- 82.Saxena R, Fatima I, Chandra V, Blesson CS, Kharkwal G, Hussain MK, Hajela K, Roy BG, Dwivedi A. Benzopyran derivative CDRI-85/287 induces G2-M arrest in estrogen receptor-positive breast cancer cells via modulation of estrogen receptors α- and β-mediated signaling, in parallel to EGFR signaling and suppresses the growth of tumor xenograft. Steroids. 2013;78:1071–1086. doi: 10.1016/j.steroids.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 83.Hajela K, Pandey J, Dwivedy A, Dhar JD, Sarkhel S, Maulik PR, Velumurugan D. Resolution, molecular structure and biological activities of the D- and L-enantiomers of potent anti-implantation agent, DL-2-[4-(2-piperidinoethoxy)phenyl]-3-phenyl-2H-1-benzopyran. Bioorg Med Chem. 1999;7:2083–2090. doi: 10.1016/s0968-0896(99)00131-5. [DOI] [PubMed] [Google Scholar]
- 84.Saxena R, Chandra V, Manohar M, Hajela K, Debnath U, Prabhakar YS, Saini KS, Konwar R, Kumar S, Megu K, Roy BG, Dwivedi A. Chemotherapeutic Potential of 2- [Piperidinoethoxyphenyl]-3-Phenyl-2H-Benzo(b)pyran in Estrogen Receptor- Negative Breast Cancer Cells: Action via Prevention of EGFR Activation and Combined Inhibition of PI-3-K/Akt/FOXO and MEK/Erk/AP-1 Pathways. PLoS One. 2013;8:e66246. doi: 10.1371/journal.pone.0066246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Jain N, Xu J, Kanojia RM, Du F, Jian-Zhong G, Pacia E, Lai MT, Musto A, Allan G, Reuman M, Li X, Hahn D, Cousineau M, Peng S, Ritchie D, Russell R, Lundeen S, Sui Z. Identification and structure-activity relationships of chromene-derived selective estrogen receptor modulators for treatment of postmenopausal symptoms. J Med Chem. 2009;52:7544–7569. doi: 10.1021/jm900146e. [DOI] [PubMed] [Google Scholar]
- 86.Jain N, Kanojia RM, Xu J, Jian-Zhong G, Pacia E, Lai MT, Du F, Musto A, Allan G, Hahn D, Lundeen S, Sui Z. Novel chromene-derived selective estrogen receptor modulators useful for alleviating hot flushes and vaginal dryness. J Med Chem. 2006;49:3056–3059. doi: 10.1021/jm060353u. [DOI] [PubMed] [Google Scholar]
- 87.Piu F, Cheevers C, Hyldtoft L, Gardell LR, Del Tredici AL, Andersen CB, Fairbairn LC, Lund BW, Gustafsson M, Schiffer HH, Donello JE, Olsson R, Gil DW, Brann MR. Broad modulation of neuropathic pain states by a selective estrogen receptor beta agonist. Eur J Pharmacol. 2009;590:423–429. doi: 10.1016/j.ejphar.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 88.Gardell LR, Hyldtoft L, Del Tredici AL, Andersen CB, Fairbairn LC, Lund BW, Gustafsson M, Brann MR, Olsson R, Piu F. Differential modulation of inflammatory pain by a selective estrogen receptor beta agonist. Eur J Pharmacol. 2008;592:158–159. doi: 10.1016/j.ejphar.2008.06.107. [DOI] [PubMed] [Google Scholar]
- 89.George S, Petit GH, Gouras GK, Brundin P, Olsson R. Nonsteroidal selective androgen receptor modulators and selective estrogen receptor β agonists moderate cognitive deficits and amyloid-β levels in a mouse model of Alzheimer’s disease. ACS Chem Neurosci. 2013;4:1537–1548. doi: 10.1021/cn400133s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McFarland K, Price DL, Davis CN, Ma JN, Bonhaus DW, Burstein ES, Olsson R. AC-186, a selective nonsteroidal estrogen receptor β agonist, shows gender specific neuroprotection in a Parkinson’s disease rat model. ACS Chem Neurosci. 2013;4:1249–1255. doi: 10.1021/cn400132u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu H, Yang Z, Lin JG, Luo SN, Shen YM. Synthesis and evaluation of fluoroethyl cyclofenil analogs: Models for potential estrogen receptor imaging agent. J Fluo Chem. 2012;139:46–52. [Google Scholar]
- 92.Maruyama K, Nakamura M, Tomoshige S, Sugita K, Makishima M, Hashimoto Y, Ishikawa M. Structure–activity relationships of bisphenol A analogs at estrogen receptors (ERs): Discovery of an ERα-selective antagonist. Bioorg Med Chem Lett. 2013;23:4031–4036. doi: 10.1016/j.bmcl.2013.05.067. [DOI] [PubMed] [Google Scholar]
- 93.Harris HA, Albert LM, Leathurby YL, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC. Evaluation of an estrogen receptor-β agonist in animal models of human disease. Endocrinology. 2003;144:4241–4249. doi: 10.1210/en.2003-0550. [DOI] [PubMed] [Google Scholar]
- 94.Study Evaluating ERB-041 With Methotrexate in Rheumatoid Arthritis. [Last Accessed 28 February 2014];A service of the US Nationa Institutes of Health. ClinicalTrial.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT00141830.
- 95.Roman-Blas JA, Castaneda S, Cutolo M, Herrero-Beaumont G. Efficacy and safety of a selective estrogen receptor-β agonist, ERB-041, in patients with rheumatoid arthritis. Arthiritis Care & Research. 2010;62:1588–1593. doi: 10.1002/acr.20275. [DOI] [PubMed] [Google Scholar]
- 96.van Vollenhoven RF, Houbries JGA, Butgereit F, Hout J, Boers M, Leij S, Kvien TK, Dijkmans BAC, Szczepanski L, Szombati I, Sierakowski S, Miltenburg AMM. The selective estrogen receptor α agonist Org 37663 induces estrogenic effects but lacks antirheumatic activity. Arthritis Rheum. 2010;62:351–358. doi: 10.1002/art.27196. [DOI] [PubMed] [Google Scholar]
- 97.Harris HA, Bruner-Tran KL, Zhang X, Osteen KG, Lyttle CR. A selective estrogen receptor-beta agonist causes lesion regression in an experimentally induced model of endometriosis. Hum Reprod. 2005;20:936–941. doi: 10.1093/humrep/deh711. [DOI] [PubMed] [Google Scholar]
- 98.Study Evaluating the Safety and Efficacy of ERB-041 on Reduction of Symptoms Associated With Endometriosis in Reproductive-Aged Women. [Last Accessed 28 February 2014];A service of the US Nationa Institutes of Health. ClinicalTrial.gov. Available at: http://www.clinicaltrials.gov/ct2/show/study/NCT00318500.
- 99.Guo SW, Evers JLH. Lack of Transparency of Clinical Trials on Endometriosis. Obstetrics & Gynecology. 2013;121:1281–1290. doi: 10.1097/AOG.0b013e318291f299. [DOI] [PubMed] [Google Scholar]
- 100.Chaudhary SC, Singh T, Talwelkar SS, Srivastava RK, Arumugam A, Weng Z, Elmets CA, Afaq F, Kopelovich L, Athar M. Erb-041, an Estrogen Receptor-β Agonist, Inhibits Skin Photocarcinogenesis in SKH-1 Hairless Mice by Downregulating the WNT Signaling Pathway. Cancer Prev Res. 2013;7:186–198. doi: 10.1158/1940-6207.CAPR-13-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Karo Bio AB. Novel Estrogen Receptor ligands. 2009127686. WO. 2009
- 102.Yakimchuck K, Iravani M, Hasni MS, Rhönnstad P, Nilsson S, Jondal M, Okret S. Effect of ligand-activated estrogen receptor β on lymphoma growth in vitro and in vivo. Leukemia. 2011;25:1103–1110. doi: 10.1038/leu.2011.68. [DOI] [PubMed] [Google Scholar]
- 103.Marzioni M, Torrice A, Saccomanno S, Rychlicki C, Agostinelli A, Pierantonelli I, Rhönnstad P, Trozzi L, Apelqvist T, Gentil R, Candelaresi A, Fava G, Semeraro R, Benedetti A, Gaudio E, Franchitto A, Onori P, De Minicis S, Carpino G, Kallin E, Alvaro D, Nilsson S. An oestrogen receptor β-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Digest Liver Dis. 2012;44:134–142. doi: 10.1016/j.dld.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 104.Manente AG, Valenti D, Pinton G, Jithesh PV, Daga A, Rossi L, Gray SG, O’Byrne KJ, Fennell DA, Vacca RA, Nilsson S, Mutti L, Moro L. Estrogen receptor βactivation impairs mitochondrial oxidative metabolism and affects malignant mesothelioma cell growth in vitro and in vivo. Oncogenesis. 2013;2:e72. doi: 10.1038/oncsis.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Angelis M, Stossi F, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Indazole estrogens: Highly selective ligands for the estrogen receptor β. J Med Chem. 2005;48:1132–1144. doi: 10.1021/jm049223g. [DOI] [PubMed] [Google Scholar]
- 106.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor beta potency-selective ligands: structure–activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 107.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor ralpha or estrogen receptor-beta. Endocrinology. 1999;140:800–804. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 108.Carroll VM, Jeyakumar M, Carlson KE, Katzenellenbogen JA. Diarylpropionitrile (DPN) Enantiomers: Synthesis and Evaluation of Estrogen Receptor β-Selective Ligands. J Med Chem. 2012;55:528–537. doi: 10.1021/jm201436k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wilkening RR, Ratcliffe RW, Tynebor EC, Wildonger KJ, Fried AK, Hammond ML, Mosley RT, Fitzgerald PM, Sharma N, McKeever BM, Nilsson S, Carlquist M, Thorsell A, Locco L, Katz R, Frisch K, Birzin ET, Wilkinson HA, Mitra S, Cai S, Hayes EC, Schaeffer JM, Rohrer SP. The discovery of tetrahydrofluorenones as a new class of estrogen receptor beta-subtype selective ligands. Bioorg Med Chem Lett. 2006;16:3489–3494. doi: 10.1016/j.bmcl.2006.03.098. [DOI] [PubMed] [Google Scholar]
- 110.Clark JA, Alves S, Gundlah C, Rocha B, Birzin ET, Cai SJ, Flick R, Hayes E, Ho K, Warrier S, Pai L, Tudkovitz J, Fleischer R, Colwell L, Li S, Wilkinson H, Schaeffer J, Wilkening R, Mattingly E, Hammons M, Roher SP. Selective estrogen receptor-beta (SERM-beta) compounds modulate raphe nuclei tryptophan hydroxylase-1 (TPH-1) mRNA expression and cause antidepressant-like effects in the forced swim test. Neuropharmacology. 2012;63:1051–1063. doi: 10.1016/j.neuropharm.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 111.Yepuru M, Eswaraka J, Kearbey JD, Barrett CM, Raghow S, Veverka KA, Miller DD, Dalton JT, Narayanan R. Estrogen Receptor-β Selective Ligands Alleviate High-Fat Diet- and Ovariectomy-Induced Obesity in Mice. J Biol Chem. 2010;285:31292–31303. doi: 10.1074/jbc.M110.147850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pedram A, Razandi M, Narayanan R, Dalton JT, McKinsey AM, Levin ER. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Moll Biol Cell. 2013;24:3805–3818. doi: 10.1091/mbc.E13-08-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Giddabasappa A, Eswaraka JR, Barrett CM, Bauler MN, Wu Z, Yepuru M, Miller DD, Dalton JT. β-LGND2, an ERβ Selective Agonist, Inhibits Pathologic Retinal Neovascularization. Invest Ophthalmol Vis Sci. 2012;53:5066–5075. doi: 10.1167/iovs.12-9627. [DOI] [PubMed] [Google Scholar]
- 114.Minutolo F, Antonello M, Bertini S, Rapposelli S, Rossello A, Sheng S, Carlson KE, Katzenellenbogen JA, Macchia M. Synthesis, binding affinity, and transcriptional activity of hydroxy- and methoxy-substituted 3,4-diarylsalicylaldoximes on estrogen receptors alpha and beta. Bioorg Med Chem. 2003;11:1247–1257. doi: 10.1016/s0968-0896(02)00640-5. [DOI] [PubMed] [Google Scholar]
- 115.Minutolo F, Bertini S, Granchi C, Marchitiello T, Prota G, Rapposelli S, Tuccinardi T, Martinelli A, Gunther JR, Carlson KE, Katzenellenbogen JA, Macchia M. Structural evolutions of salicylaldoximes as selective agonists for estrogen receptor β. J Med Chem. 2009;52:858–867. doi: 10.1021/jm801458t. [DOI] [PubMed] [Google Scholar]
- 116.Bertini S, De Cupertinis A, Granchi C, Bargagli B, Tuccinardi T, Martinelli A, Macchia M, Gunther JR, Carlson KE, Katzenellenbogen JA, Minutolo F. Selective and potent agonists for estrogen receptor beta derived from molecular refinements of salicylaldoximes. Eur J Med Chem. 2011;46:2453–2462. doi: 10.1016/j.ejmech.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liao ZQ, Dong C, Carlson KE, Srinivasan S, Nwachukwu JC, Chesnut RW, Sharma A, Nettles KW, Katzenellenbogen JA, Zhou H-B. Triaryl-Substituted Schiff Bases Are High-Affinity Subtype-Selective Ligands for the Estrogen Receptor. J Med Chem. 2014;57:3532–3545. doi: 10.1021/jm500268j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ohta K, Ogawa T, Kaise A, Oda A, Endo Y. Aliphatic Substitution of o-Carboranyl Phenols Enhances Estrogen Receptor Beta Selectivity. Chem Pharm Bull. 2014;62:386–391. doi: 10.1248/cpb.c13-00796. [DOI] [PubMed] [Google Scholar]
- 119.Ohta K, Ogawa T, Kaise A, Endo Y. Enhanced estrogen receptor beta (ERβ) selectivity of fluorinated carborane-containing ER modulators. Bioorg Med Chem Lett. 2013;23:6555–6558. doi: 10.1016/j.bmcl.2013.10.067. [DOI] [PubMed] [Google Scholar]
- 120.Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JÅ. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 121.Velders M, Solzbacher M, Schleipen B, Laudenbach U, Fritzemeier KH, Diel P. Estradiol and genistein antagonize the ovariectomy effects on skeletal muscle myosin heavy chain expression via ER-beta mediated pathways. J Steroid Biochem Mol Biol. 2010;120:53–59. doi: 10.1016/j.jsbmb.2010.03.059. [DOI] [PubMed] [Google Scholar]
- 122.Bagheri M, Joghataei MT, Mohseni S, Roghani M. Genistein ameliorates learning and memory deficits in amyloid β(1–40) rat model of Alzheimer’s disease. Neurobiol Learn Mem. 2011;95:270–276. doi: 10.1016/j.nlm.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 123.Li R, He P, Cui J, Staufenbiel M, Harada N, Shen Y. Brain endogenous estrogen levels determine responses to estrogen replacement therapy via regulation of BACE1 and NEP in female Alzheimer’s transgenic mice. Mol Neurobiol. 2013;47:857–867. doi: 10.1007/s12035-012-8377-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pisani SL, Neese SL, Doerge DR, Helferich WG, Schantz SL, Korol DL. Acute genistein treatment mimics the effects of estradiol by enhancing place learning and impairing response learning in young adult female rats. Horm Behav. 2012;62:491–499. doi: 10.1016/j.yhbeh.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Donzelli A, Braida D, Finardi A, Capurro V, Valsecchi AE, Colleoni M, Sala M. Neuroprotective effects of genistein in Mongolian gerbils: estrogen receptor-β involvement. J Pharmacol Sci. 2010;114:158–167. doi: 10.1254/jphs.10164fp. [DOI] [PubMed] [Google Scholar]
- 126.Schleipen B, Hertrampf T, Fritzemeier KH, Kluxen FM, Lorenz A, Molzberger A, Velders M, Diel P. ERβ-specific agonists and genistein inhibit proliferation and induce apoptosis in the large and small intestine. Carcinogenesis. 2011;32:1675–1683. doi: 10.1093/carcin/bgr188. [DOI] [PubMed] [Google Scholar]
- 127.Slusarz A, Jackson GA, Day JK, Shenouda NS, Bogener JL, Browning JD, Fritsche KL, MacDonald RS, Besch-Williford CL, Lubahn DB. Aggressive prostate cancer is prevented in ERαKO mice and stimulated in ERβKO TRAMP mice. Endocrinology. 2012;153:4160–4170. doi: 10.1210/en.2012-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nakamura H, Wang Y, Xue H, Romanish MT, Mager DL, Helgason CD, Wang Y. Genistein versus ICI 182, 780: an ally or enemy in metastatic progression of prostate cancer. Prostate. 2013;73:1747–1760. doi: 10.1002/pros.22712. [DOI] [PubMed] [Google Scholar]
- 129.Mahmoud AM, Zhu T, Parray A, Siddique HR, Yang W, Saleem M, Bosland MC. Differential effects of genistein on prostate cancer cells depend on mutational status of the androgen receptor. PLoS One. 2013;8:e78479. doi: 10.1371/journal.pone.0078479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim H, Peterson TG, Barnes S. Mechanisms of action of the soy isoflavone genistein: emerging role for its effects via transforming growth factor beta signaling pathways. Am J Clin Nutr. 1998;68:1418S–1425S. doi: 10.1093/ajcn/68.6.1418S. [DOI] [PubMed] [Google Scholar]
- 131.Yu SB, Xing XH, Dong GY, Weng XL, Wang MQ. Excess genistein suppresses the synthesis of extracellular matrix in female rat mandibular condylar cartilage. Acta Pharmacol Sin. 2012;33:918–923. doi: 10.1038/aps.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hayashi K, Handa K, Koike T, Saito T. The possibility of genistein as a new direct pulp capping agent. Dent Mater J. 2013;32:976–985. doi: 10.4012/dmj.2013-091. [DOI] [PubMed] [Google Scholar]
- 133.Sullivan AW, Hamilton P, Patisaul HB. Neonatal agonism of ERβ impairs male reproductive behavior and attractiveness. Horm Behav. 2011;60:185–194. doi: 10.1016/j.yhbeh.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gilbert ER, Liu D. Anti-diabetic functions of soy isoflavone genistein: mechanisms underlying its effects on pancreatic β-cell function. Food Funct. 2013;4:200–212. doi: 10.1039/c2fo30199g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weigt C, Hertrampf T, Kluxen FM, Flenker U, Hülsemann F, Fritzemeier KH, Diel P. Molecular effects of ER alpha- and beta-selective agonists on regulation of energy homeostasis in obese female Wistar rats. Mol Cell Endocrinol. 2013;377:147–158. doi: 10.1016/j.mce.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 136.Pugalendhi P, Manoharan S. Chemopreventive potential of genistein and daidzein in combination during 7,12-dimethylbenz [a]anthracene (DMBA) induced mammary carcinogenesis in Sprague-Dawley rats. Pak J Biol Sci. 2010;13:279–286. doi: 10.3923/pjbs.2010.279.286. [DOI] [PubMed] [Google Scholar]
- 137.Zhao L, Mao Z, Brinton RD. A select combination of clinically relevant phytoestrogens enhances estrogen receptor beta-binding selectivity and neuroprotective activities in vitro and in vivo. Endocrinology. 2009;150:770–783. doi: 10.1210/en.2008-0715. [DOI] [PubMed] [Google Scholar]
- 138.Zhao L, Mao Z, Schneider LS, Brinton RD. Estrogen receptor β-selective phytoestrogenic formulation prevents physical and neurological changes in a preclinical model of human menopause. Menopause. 2011;18:1131–1142. doi: 10.1097/gme.0b013e3182175b66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhao L, Mao Z, Chen S, Schneider LS, Brinton RD. Early intervention with an estrogen receptor β-selective phytoestrogenic formulation prolongs survival, improves spatial recognition memory, and slows progression of amyloid pathology in a female mouse model of Alzheimer’s disease. J Alzheimer’s Dis. 2013;37:403–419. doi: 10.3233/JAD-122341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yao J, Zhao L, Mao Z, Chen S, Wong KC, To J, Brinton RD. Potentiation of brain mitochondrial function by S-equol and R/S-equol estrogen receptor β-selective phytoSERM treatments. Brain Res. 2013;1514:128–141. doi: 10.1016/j.brainres.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Castro CC, Pagnussat AS, Orlandi L, Worm P, Moura N, Etgen AM, Alexandre Netto C. Coumestrol has neuroprotective effects before and after global cerebral ischemia in female rats. Brain Res. 2012;1474:82–90. doi: 10.1016/j.brainres.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 142.Castro CC, Pagnussat AS, Moura N, da Cunha MJ, Machado FR, Wyse AT, Netto CA. Coumestrol treatment prevents Na+, K+-ATPase inhibition and affords histological neuroprotection to male rats receiving cerebral global ischemia. Neurol Res. 2014;36:198–206. doi: 10.1179/1743132813Y.0000000286. [DOI] [PubMed] [Google Scholar]
- 143.Cvoro A, Paruthiyil S, Jones JO, Tzagarakis-Foster C, Clegg NJ, Tatomer D, Medina RT, Tagliaferri M, Schaufele F, Scanlan TS, Diamond MI, Cohen I, Leitman DC. Selective activation of estrogen receptor-beta transcriptional pathways by an herbal extract. Endocrinology. 2007;148:538–547. doi: 10.1210/en.2006-0803. [DOI] [PubMed] [Google Scholar]
- 144.http://www.mf101.com/
- 145.Bionovo Inc. Composition for treatment of menopause. 2006222721. US. 2006
- 146. [Accessed 28 March 2014]; http://www.clinicaltrials.gov.
- 147.Sareddy GR, Nair BC, Gonugunta VK, Zhang QG, Brenner A, Brann DW, Tekmal RR, Vadlamudi RK. Therapeutic significance of estrogen receptor β agonists in gliomas. Mol Cancer Ther. 2012;11:1174–1182. doi: 10.1158/1535-7163.MCT-11-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tolba MF, Azab SS, Khalifa AE, Abdel-Rahman SZ, Abdel-Naim AB. Caffeic acid phenethyl ester, a promising component of propolis with a plethora of biological activities: a review on its anti-inflammatory, neuroprotective, hepatoprotective, and cardioprotective effects. IUBMB Life. 2013;65:699–709. doi: 10.1002/iub.1189. [DOI] [PubMed] [Google Scholar]
- 149.Jung BI, Kim MS, Kim HA, Kim D, Yang J, Her S, Song YS. Caffeic acid phenethyl ester, a component of beehive propolis, is a novel selective estrogen receptor modulator. Phytother Res. 2010;24:295–300. doi: 10.1002/ptr.2966. [DOI] [PubMed] [Google Scholar]
- 150.Bhargavan B, Singh D, Gautam AK, Mishra JS, Kumar A, Goel A, Dixit M, Pandey R, Manickavasagam L, Dwivedi SD, Chakravarti B, Jain GK, Ramachandran R, Maurya R, Trivedi A, Chattopadhyay N, Sanyal S. Medicarpin, a legume phytoalexin, stimulates osteoblast differentiation and promotes peak bone mass achievement in rats: evidence for estrogen receptor β-mediated osteogenic action of medicarpin. J Nutr Biochem. 2012;23:27–38. doi: 10.1016/j.jnutbio.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 151.Tyagi AM, Srivastava K, Kureel J, Kumar A, Raghuvanshi A, Yadav D, Maurya R, Goel A, Singh D. Premature T cell senescence in Ovx mice is inhibited by repletion of estrogen and medicarpin: a possible mechanism for alleviating bone loss. Osteoporos Int. 2012;23:1151–1161. doi: 10.1007/s00198-011-1650-x. [DOI] [PubMed] [Google Scholar]
- 152.Takemura H, Shim JY, Sayama K, Tsubura A, Zhu BT, Shimoi K. Characterization of the estrogenic activities of zearalenone and zeranol in vivo and in vitro. J Steroid Biochem Mol Biol. 2007;103:170–177. doi: 10.1016/j.jsbmb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 153.Wang DF, Zhang NY, Peng YZ, Qi DS. Interaction of zearalenone and soybean isoflavone on the development of reproductive organs, reproductive hormones and estrogen receptor expression in prepubertal gilts. Anim Reprod Sci. 2010;122:317–323. doi: 10.1016/j.anireprosci.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 154.Seidlová-Wuttke D, Becker T, Christoffel V, Jarry H, Wuttke W. Silymarin is a selective estrogen receptor beta (ERbeta) agonist and has estrogenic effects in the metaphysis of the femur but no or antiestrogenic effects in the uterus of ovariectomized (ovx) rats. J Steroid Biochem Mol Biol. 2003;86:179–188. doi: 10.1016/s0960-0760(03)00270-x. [DOI] [PubMed] [Google Scholar]
- 155.Barone M, Tanzi S, Lofano K, Scavo MP, Pricci M, Demarinis L, Papagni S, Guido R, Maiorano E, Ingravallo G, Comelli MC, Francavilla A, Di Leo A. Dietary-induced ERβ upregulation counteracts intestinal neoplasia development in intact male ApcMin/+ mice. Carcinogenesis. 2010;31:269–274. doi: 10.1093/carcin/bgp275. [DOI] [PubMed] [Google Scholar]
- 156.Demirci B, Dost T, Gokalp F, Birincioglu M. Silymarin Improves Vascular Function of Aged Ovariectomized Rats. Phytother Res. 2013;28:868–872. doi: 10.1002/ptr.5067. [DOI] [PubMed] [Google Scholar]
- 157.El-Shitany NA, Hegazy S, El-Desoky K. Evidences for antiosteoporotic and selective estrogen receptor modulator activity of silymarin compared with ethinylestradiol in ovariectomized rats. Phytomedicine. 2010;17:116–125. doi: 10.1016/j.phymed.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 158.Kim JL, Kim YH, Kang MK, Gong JH, Han SJ, Kang YH. Antiosteoclastic activity of milk thistle extract after ovariectomy to suppress estrogen deficiency-induced osteoporosis. BioMed Res Int. 2013:Article ID 919374. doi: 10.1155/2013/919374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Baluchnejadmojarad T, Roghani M, Mafakheri M. Neuroprotective effect of silymarin in 6-hydroxydopamine hemi-parkinsonian rat: involvement of estrogen receptors and oxidative stress. Neurosci Lett. 2010;480:206–210. doi: 10.1016/j.neulet.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 160.Suksamrarn A, Ponglikitmongkol M, Wongkrajang K, Chindaduang A, Kittidanairak S, Jankam A, Yingyongnarongkul BE, Kittipanumat N, Chokchaisiri R, Khetkam P, Piyachaturawat P. Diarylheptanoids, new phytoestrogens from the rhizomes of Curcuma comosa: Isolation, chemical modification and estrogenic activity evaluation. Bioorg Med Chem. 2008;16:6891–6902. doi: 10.1016/j.bmc.2008.05.051. [DOI] [PubMed] [Google Scholar]
- 161.Winuthayanon W, Suksen K, Boonchird C, Chuncharunee A, Ponglikitmongkol M, Suksamrarn A, Piyachaturawat P. Estrogenic activity of diarylheptanoids from Curcuma comosa Roxb. Requires metabolic activation. J Agric Food Chem. 2009;57:840–845. doi: 10.1021/jf802702c. [DOI] [PubMed] [Google Scholar]
- 162.Winuthayanon W, Piyachaturawat P, Suksamrarn A, Ponglikitmongkol M, Arao Y, Hewitt SC, Korach KS. Diarylheptanoid phytoestrogens isolated from the medicinal plant Curcuma comosa: biologic actions in vitro and in vivo indicate estrogen receptor-dependent mechanisms. Environ Health Perspect. 2009;117:1155–1161. doi: 10.1289/ehp.0900613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Winuthayanon W, Piyachaturawat P, Suksamrarn A, Burns KA, Arao Y, Hewitt SC, Pedersen LC, Korach KS. The natural estrogenic compound diarylheptanoid (D3): in vitro mechanisms of action and in vivo uterine responses via estrogen receptor α. Environ Health Perspect. 2013;121:433–439. doi: 10.1289/ehp.1206122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Intapad S, Saengsirisuwan V, Prasannarong M, Chuncharunee A, Suvitayawat W, Chokchaisiri R, Suksamrarn A, Piyachaturawat P. Long-term effect of phytoestrogens from Curcuma comosa Roxb. on vascular relaxation in ovariectomized rats. J Agric Food Chem. 2012;60:758–764. doi: 10.1021/jf203173b. [DOI] [PubMed] [Google Scholar]
- 165.van Kruchten M, de Vries EGE, Brown M, de Vries EFJ, Glaudemans AWJM, Dierckx RAJO, Schroeder CP, Hospers GAP. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncology. 2013;14:e465–e475. doi: 10.1016/S1470-2045(13)70292-4. [DOI] [PubMed] [Google Scholar]
- 166.Yoo J, Dence CS, Sharp TL, Katzenellenbogen JA, Welch MJ. Synthesis of an estrogen receptor β-selective radioligand: 5-[18F]fluoro-(2R,3S)-2,3-bis(4-hydroxyphenyl) pentanenitrile and comparison of in vivo distribution with 16α-[18F]fluoro-17β-estradiol. J Med Chem. 2005;48:6366–6378. doi: 10.1021/jm050121f. [DOI] [PubMed] [Google Scholar]
- 167.Moon BS, Katzenellenbogen JA, Cheon GJ, Chi DY, Lee KC, An GI. Synthesis and evaluation of estrogen receptor β-selective ligands: Fluoroalkylated indazole estrogens. Bull Korean Chem Soc. 2008;29:1107–1114. [Google Scholar]
- 168.Moon BS, Carlson KE, Katzenellenbogen JA, Choi TH, Chi DY, Kim JY, Cheon GJ, Koh KY, Lee KC, An G. Synthesis and evaluation of aryl-substituted diarylproprionitriles, selective ligands for estrogen receptor b, as positron-emission tomographic imaging agents. Bioorg Med Chem. 2009;17:3479–3488. doi: 10.1016/j.bmc.2009.02.064. [DOI] [PubMed] [Google Scholar]
- 169.Zhou D, Zhou H, Jenks CC, Lewis JS, Katzenellenbogen JA, Welch MJ. Bromination from the macroscopic level to the tracer radiochemical level: 76Br radiolabeling of aromatic compounds via electrophilic substitution. Bioconj Chem. 2009;20:808–816. doi: 10.1021/bc800313c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lee JH, Peters O, Lehmann L, Dence CS, Sharp TL, Carlson KE, Zhou D, Jeyakumar M, Welch MJ, Katzenellenbogen JA. Synthesis and biological evaluation of two agents for imaging estrogen receptor β by positron emission tomography: challenges in PET imaging of a low abundance target. Nucl Med Biol. 2012;39:1105–1116. doi: 10.1016/j.nucmedbio.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Dehdashti F, Mortimer JE, Trinkaus K, Naughton MJ, Ellis M, Katzenellenbogen JA, Welch MJ, Siegel BA. PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res Treat. 2009;113:509–517. doi: 10.1007/s10549-008-9953-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fowler A, Chan SR, Sharp TL, Fettig NM, Zhou D, Dence CS, Carlson KE, Jeyakumar M, Katzenellenbogen JA, Schreiber RD, Welch MG. Small-Animal PET of Steroid Hormone Receptors Predicts Tumor Response to Endocrine Therapy Using a Preclinical Model of Breast Cancer. J Nucl Med. 2012;53:1119–1126. doi: 10.2967/jnumed.112.103465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lathe R, Kotelevtsev Y. Steroid signaling: Ligand-binding promiscuity, molecular symmetry, and the need for gating. Steroids. 2014;82:14–22. doi: 10.1016/j.steroids.2014.01.002. [DOI] [PubMed] [Google Scholar]