

Abstract
Angiotensin (Ang) II is a potent mediator of both hypertension and cardiac damage however the mechanisms by which this occur remains unclear. B-cell lymphoma/leukemia 10 (Bcl10) is a member of the CBM signalosome, which links Ang II and NF-κB signaling. We hypothesized that Bcl10 is pivotal in the pathogenesis of Ang II-induced cardiac damage. Ang II infusion in mice lacking Bcl10 resulted in reduced cardiac fibrosis, less cellular infiltration and improved arrhythmogenic electrical remodeling, despite a similar degree of hypertension or cardiac hypertrophy. Adoptive transfer of bone marrow (BM), whereby Bcl10 KO or wildtype BM was transferred to their opposite genotype recipients, revealed the dual-importance of Bcl10 within both cardiac and immune cells. Loss of Bcl10 in cardiac cells resulted in reduced expression of genes important for the adhesion and recruitment of immune cells. In vitro experiments demonstrated that adhesion of monocytes to Ang II-treated endothelial cells also required Bcl10. Additionally, Bcl10 deficiency in macrophages reduced their intrinsic migratory ability. To address the role of BM-derived fibroblasts in the formation of cardiac fibrosis, we explored if Bcl10 is also important for the infiltration of BM-derived (myo)fibroblasts into the heart. The transfer of GFP+ wildtype BM into Bcl10 KO recipient mice revealed a reduced number of non-cardiac (myo)fibroblasts compared to those wildtype recipients. Our results demonstrate the significant role of Bcl10 in multiple cell types important for the generation of Ang II-induced cardiac damage and electrical remodeling, and may provide a new avenue for therapeutic intervention.
Keywords: angiotensin II, fibrosis, cardiac arrhythmia, Bcl10, immune system
INTRODUCTION
Hypertension promotes cardiomyocyte growth, cardiac hypertrophy, and arrhythmias.1,2 In several hypertension models, monocytes/macrophages and T lymphocytes infiltrate the perivascular region of the heart and initiate perivascular and interstitial extracellular matrix formation.3 More recently, macrophage and T-cell subsets have been implicated in the pathogenesis of hypertension and cardiovascular remodeling.4,5,6 Angiotensin (Ang) II-initiated inflammation is involved in these processes,7,8,9 particularly in the heart.10 Ang II activates the nuclear factor kappa light chain enhancer of activated B cells (NF-κB), a major transcription factor regulating various aspects of inflammatory responses.11 We have shown previously that NF-κB is upregulated in Ang II-dependent target-organ damage12 and that pharmacological NF-κB inhibition,12 or endothelial-specific NF-κB inhibition reduced target-organ damage independent of blood pressure.13
Despite the large amount of knowledge that connects Ang II and NF-κB signaling, the exact molecular mechanism as to how the activated Ang II receptor signals to NF-κB remains unclear. A previously undescribed signaling pathway has recently been shown to mediate Ang II-dependent activation of NF-κB. In this signaling pathway, three major proteins are involved: caspase recruitment domain (CARD) 10 (also known as CARMA3, for CARD-MAGUK (membrane-associated guanylate kinase)), B-cell lymphoma/leukemia 10 (Bcl10), and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1). Together, these three proteins comprise the CBM signalosome.14 Originally Bcl10 was identified as a target of a chromosomal translocation in MALT lymphomas15 and was linked to normal lymphocyte function as a member of the CARMA1-containing CBM signalosome utilized by lymphocytes.16 However, Bcl10 functions as part of a CARMA3-containing CBM signalosome outside of the immune system. Of particular relevance to cardiovascular biology, Bcl10 deficient ApoE−/− mice were protected from developing Ang II-dependent atherosclerosis and aortic aneurysms.17
Until now, no studies have investigated the role of the CBM signalosome in Ang II-mediated cardiac damage. In this study, we investigated the role of Bcl10, the bridging molecule of the CBM signalosome, and further aimed to discriminate its role in immune cells and in the heart in Ang II/hypertension-induced cardiac damage, including cardiac fibrosis and electrical remodeling.
METHODS
Detailed description of methods is available in the online Data Supplement.
RESULTS
CBM signalosome expression in target organs
We first confirmed the expression of the CBM signalosome components in Ang II responsive tissues in WT mice. Similarly to published data,14 we found high expression of CARMA3, Bcl10 and MALT1 in the heart, kidney and liver, whereas CARMA1 was restricted to lymphoid organs such as the spleen (Figure S1). Infusion of Ang II in any of our study groups did not change the expression of the signalosome in the heart (Figure S2).
Lacking Bcl10 leads to reduced NF-κB activation in the heart
Intraperitoneal injection of Ang II led to increased NF-κB activity in the heart of WT mice, while this increase was reduced in the heart of the Bcl10 KO mice as measured by EMSA (Figure S3A–C).
Bcl10 deficiency is protective despite hypertension
Both WT and Bcl10 KO mice were normotensive and showed no difference in blood pressure at baseline measured by radiotelemetry. Ang II infusion led to similarly high blood pressure in both WT and Bcl10 KO animals (Figure 1A). Accordingly, both groups of mice developed a similar degree of cardiac hypertrophy defined by echocardiography (Figure 1B and Table S2), heart weight-to-body weight ratio (Figure 1C), heart weight-to-tibia length ratio (Figure S4A) and by the hypertrophy markers brain natriuretic peptide (BNP; Figure 1D) and atrial natriuretic peptide (ANP; Figure S4B). Despite the high blood pressure, Ang II-treated Bcl10 KO mice showed less CD4+ and CD8+ T cell and macrophage infiltration (Figure 1E) and developed significantly less perivascular (Figure 1F) and interstitial (Figure 1G) cardiac fibrosis compared to Ang II-infused WT counterparts. Quantification confirmed the histological results (Figure 1H and 1I). Next, we determined the expression levels of genes known to mediate cardiovascular remodeling including fibrosis. Ang II infusion led to increased cardiac expression of transforming growth factor beta (TGF-β) (Figure S5A) connective tissue growth factor (CTGF) (Figure S5B) and interleukin (IL)-6 (Figure S6A) in both WT and Bcl10 KO mice, whereby TGF-β was not but CTGF and IL-6 were significantly reduced in mice lacking Bcl10. More interestingly, short-term (30 min) Ang II treatment led to an immediate elevation in IL-6 expression, but not in Bcl10 KO mice (Figure S6B). These data indicate that Ang II-mediated IL-6 expression by Bcl10 is controlled not only at transcriptional level but at the mRNA level too. It has recently been shown that IL-6 is destabilized by a novel ribonuclease (RNase) named Regnase-1, which is partly under the control of the Bcl10.18 Nonetheless, neither short-term Ang II infusion did not lead to Regnase-1 degradation in the heart nor Ang II treatment of mouse embryonic fibroblast cells (NIH3T3) overexpressing the type 1 angiotensin II receptor (Figure S6C–D). These data suggest that Bcl10 dependent expression of IL-6 in the heart may be regulated by other RNase(s).
Figure 1.

Effect of Bcl10 deficiency on mean arterial pressure (MAP) and angiotensin (Ang) II-induced cardiac damage. (A) Telemetric MAP was elevated during Ang II in both groups. Cardiac hypertrophy assessed by (B) echocardiography as the sum of septal (IVSd) and posterior wall (LVPWd) thickness, (C) heart weight-to-body weight ratio or (D) cardiac hypertrophy marker brain natriuretic peptide (BNP) after 2 weeks of saline or Ang II treatment in the heart of wildtype (WT) and Bcl10 knockout (KO) mice. Number of infiltrating (E, left) CD4+ T cells, (middle) CD8+ T cells and (right) F4/80+ macrophages in the same animals. (F) Perivascular collagen I and (G) interstitial fibronectin and quantification (H and I) of the images of the study animals. Results are mean±SEM. *P<0.05 versus WT and Bcl10 KO, #P<0.05 versus WT+Ang II, one-way ANOVA using Tukey’s post-hoc test, n=4 in untreated groups, n=5 in Bcl10±Ang II and n=9 in WT±Ang II groups.
Arrhythmogenic electrical remodeling is improved in the absence of Bcl10
To investigate the pathophysiological relevance of reduced infiltration of immune cells and reduced fibrosis in Ang II-treated Bcl10 KO mice, we performed electrophysiological studies. Arrhythmias (Figure 2A) during programmed electrical stimulation could be induced in all Ang II-infused WT mice (5/5), whereas only in 25% of Ang II-treated Bcl10 KO mice (1/4) (Figure S7A). The amount of the inducible non-sustained ventricular arrhythmia protocols (as detailed in the online only Data Supplement) was significantly higher in Ang II-infused WT (67%) compared to Bcl10 KO (8%) mice (Figure 2B). The ventricular effective refractory period (VERP) was also assessed. Bcl10 KO mice treated with Ang II showed higher VERP compared to Ang II-treated WT controls (37.3±3.8 ms vs. 32.0±1.3 ms, Figure S7B). Connexin-43 (Cx43), the major cardiac gap junction protein in intercalated disks, was redistributed towards the lateral borders of cardiomyocytes in Ang II-treated WT mice (Figure 2C). In contrast, Ang II-treated Bcl10 KO mice showed almost unaltered Cx43 protein localization with restriction to the intercalated discs similar to WT and Bcl10 KO control mice (Figure S7C). Furthermore, Ang II-treated Bcl10 KO mice displayed significantly less severe QRS interval prolongation compared to Ang II-infused WT controls (12.1±0.2 ms vs. 13.7±0.3 ms; Figure S5D); other ECG parameters were similar.
Figure 2.

Bcl10 deficiency and angiotensin (Ang) II-induced cardiac electrical remodeling. (A) Representative ECG recordings of both Ang II-treated wildtype (WT) and Bcl10 knockout (KO) mice. (B) Ventricular arrhythmia inducibility of the same animals. (C) Representative connexin-(Cx) 43 (green) and N-cadherin (red) double immunostained heart sections of the study animals. *P<0.05 versus WT+Ang II, Mann-Whitney’s U-test, n=4 in Ang II-treated Bcl10 and n=5 in WT group.
Bcl10 in BM and heart affects inflammation and fibrosis
To understand which cell types were pivotal in the amelioration of cardiac damage in the Ang II-infused Bcl10 KO mice, we performed BM transplantation studies (as depicted on Figure 3A; verification of successful BM transplantation is shown on Figure S8). All Ang II-treated transplanted groups developed a similar level of cardiac hypertrophy defined by echocardiography (Figure 3B and Table S3), heart weight-to-body weight ratio (Figure 3C), heart weight-to-tibia length ratio (Figure S9A) and by hypertrophy markers BNP (Figure 3D) and ANP (Figure S9B) confirming the results in complete Bcl10 KO with Ang II. Surprisingly, after Ang II treatment both WT mice that received Bcl10 KO BM, or Bcl10 KO mice that received WT BM, showed less macrophage, CD4+, and CD8+ cell infiltration (Figure 3E), as well as less perivascular collagen I (Figure 3F) and reduced interstitial fibronectin deposition (Figure 3G) compared to control WT mice receiving WT BM. Quantification of collagen I staining (Figure 3H) and fibronectin (Figure 3I) was confirmatory.
Figure 3.

Bone marrow (BM) transplantation studies, angiotensin (Ang) II-induced cardiac hypertrophy, inflammation and fibrosis. (A) Wildtype (WT) and Bcl10 knockout (KO) BM were transplanted into irradiated WT or Bcl10 KO as depicted. Cardiac hypertrophy assessed by (B) echocardiography as the sum of septal (IVSd) and posterior wall (LVPWd) thickness and measured as (C) heart weight-to-body weight ratio or (D) by the expression of brain natriuretic peptide (BNP) after 2 weeks of Ang II infusion in the BM-transplanted animals. Number of infiltrating (E, left) CD4+ T cells, (middle) CD8+ T cells and (right) F4/80+ macrophages in the groups depicted. (F) Perivascular collagen I expression and (G) interstitial fibronectin deposition in the BM-transplanted and Ang II-infused animals. (H and I) Quantification of immunohistochemistry. Results are mean±SEM. *P<0.01 versus WT BM→WT+Ang II, one-way ANOVA using Tukey’s post-hoc test, n=9 in WT BM→WT, n=7 in Bcl10 KO BM→WT and n=4 in WT BM→Bcl10 KO group.
Bcl10 is required for cell adhesion and chemotaxis
To pursue the mechanisms as to how Bcl10 deficiency in the heart reduces immune cell infiltration, we next analyzed the expression vascular cell adhesion molecule-1 (VCAM-1), which is a target gene of NF-κB. Immunohistological analysis revealed that after Ang II treatment, Bcl10 KO mice transplanted with WT BM expressed significantly less vascular VCAM-1, as measured by a lower number of affected vessels as well as a lower magnitude of VCAM-1 expression in the respective endothelium (Figure 4A). These results were confirmed on the mRNA level (Figure 4B). Other target genes of NF-κB which are important for cell adhesion and for chemotaxis also had reduced expression in Bcl10 KO mice recipient of WT BM, as compared to the two other groups (Figure S10A-C). To further test the hypothesis that Bcl10 mediates Ang II-dependent cell adhesion to endothelial cells, we performed an in vitro monocyte/endothelial adhesion assay. Endothelial cells transfected with control siRNA showed a more than 20-fold increase in monocyte attachment after Ang II treatment, whereas those transfected with Bcl10 siRNA showed only a 4-fold increase (Figure 4C–E).
Figure 4.

Adhesion molecule expression and angiotensin (Ang) II-induced monocyte/endothelial adhesion. (A) Expression of vascular cell adhesion molecule-1 (VCAM-1) on the endothelium of the different bone marrow (BM)-transplanted animals after Ang II infusion. (B) Cardiac VCAM-1 mRNA in the same animals. SVEC4–10 cells were transfected with control or Bcl10 siRNA and were treated with Ang II as described in the methods. (C) Endothelial cell lysates were also analyzed for Bcl10 knockdown by Western blotting and showed nearly complete Bcl10 knockdown. WEHI-274.1 mouse monocytes were added, washed and counted as described. (D) Representative bright field micrographs of individual 20x fields. (E) Fold increase in monocyte adherence upon Ang II treatment in Bcl10 and control siRNA treated endothelial cells. The results represent the averages of 3 independent determinations performed in triplicate. *P<0.01 versus WT BM→WT+Ang II and Bcl10 KO BM→WT+Ang II, one-way ANOVA using Tukey’s post-hoc test, n=9 in WT BM→WT, n=7 in Bcl10 KO BM→WT and n=4 in WT BM→Bcl10 KO group.
Bcl10 is required for monocyte/macrophage migration
Recently, Bcl10 was shown to play a role in F-actin polymerization within cells of the immune system,19 a process required for cell motility.20 To further understand as to why WT mice recipient of Bcl10 KO BM had reduced immune cell infiltration, despite high expression of adhesion molecules and chemokines (Figure S10A-C, white bar), we performed an in vitro chemotaxis assay. Chemotaxis toward monocyte chemotactic protein-1 (MCP-1) was significantly higher in WT BM-derived macrophages (BMDM) compared to Bcl10 KO BMDM (Figure 5A–B). These results were further verified in vivo. Intraperitoneal injection of MCP-1 into WT mice significantly increased the number of peritoneal CD11b+ cells, whereas Bcl10 KO mice showed no significant increase in response to MCP-1 (Figure 5C–D), despite the number of circulating immune cells do not differ from WT animal.16
Figure 5.

In vitro and in vivo macrophage chemotaxis toward monocyte chemotactic protein-1 (MCP-1). Chemotactic response of bone marrow-derived macrophages (BMDM) from wildtype (WT) and Bcl10 knockout (KO) mice was evaluated using MCP-1. Migrated cells were fixed, stained and counted manually under the microscope (average of 5 20x random high-power fields each well, 6 wells/group, 3 independent experiment). (A) Representative images. (B) Relative amount of the migrated WT and Bcl10 KO BMDM at the indicated time points. Number of migrated WT BMDM at 4 hours of each independent experiment was arbitrarily set at 100% and relative amount was calculated for the groups. Furthermore, WT and Bcl10 KO mice were injected with MCP-1 or PBS. After 6 hours peritoneal cavity was lavaged with PBS, containing equal amounts of PE-conjugated microbeads. Mixture of cells and microbeads was harvested and cells were labeled with PE-conjugated anti-mouse CD11b antibody and counted by flow cytometry. (C) CD11b+ cells were gated (R2) and counted until 103 PE+ microbeads in R1 had been counted simultaneously. (D) Quantification of the flow cytometry data. *P<0.05 vs Bcl10 KO, unpaired t-test, #P<0.05 vs WT+PBS, n=4 in case of PBS-treated groups and n=7 in MCP-1-treated group.
Infiltration of BM-derived (myo)fibroblasts are reduced by Bcl10 deficiency
The origin of cardiac fibroblasts which promote fibrosis is still not completely understood.21 Recent data suggests that BM-derived cells could represent an important source of such cardiac fibroblasts.22 We hypothesized that the cardiac recruitment of these BM-derived fibroblasts could be altered by the lack of Bcl10. To this end, GFP+ WT BM was transplanted into irradiated WT and Bcl10 KO recipients, and the mice were infused with Ang II as previously described. Ang II infusion led to a rise in the number of fibroblast-specific protein 1 positive (FSP1+) cells in the heart of WT and Bcl10 KO mice, however Bcl10 KO heart samples had significantly less number of FSP+ cells (Figure 6A–B). In the hearts of WT mice receiving GFP+ WT BM, we detected GFP/FSP1 double positive cells (Figure 6A) indicating a BM origin of the FSP1+ cells. Importantly, semi-quantification of GFP/FSP1 double positive cells showed significantly fewer such cells in the hearts of Bcl10 KO recipients compared to WT recipients (Figure 6B). Furthermore, FSP1 co-staining with myofibroblast marker αSMA indicates that these fibroblasts are able to produce extracellular matrix (Figure S11).
Figure 6.
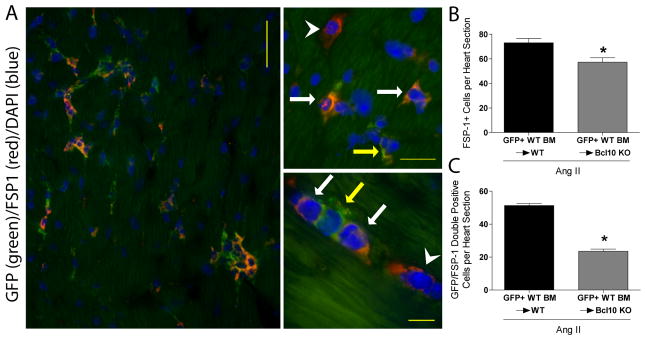
Assessment of bone marrow (BM)-derived cardiac fibroblast. BM of green fluorescent protein (GFP) expressing wildtype (WT) mice were transplanted in irradiated WT and Bcl10 knockout (KO) and infused with Ang II. (A) Representative photomicrographs of infiltrating cells (left photomicrograph). On the right side (upper and lower photomicrographs) higher magnification is shown. We detected GFP/fibroblast specific protein (FSP) 1 double-positive cells (orange cells; indicated by the white arrow), FSP1+/GFP- single-positive cells (red; indicated by the white arrowhead) as wells as FSP1-/GFP+ single-positive leukocytes (green; indicated by the yellow arrow). (B) Number of FSP1 single-positive cells and (C) infiltrating GFP/FSP1 double positive cells. *P<0.01 versus GFP+ WT BM→WT, unpaired t-test, n=4/group.
DISCUSSION
The present study highlights the importance of Bcl10, a major component of the recently described non-immune cell CBM signalosome, in Ang II-mediated cardiac damage. The current results clearly show that deletion of Bcl10 is enough to circumvent severe fibrosis, inflammation and susceptibility to ventricular arrhythmias, despite a similar elevation in blood pressure and cardiac hypertrophy. Moreover, this study shows that Bcl10 plays an important role both in bone marrow-derived cells and in the heart, in the development of Ang II-mediated cardiac damage; albeit in different ways.
Several studies have provided evidence for an important role of NF-κB in the development of cardiac end-organ damage in various hypertension models, including Ang II.12,23,24 However, the exact molecular mechanism as to how Ang II activates NF-κB is not completely understood. Recently, a complex of proteins, including CARMA3, Bcl10, and MALT1, was shown to be involved in the transmission of the signal from the AT1R to NF-κB.14 We hypothesized that Bcl10 deficiency would play an important role in Ang II-induced cardiac damage and electrical remodeling. In our study, Ang II-treated Bcl10 KO mice developed similar elevations in blood pressure and cardiac hypertrophy, compared to WT mice. However, cardiac fibrosis and inflammatory cell infiltration was reduced in the infused Bcl10 KO mice. Fibrosis and inflammation are factors known to play an important role in electrical remodeling of the heart, which increases susceptibility to arrhythmias.25 Thus, patients with hypertension-induced cardiac damage are prone to arrhythmias, which contributes to the increased cardiovascular mortality in these patients.26 Our programmed electrical stimulations revealed that Ang II-treated Bcl10 KO mice were less vulnerable for the inducibility of arrhythmias compared to their Ang-II treated WT controls. This improvement was accompanied by higher VERP, which also decreases the likelihood of ventricular arrhythmias.27 Ang II-induced cardiac hypertrophy was also associated with QRS prolongation in WT and Bcl10 KO mice. The duration of QRS complex in untreated animals of the same age under the same conditions lies between 10 und 11 ms. Bcl10 KO mice displayed significantly less severe QRS interval prolongation. QRS prolongation indicates slowing of ventricular conduction, and is often associated with hypertrophy, structural and electrical remodeling including fibrosis, as well as Cx43 delocalization, and has been considered as a predictor of mortality in congestive heart failure patients.28, 29 Consistent with the less severe QRS interval prolongation, we found that Bcl10 KO mice showed normal Cx43 localization at the intercalated disc regions after 2 weeks of Ang II-infusion, whereas in Ang II-treated WT mice, Cx43 was partially redistributed towards the lateral borders of cardiomyocytes.
To determine whether the ameliorated phenotype of Bcl10 KO mice was due to resident cardiac cells or alternatively due to the infiltration of non-cardiac cells, we performed a series of BM transplantation studies. Whilst cardiac hypertrophy was not altered in any of the transplant groups, Ang II-induced cardiac fibrosis and immune cell infiltration was reduced both in WT mice recipient of Bcl10 KO BM, and in Bcl10 KO mice recipient of WT BM. These results suggest that Bcl10 has a multi-factorial role in both BM and cardiac compartments.
Firstly, our findings with Bcl10 KO mice recipient of WT BM, suggested that the CBM signalosome is playing an important role in resident cells of the heart. Accordingly, we found evidence that Bcl10 is required for Ang II-dependent induction of the adhesion molecule VCAM-1 within endothelial cells, and for cardiac expression of the chemokines such as MCP-1 and CCL5, which play an essential role in recruiting immune cells to the heart. We also provide the first evidence that Ang II-dependent adhesion of monocytes to endothelial cells requires an intact CBM signalosome. These data are in line with the findings that Bcl10 KO mice are protected from developing Ang II-dependent atherosclerotic lesions and aortic aneurysms.17 Together these data suggest that Bcl10 specifically in endothelial cells - through coordination of adhesion molecules and chemokine expression - regulates susceptibility to fibrosis.
Secondly, our findings with WT mice, which were recipients of Bcl10 KO BM, suggested that the CBM signalosome plays an equally important role in the bone marrow, in regards to the induction of cardiac damage. We found that Bcl10 KO monocyte/macrophage migrate less toward chemotactic stimuli like MCP-1 in vitro and in vivo. One putative mechanism to explain the role of Bcl10 in the BM stems from the recent finding that Bcl10 controls TCR and FCγR-induced F-actin polymerization in monocytes/macrophages and in T cells.19 F-actin rearrangement is also a prerequisite of transendothelial migration of immune cells.20 Whilst the reduced presence of immune cells could explain the reduction in cardiac fibrosis in Bcl10 KO mice, we have also considered other mechanisms. BM-derived cells could also represent an important source for fibroblasts in pathological fibrosis. This concept is supported by previous work wherein BM-derived cells were shown to contribute to FSP1+ fibroblasts in an aortic banding model.30 In non-banded hearts, the number of FSP1+ cells was found to be very low.30 Similarly, we found very few FSP1+ cells in untreated mice, but Ang II infusion led to a significant rise in the number of FSP1+ cells in the heart, which was found to be lower in Bcl10 KO hearts. By transplanting WT and Bcl10 KO mice with GFP+ WT BM, we were able to demonstrate that a substantial number (in the case of transplanted WT mice 70±4%, and in the case of the Bcl10 KO mice 42±7%) of the cardiac FSP1+ cells were also GFP+, and therefore BM-derived. Furthermore, these cells expressed the myofibroblast marker αSMA and were observed to be localized to the site of fibrotic areas, which supports their involvement in the development of fibrosis. This finding indicates that there are substantial numbers of non-cardiac fibroblasts that may also contribute to cardiac fibrosis after Ang II infusion. Importantly, Bcl10 KO mice recipient of GFP+ WT BM showed a more than 50% reduction in GFP/FSP1 double positive fibroblasts, as compared to WT controls. We believe that in our experimental setting, the lower number of GFP+/FSP1+ cells in hearts of Bcl10 KO mice is due to the lower expression of chemokines, such as MCP-1, as these were shown to regulate the mobilization and incorporation of BM-derived fibroblasts into the tissue.22
Our data provided the first evidence that Bcl10, a protein which plays a major role in the assembly of NF-κB activator CBM signalosome, plays a substantial role in Ang II-induced cardiac fibrosis, cardiac inflammation and electrical remodeling. The lack of Bcl10 in both BM-derived cells and cardiac compartments led to a reduced infiltration of immune cells and BM-derived FSP1+ (myo)fibroblasts into the heart, which both contribute to the reduced cardiac fibrosis and enhanced electrical remodeling. These findings implicate the CBM signalosome as a target for pharmaceutical development in the management of cardiac fibrosis and arrhythmias.
PERSPECTIVES
High blood pressure is a major cause of cardiovascular morbidity and mortality. Ang II is one of the major mediators of high blood pressure-caused cardiovascular damage. Nonetheless, the molecular mechanism of action is still not completely understood. Recently, a previously undescribed protein complex has been shown to mediate the Ang II-dependent activation of NF-κB; a transcription factor that has been implicated in Ang II-mediated cardiovascular damage. Bcl10 serves as a bridging molecule in the assembly of this complex. Here we show that Bcl10 both in cardiac and in immune cells plays a substantial role in Ang II-induced cardiac fibrosis, cardiac inflammation and electrical remodeling by regulating the migration of immune cells, ultimately leading to arrhythmogenic electrical remodeling. These and previous findings strongly suggest that inhibition of this protein complex ameliorates Ang II-mediated NF-κB activation and cardiovascular damage and therefore can serve as a new target for pharmacological intervention.
Supplementary Material
Novelty and Significance.
What Is New?
This study shows that B-cell lymphoma/leukemia (Bcl) 10, a bridging molecule of a recently discovered complex for NF-κB activation, mediates angiotensin (Ang) II-induced cardiac damage and arrhythmogenic electrical remodeling.
What Is Relevant?
Bcl10 plays a pivotal in immune and other bone marrow-derived cells, as well as in cardiac cells by regulating the migration of immune cells leading to cardiac inflammation, cardiac fibrosis and arrhythmias.
Summary
Our findings, together with previous findings on Ang II-mediated atherosclerosis, show that Bcl10 is a powerful regulator of Ang II-mediated cardiovascular damage and thus is a promising new target for pharmacological intervention.
Acknowledgments
We thank Dr. Andrea Oeckinghaus (Columbia University, New York, NY) and Dr. Daniel Krappmann (Helmholtz Center, Munich, Germany) for fruitful discussions. We thank Prof. Osamu Takeuchi for providing the Regnase-1 antibody (Institute for Virus Research, Kyoto University, Kyoto, Japan). We thank Gabriele N’diaye, May-Britt Köhler, Ilona Kamer, Dr. Sabine Bartel, Juliane Anders, Hanna Schmidt, Jana Czychi, Martin Taube and Petra Berkefeld for their excellent technical assistance.
SOURCE OF FUNDING
The KFH Foundation for Preventive Medicine supported N.H. The German Center for Cardiovascular Research (DZHK; 81Z1100111) supported D.N.M. NHLBI of the National Institutes of Health (R01HL082914) supported I.A, K.O-W., P.C.L., and L.M.M-L. K.J.B. is the recipient of an Australian National Health and Medical Research Council C. J. Martin Fellowship (APP1037633).
Footnotes
CONFLICTS OF INTEREST/DISCLOSURES: None.
References
- 1.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 2.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 3.Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J, Michel JB. Inflammatory cells and myocardial fibrosis: Spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res. 1996;32:1096–1107. doi: 10.1016/s0008-6363(96)00158-7. [DOI] [PubMed] [Google Scholar]
- 4.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory t cells ameliorate angiotensin ii-induced cardiac damage. Circulation. 2009;119:2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 7.Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM. Angiotensin ii regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. 1999;104:1693–1701. doi: 10.1172/JCI7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, Coffman TM. Stimulation of lymphocyte responses by angiotensin ii promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295:F515–524. doi: 10.1152/ajprenal.00527.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luft FC, Dechend R, Muller DN. Immune mechanisms in angiotensin ii-induced target-organ damage. Ann Med. 2012;44:S49–54. doi: 10.3109/07853890.2011.653396. [DOI] [PubMed] [Google Scholar]
- 10.Phillips MI, Kagiyama S. Angiotensin ii as a pro-inflammatory mediator. Curr Opin Investig Drugs. 2002;3:569–577. [PubMed] [Google Scholar]
- 11.Hayden MS, West AP, Ghosh S. Nf-kappab and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 12.Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. Nf-kappab inhibition ameliorates angiotensin ii-induced inflammatory damage in rats. Hypertension. 2000;35:193–201. doi: 10.1161/01.hyp.35.1.193. [DOI] [PubMed] [Google Scholar]
- 13.Henke N, Schmidt-Ullrich R, Dechend R, Park JK, Qadri F, Wellner M, Obst M, Gross V, Dietz R, Luft FC, Scheidereit C, Muller DN. Vascular endothelial cell-specific nf-kappab suppression attenuates hypertension-induced renal damage. Circ Res. 2007;101:268–276. doi: 10.1161/CIRCRESAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 14.McAllister-Lucas LM, Ruland J, Siu K, Jin X, Gu S, Kim DS, Kuffa P, Kohrt D, Mak TW, Nuñez G, Lucas PC. Carma3/bcl10/malt1-dependent nf-kappab activation mediates angiotensin ii-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci. 2007;104:139–144. doi: 10.1073/pnas.0601947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willis TG, Jadayel DM, Du MQ, Peng H, Perry AR, Abdul-Rauf M, Price H, Karran L, Majekodunmi O, Wlodarska I, Pan L, Crook T, Hamoudi R, Isaacson PG, Dyer MJ. Bcl10 is involved in t(1;14)(p22;q32) of malt b cell lymphoma and mutated in multiple tumor types. Cell. 1999;96:35–45. doi: 10.1016/s0092-8674(00)80957-5. [DOI] [PubMed] [Google Scholar]
- 16.Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A, Ohashi PS, Mak TW. Bcl10 is a positive regulator of antigen receptor-induced activation of nf-kappab and neural tube closure. Cell. 2001;104:33–42. doi: 10.1016/s0092-8674(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 17.McAllister-Lucas LM, Jin X, Gu S, Siu K, McDonnell S, Ruland J, Delekta PC, Van Beek M, Lucas PC. The carma3-bcl10-malt1 signalosome promotes angiotensin ii-dependent vascular inflammation and atherogenesis. J Biol Chem. 2010;285:25880–25884. doi: 10.1074/jbc.C110.109421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, Satoh T, Mino T, Suzuki Y, Standley DM, Tsujimura T, Rakugi H, Isaka Y, Takeuchi O, Akira S. Malt1-induced cleavage of regnase-1 in cd4(+) helper t cells regulates immune activation. Cell. 2013;153:1036–1049. doi: 10.1016/j.cell.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 19.Rueda D, Gaide O, Ho L, Lewkowicz E, Niedergang F, Hailfinger S, Rebeaud F, Guzzardi M, Conne B, Thelen M, Delon J, Ferch U, Mak TW, Ruland J, Schwaller J, Thome M. Bcl10 controls tcr- and fcgammar-induced actin polymerization. J Immunol. 2007;178:4373–4384. doi: 10.4049/jimmunol.178.7.4373. [DOI] [PubMed] [Google Scholar]
- 20.Sandig M, Negrou E, Rogers KA. Changes in the distribution of lfa-1, catenins, and f-actin during transendothelial migration of monocytes in culture. J Cell Sci. 1997;110:2807–2818. doi: 10.1242/jcs.110.22.2807. [DOI] [PubMed] [Google Scholar]
- 21.Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107:1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML. Monocytic fibroblast precursors mediate fibrosis in angiotensin-ii-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dechend R, Fiebeler A, Park JK, Muller DN, Theuer J, Mervaala E, Bieringer M, Gulba D, Dietz R, Luft FC, Haller H. Amelioration of angiotensin ii-induced cardiac injury by a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor. Circulation. 2001;104:576–581. doi: 10.1161/hc3001.092039. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Arrigo AP, Currie RW. Heat shock treatment suppresses angiotensin ii-induced activation of nf-kappab pathway and heart inflammation: A role for ikk depletion by heat shock? Am J Physiol Heart Circ Physiol. 2004;287:H1104–1114. doi: 10.1152/ajpheart.00102.2004. [DOI] [PubMed] [Google Scholar]
- 25.de Jong S, van Veen TA, van Rijen HV, de Bakker JM. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011;57:630–638. doi: 10.1097/FJC.0b013e318207a35f. [DOI] [PubMed] [Google Scholar]
- 26.Diamond JA, Phillips RA. Hypertensive heart disease. Hypertens Res. 2005;28:191–202. doi: 10.1291/hypres.28.191. [DOI] [PubMed] [Google Scholar]
- 27.Maguire CT, Wakimoto H, Patel VV, Hammer PE, Gauvreau K, Berul CI. Implications of ventricular arrhythmia vulnerability during murine electrophysiology studies. Physiol Genomics. 2003;15:84–91. doi: 10.1152/physiolgenomics.00034.2003. [DOI] [PubMed] [Google Scholar]
- 28.Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. doi: 10.1016/j.cardiores.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Kashani A, Barold SS. Significance of qrs complex duration in patients with heart failure. J Am Coll Cardiol. 2005;46:2183–2192. doi: 10.1016/j.jacc.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 30.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.