

Abstract
Aims
Dissection and rupture of the ascending aorta are life-threatening conditions resulting in 80% mortality. Ascending aortic replacement in patients presenting with thoracic aortic aneurysm (TAA) is determined by metric measurement. However, a significant number of dissections occur outside of the parameters suggested by the current guidelines. We investigate the correlation among altered haemodynamic condition, oxidative stress, and vascular smooth muscle cell (VSMC) phenotype in controlling tissue homoeostasis.
Methods and results
We demonstrate using finite element analysis (FEA) based on computed tomography geometries that TAA patients have higher wall stress in the ascending aorta than non-dilated patients. We also show that altered haemodynamic conditions are associated with increased levels of reactive oxygen species (ROS), direct regulators of the VSMC phenotype in the microregional area of the ascending aorta. Using in vitro and ex vivo studies on human tissues, we show that ROS accumulation correlates with media layer degeneration and increased connective tissue growth factor (CTGF) expression, which modulate the synthetic VSMC phenotype. Results were validated by a murine model of TAA (C57BL/6J) based on Angiotensin II infusion showing that medial thickening and luminal expansion of the proximal aorta is associated with the VSMC synthetic phenotype as seen in human specimens.
Conclusions
Increased peak wall stress correlates with change in VSMC towards a synthetic phenotype mediated by ROS accumulation via CTGF. Understanding the molecular mechanisms that regulate VSMC towards a synthetic phenotype could unveil new regulatory pathways of aortic homoeostasis and impact the risk-stratification tool for patients at risk of aortic dissection and rupture.
Keywords: Thoracic aortic aneurysm, ROS, CTGF, VSMC phenotype
1. Introduction
Dissection of the ascending aorta is associated with excessive mortality.1–3 Aortic dissection and thoracic aortic aneurysm (TAA) are highly interrelated, as the only preventive treatment for aortic dissection is elective aortic replacement in patients with dilated ascending aortas.2,4–7 The current American College of Cardiology (ACC)/American Heart Association (AHA) guidelines recommend replacing the ascending aorta at the time of aortic valve replacement if the ascending aorta is >5.5 cm.1,2,8,9 However, the optimal timing of aortic surgery in TAA patients remains uncertain.1–7,10,11 In a recent study, we reported that >60% of patients with acute type A aortic dissection presented with aortic diameters < 5.5 cm.2,4–7 Therefore, the current surgical guidelines for ascending aortic aneurysm repair would fail to prevent the majority of acute aortic dissections seen in our cohort of patients. Although metric measurements are the current indications for elective surgical intervention, they are imperfect predictors of aortic dissection and rupture1,2,8,9,12,13 as they do not consider the status of cellular and extracellular components of the aortic tissues.
Biomechanical studies including our recent work have shown that aortic dissection or rupture occur when aortic wall stress exceeds aortic wall strength.14–16 We recently characterized the role that geometric indices and aortic wall stress distribution might play in the pathogenesis of thoracic aortopathies.16,17 In that prior work, fine element analysis (FEA) was applied to normal human thoracic aortas reconstructed from electrocardiogram-gated computed tomography angiography (CTA) scans. We demonstrated the presence of increased wall stress in the human thoracic aorta above the sinotubular junction and distal to the origin of the left subclavian artery (the two locations where the tears resulting in aortic dissections typically occur).16 Although factors that increase aortic wall stress—such as aortic diameter and hypertension—or decrease wall strength—have been identified as risk factors for aortic dissection and rupture, there remains neither a clear understanding of the cellular pathophysiological mechanisms of aortic dilatation nor molecular mechanisms linking aortic wall stress with altered tissue homoeostasis. Therefore, we undertook to correlate computationally predicted wall stresses with changes in the local arterial wall homoeostasis.
Recent studies show that reactive oxygen species (ROS) are modulated by altered haemodynamic conditions and directly regulate cellular and extracellular components of the aortic wall.18,19 Vascular smooth muscle cells (VSMCs) are the main source of extracellular matrix (ECM) proteins in the aortic media, and the interplay between aortic wall stress, VSMCs, and ECM proteins is critical for the structural and functional integrity of the proximal aorta.20 At homoeostasis, mature VSMCs maintain a quiescent differentiated state directed towards contractile function. In vascular pathologies contractile, VSMCs can change dramatically to a synthetic, fibroblast-like cell expressing abundant ECM proteins, and proteases.1–3,21–23 Therefore, VSMC phenotypic modulation in response to arterial stress potentiates the metabolic deregulation of ECM proteins. Here, we demonstrate that ROS, modulated by altered haemodynamic conditions, directly regulate the VSMC phenotype via connective tissue growth factor (CTGF). CTGF is a matricellular protein belonging to the transforming growth factor beta superfamily involved in ECM biosynthesis and VSMC proliferation and apoptosis.2,4–7,24 Since matricellular proteins are a class of non-structural and secreted proteins that regulate the bi-directional interaction between VSMCs and ECM proteins and act as mediators of biomechanical forces, we investigated CTGF role with respect to development of TAA in human surgically resected specimens.1,2,8,24,25 We therefore aimed to test the hypothesis that human ascending aortic aneurysms have higher wall stress that correlates with increased ROS-injury and change in the VSMC phenotype driven by CTGF.
2. Methods
2.1. Patients population and tissue collection
From January 2010 to February 2011, 106 patients were enrolled according to the approved IRB protocol #809349. This study conforms to the principles outlined in the Declaration of Helsinki. A detailed description of the subjects enrolled, the patients’ demographics, and the exclusion criteria are summarized in Supplementary material online, Table S1 and S2.
2.2. Finite element analysis
Wall stress was calculated using the finite element method, where complex geometry is parsed into small elements upon which numerical simulations are more readily carried out. A uniform pressure of 120 mmHg was applied to the luminal surface and a non-linear large deformation model was used. The aortic wall was modelled with first-order triangular shell elements. A uniform wall thickness of 1.7 mm was used based on echocardiography data.13,26 The arterial wall was assumed to be hyperelastic, homogeneous, and isotropic. A detailed description of the method is summarized in the Supplementary material online.
2.3. H2O2 and CTGF treatment of VSMC
A detailed description of the method is summarized in the Supplementary material online.
2.4. Statistical analysis
The data were analysed using the SPSS software (version 15; SPSS). Continuous variables were expressed as means ± standard deviation. Comparisons of continuous variables between groups were performed with Student's t test or non-parametric (Mann–Whitney U test) tests as appropriate, depending upon normal distribution. Statistical differences between non-dilated and TAA patients and between treated cells compared with untreated, were analysed by Student's t-test. Differences were considered statistically significant at values of P < 0.05.
3. Results
3.1. Altered wall stress correlates with accumulation of ROS and dysfunctional structure of the proximal aorta
A subset of 16 patients who underwent chest CT was retrospectively identified and analysed for image segmentation and aneurysm reconstruction followed by FEA. Figure 1A and B show that the average peak wall stress for patients with an aneurysmal ascending aorta was significantly higher than in patients with a normal ascending aorta (384 ± 34 vs. 236 ± 27 kPA, P < 0.001).
Figure 1.
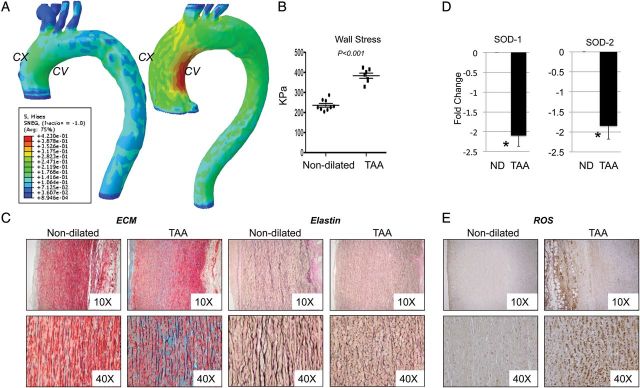
Altered wall stress correlates with accumulation of ROS and dysfunctional microstructure of the aorta. (A) Stress contour plots of a patient with a normal ascending aorta (left) compared with one with an aneurysmal ascending aorta (right). Concave and Convex sides are indicated by CV and CX, respectively. (B) Graph showing walls stress values (C) Modified Movat Pentachrome staining and Verhoeff-Van Gieson staining on ascending aorta tissues from non-dilated and aneurysmal patients. Images are representative of staining performed on n = 3 tissues/group of patients; ×10 and ×40 magnification. (D) Fold change gene expression for superoxide dismutase 1, and 2 (SOD1, 2). qPCR data were normalized against actinB (E) Nitrotyrosine staining in aortic media from patients with and without aneurysm (magnification ×10 and ×40).
In the same patient population oxidative stress was analysed by staining for nitrotyrosine. Our results show intense staining in the medial layer of the TAA aorta (Figure 1F). Furthermore, antioxidative enzymes superoxide dismutases 1 and 2 (SOD1–2) gene expression were reduced in aneurysmal tissues by 2.2 ± 0.3 and 1.9 ± 0.5 fold, respectively, when compared with non-dilated (Figure 1D) (P < 0.05). Movat Pentachrome staining was then used to analyse the distribution and integrity of collagen fibres and proteoglycans. Verhoeff-Van Gieson staining was used to detect elastin organization. Staining on three sections/group was quantified using a score from 0 to 3. The aorta of TAA subjects shows increased proteoglycan and collagen deposition (blue and yellow staining, respectively, score = 2.5 ± 0.8) compared with non-dilated aortas (score 0.3 ± 0.5), together with dramatic loss of elastin fibres (black staining) and fragmentation (score = 2.3 ± 0.6 vs. score = 0.3 ± 0.3) (Figure 1C).
3.2. Synthetic VSMC phenotype and thoracic aortic aneurysm
To determine the impact of altered wall stress and tissue microstructure on cellular components, we analysed differences in the VSMC population of the aortic media in the region at high (concave) and low (convex) wall stress by the expression of a spectrum of markers: those of the contractile phenotype included SMA, myocardin, and smoothelin B.16,17,22,27 The synthetic phenotype was characterized by the decrease of contractile markers and up-regulated expression of osteopontin (OPN), MMP-9, ELK-1, and vimentin.16,22 We show that human, surgically resected specimens from dilated and non-dilated aortas show a differential expression of SMA, vimentin and smoothelin B by western blotting and immunohistochemistry (Figure 2A–C). Smoothelin B and SMA are expressed abundantly in non-dilated aortas, both at the convex and concave side, whereas vimentin is minimally detectable. In contrast in the dilated aorta, smoothelin B and SMA levels are diminished in particular in the concave side of the ascending aorta (high wall stress), whereas vimentin is up-regulated. The expression of these markers was quantified by densitometry and the difference between their expression at the concave and convex side of the ascending aorta was plotted against CT scan measures of the ascending aorta diameter. As shown in Figure 2B, we observed a linear correlation between ascending aorta diameter and the expression of VSMC markers of phenotype. To validate the results, we performed real-time PCR using total mRNA extracted from surgically resected specimens. This analysis confirmed that in the dilated aorta; there is a decrease in contractile markers, such as SMA (−2.2 ± 0.4), and accumulation of markers of the synthetic phenotype such as OPN (2.04 ± 0.3) and MMP-9 1.7 ± 0.4. The myocardin-serum response factor (SRF)-Elk-1 complex has been shown to regulate the expression of SMC plasticity at the transcription level. SRF activates genes involved in smooth muscle differentiation and proliferation by recruiting muscle-restricted myocardin, as well as ternary complex factors of the ETS-domain family. Several growth signals have been shown to repress smooth muscle genes by triggering the displacement of myocardin from SRF by Elk-1. The opposing influences of myocardin and Elk-1 on smooth muscle gene expression are mediated by structurally related SRF-binding motifs that compete for a common binding site on SRF. In the tissues analysed ELK-1 was undetectable in control non-dilated aorta, but is significantly induced in TAA specimens (Figure 2E and F), whereas myocardin expression was not changed. ELK-1 and SRF expression were also assayed by immunofluorescence on primary human VSMCs harvested from non-dilated and aneurismal patients. Once again, ELK-1 expression was higher in TAA isolated VSMCs when compared with controls and co-localized in the nuclei of the cells with SRF (Figure 2G).
Figure 2.

Synthetic VSMC phenotype and thoracic aortic aneurysm. (A) Western blotting for smoothelin B, smooth muscle actin (SMA) and vimentin (VIM) from dilated and non-dilated patients. GAPDH as loading control. (B) Densitometry analysis over ascending aorta diameter (measured by CT scan) of the relative patient. (C) Immunohistochemistry for smoothelin B and vimentin on tissue sections. Images are representatives of staining performed on n = 6 tissues/group of patients. (*P < 0.05). Magnification ×40. (D) Fold change gene expression for SMA, OPN, MMP-9 in ascending aorta tissues (n = 4 patients/group) normalized against actinB. (E) Myocardin and Elk-1 expression tested by western blotting in the ascending aorta tissues from non-dilated and dilated patients and (F) relative densitometry. (G) Immunofluorescence staining for Elk-1 (green) and SRF (red) on isolated cells from two groups of patients. Magnification ×60.
3.3. Overexpression of connective tissue growth factor is associated with thoracic aortic aneurysm
We then investigated possible downstream effectors of ROS-mediated VSMC regulations. RT2 PCR Array for 84 matricellular and cellular adhesion protein genes was performed in a total of 16 patients followed by validation in a total of 20 patients (Figure 3A). A complete list of tested molecules is provided in Supplementary material online, Table S3. Among these genes, we found CTGF to be associated with TAA in human specimens. CTGF up-regulation was then validated by immunohistochemistry, real-time PCR and western blotting (Figure 3B–E). As shown in Figure 3E both nitrotyrosine and CTGF are up-regulated in aneurismal tissues with no significant difference in the concave and convex side of the ascending aorta. CTGF and nitrotyrosine quantification revealed that there is a linear correlation between ascending aorta diameter and the expression of these markers (Figure 3F).
Figure 3.

Overexpression of connective tissue growth factor is associated with thoracic aortic aneurysm. (A) RT2 PCR Array performed in n = 4 tissue/group for ECM and cellular adhesion molecules. Table represents genes up or down-regulated more than two fold in aneurismal tissues compared with non-dilated. (B) Immunohistochemistry showing CTGF protein levels in non-dilated and aneurismal tissues; ×40 magnification. (C) CTGF expression validation by qPCR performed on n = 10 tissues/group of patients normalized against actinB. (D) Nitrotyrosine dot blot and CTGF western blotting of whole cell extracts (E) Dot blot for nitrotyrosine and CTGF using tissue extracts from the ascending aorta [convex (CX) and concave (CV) side] and (F) relative densitometry analysis over ascending aorta diameter of each patient.
3.4. CTGF modulates VSMC synthetic phenotype in vitro
To test the effect of CTGF as a direct modulator of the VSMC phenotype and ECM biosynthesis primary human VSMCs were treated with hrCTGF in the presence or absence of a CTGF neutralizing antibody. Figure 4A shows that CTGF induces vimentin expression, which was blocked by CTGF neutralizing antibody. Increasing concentration of hrCTGF (from 20 to 80 ng/mL), doses previously reported to modulate collagen synthesis in vitro,18,19,28 was used to test a dose-dependent response of vimentin under CTGF treatment by real-time PCR. As shown in Figure 4B, CTGF induces an up-regulation of the synthetic marker vimentin, 15- and 25-fold, respectively. Notably, CTGF did not significantly alter smoothelin B expression. Also, consistent with a phenotypic transition towards a synthetic phenotype we observed a significant up-regulation of the COL1, FN, ITGα5, and ITGβ1, MMP-2, -9, -14 (P < 0.05) in response to CTGF treatment (Figure 4C and D). Then, we isolated aortic VSMCs from dilated and non-dilated ascending aorta according to previously described protocol.4,20 To test the ability of human isolated VSMCs to modulate their phenotype in response to CTGF stimulation, we treated the cells with hrCTGF and assayed smoothelin B and vimentin expression. In response to CTGF, VSMCs up-regulated the expression of synthetic markers of phenotype (Figure 4E).
Figure 4.
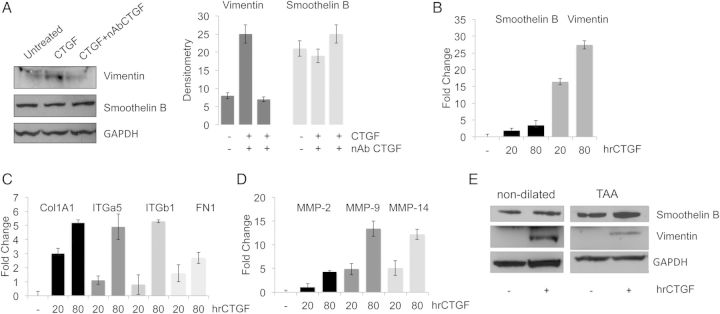
CTGF modulates VSMC synthetic phenotype in vitro. (A) vimentin and smoothelin B expression in VSMCs treated with hrCTGF in the presence or absence of CTGF neutralizing antibody and relative densitometry. (B) vimentin and smoothelin B expression assayed by qPCR (*P < 0.05). (C and D) In vitro hrCTGF treatment of VSMCs on ECM components analysed by qPCR (*P < 0.05). (E) Smoothelin B and vimentin expression in VSMC isolated from non-dilated and dilated tissues treated with 80 ng/mL of hrCTGF. GAPDH as a loading control. Data are means ± SD.
3.5. Oxidative stress mediates the expression of synthetic VSMC markers via CTGF
To test whether the accumulation of synthetic VSMCs is mediated by ROS via CTGF, human patient-derived VSMCs were treated with H2O2 in the presence or absence of 30 μM SOD mimetic MnTMPyP and then CTGF expression was quantified (Figure 5A). We observed that MnTMPyP blocks H2O2-induced CTGF expression in VSMCs. To confirm the modulation of VSMC phenotype towards a synthetic phenotype we assayed the expression of vimentin (+3.91), MMP-2 (+1.61), MMP-9 (+7,14), and CTGF (+2.96) in patient-derived VSMC treated with 100 or 200 μM H2O2 (P < 0.05) (Figure 5B and C). We then tested whether CTGF neutralizing antibody was able to modulate vimentin expression under H2O2 treatment. Figure 5D shows a reduction of vimentin when VSMCs were pre-treated with CTGF neutralizing antibody (P < 0.05).
Figure 5.

Oxidative stress mediates the expression of synthetic VSCM markers via CTGF. (A) Western blotting and densitometry showing CTGF expression in VSMC treated with H2O2 in the presence or absence of MnTMPyP. GAPDH was used as a loading control. (B and C) CTGF, vimentin, MMP-2, and MMP-9 expression in VSMC treated with 100 or 200 μM H2O2 assayed by qPCR. Fold change were normalized against actinB (*P < 0.05). (D) Western blotting and densitometry of VSMCs treated with 100 μM H2O2 assayed for vimentin expression in the presence or absence of CTGF neutralizing antibody. Data are means ± SD.
3.6. Angiotensin II infusion provokes media thickness, aortic aneurysm development, nitrotyrosine, and vimentin accumulation
Finally, to examine the role of ROS on CTGF and VSMC phenotype in vivo, we used an Angiotensin II (Ang II) infusion model in wild-type mice (C57BL/6J) under a hypercholesterolaemic diet.29,30 Seven-week-old male mice were fed with a hypercholesterolaemic diet and infused with saline (Controls), or Ang II (1000 ng/kg/min) using osmotic pumps, for 28 days, as previously described. Twenty-eight treated and 12 untreated mice were used. Aortic aneurysms were observed in 50% of the Ang II-treated group compared with the control (saline) group, none of which developed aortic pathology. Eight per cent of AngII-treated mice died from aortic dissection or rupture before the 28th day of treatment. During the course of Ang II infusion luminal expansion was observed throughout the entire ascending aorta that progressed during prolonged treatment. Animals subjected to chronic Ang II infusion show increased luminal diameter 1.12 ± 0.02 vs. 1.81 ± 0.06 mm P < 0.01(Figure 6A–C) and increase in systolic blood pressure (201 ± 13 vs. 127 ± 10 mmHg). Pathological medial changes were present concentrically and include wall thickening (6.2 vs. 14.1 mm, P < 0.01) and extensive elastin fragmentation (Figure 6C). Concomitantly with luminal expansion, we observed nitrotyrosine accumulation and up-regulation of both CTGF and vimentin further corroborating the observation in aneurysmal human tissues (Figure 6D and E). To evaluate whether CTGF and vimentin up-regulation were indeed related to ascending aorta dilatation real-time PCR was performed using RNA extracted from both the ascending and descending aorta of saline and AngII-treated mice (Figure 6D).
Figure 6.
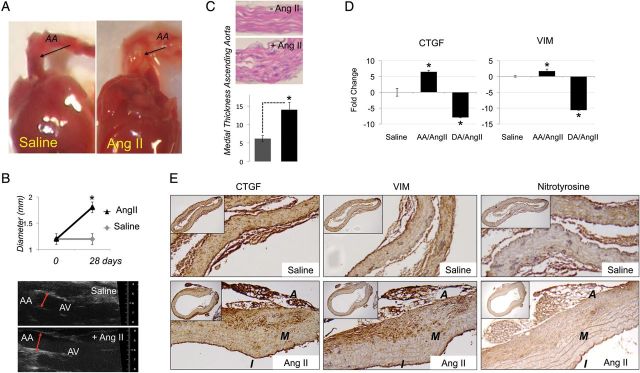
Angiotensin II infusion provokes media thickness, formation of aortic aneurysms, and vimentin accumulation. (A) Ang II-induced ascending aortic aneurysm (B) AA expansion (in mm) in mice with Ang II-induced TAA compared with saline-infused controls measured by TEE. (C) Histology of the ascending aorta showing medial thickness and measure of luminal diameter (mm). (D) CTGF and vimentin quantification by qPCR performed using RNA obtained from the ascending (AA) and descending (DA) aorta. (E and F) Immunohistochemistry showing CTGF, nitrotyrosine and vimentin expression in saline vs. ANGII-induced aneurysmal aorta. Magnification ×10–40. Data are means ± SD.*P < 0.05.
4. Discussion
The current indications for surgical intervention of a dilated ascending aorta are imperfect predictors of adverse aortic events, as metric measurements do not consider the cellular and molecular characteristic of the aortic tissues. The data reported in this paper addresses directly the problem of aortic tissue ‘quality’ in patients with dilatation of the proximal aorta linking wall stress to oxidative injury and VSMC activation. This work provides several new insights into the pathophysiology of thoracic aortic aneurysm and dissection:
The function of the thoracic aorta is the result of a dynamic, regulated and highly preserved micro-structural organization, which correlates with both haemodynamic conditions and the cellular and extracellular components of the aortic wall. Growing evidence suggests that altered wall stress distribution and predisposing genetic background participate in the overall risk of thoracic aortopathies.1,31–33 Here, we report for the first time an analysis of peak wall stress of non-dilated compared with dilated ascending aortas. Our results show that aneurysmal aortas have higher peak wall stress compared with non-aneurysmal. Aortic wall stress can predict aortic dilation,34 and by extension rupture and dissection. In addition, hydrostatic pressure-induced wall stress influences VSMC physiological responses in the aortic wall further supporting this putative relationship.
Despite its clinical importance, the cellular and molecular determinants of aortic wall structure are relatively unexplored in human pathological specimens. Our data and recent studies show that ROS are induced by altered haemodynamic conditions and directly regulate the cellular and extracellular components of the aortic wall.18,19 The higher level of nitrotyrosine in the TAA aorta is also in accordance with the marked inflammation previously observed in these types of tissues.35,36 Furthermore, both our human and murine studies revealed that dilatation of the proximal aorta is associated with induction of the VSMC synthetic phenotype markers including vimentin, MMPs and ELK-1/SRF pathways along with disorganization of the ECM structure. According to previous reports the dysfunctional organization of the media, along with increased oxidative stress, could be responsible for the increased propensity to dissect and rupture in patients with dilated aortas. Notably, chronic infusion of Ang II was previously used in ApoE−/− mice to generate both thoracic and adnominal aortic aneurysm.29,37 Growing evidence indicate that Ang II induces its pleiotropic vascular effects through NADPH-driven generation of ROS2,4–6,11,29,37,38 and it has been reported that NADPH activity shifts VSMCs towards a synthetic phenotype and increases collagen I production in prosthetic vascular graft.2,39 Our results show that AngII infusion in mice induces medial thickening and luminal expansion of the proximal aorta together with accumulation of synthetic VSMC as seen in human TAA specimens. In addition our data reveals a specific dilatation of the ascending thoracic aorta using Ang II chronic infusion on wild-type hypercholesterolaemic mice (C57BL/6J).
We reported that CTGF expression is up-regulated in human and murine thoracic aneurysmal aorta, it is induced by ROS, and directly regulates VSMC synthetic markers. In response to vascular injury VSMC become synthetic and increase their ability to produce ECM playing in this way a critical role in vascular repair. However, the negative effect of VSMC plasticity is that it can predispose to an abnormal response that ultimately contributes to the progression of vascular disease. Here, we demonstrate that CTGF induces the expression VSMC synthetic markers both directly and indirectly mediating ROS effect. As reported in Figure 1, the presence of dilatation of the ascending aorta is associated with reduced SOD 1 and 2 expressions. We, therefore, tested in vitro whether the exogenous delivery of SOD mimetics could revert the phenotypic differentiation of primary isolated H202-treated VSMC. We decided to use MnTMPyP for his stability over the use of SOD adenovirus.40–42 CTGF has previously been reported to be up-regulated in aneurismal and dissected arteries where the stimulation of collagen production was associated with the weakening of the aortic wall and the predisposition to the dissection.28 Our results show that CTGF regulates the ECM composition acting on VSMC not only by stimulating collagen production but also inducing the expression of MMP-2, MMP-9, MMP-14, FN, Integrin α5 and β1. Taken together our results suggest that CTGF could be used to follow VSMC phenotypic transition towards a synthetic phenotype in TAA patients and mice. Increasing evidences suggest that CTGF levels in plasma, serum, and urine could potentially be promising biomarkers for fibrotic disorders such as hepatitis, diabetes, and renal transplantation. It is reasonable to hypothesize that CTGF could be measured in the patient serum as a biomarker for aneurismal diseases as well. As of now our preliminary data do not provide strong evidences that serum CTGF could be associated with the presence of the aneurismal ascending aorta. However, further studies need to be performed to clarify whether or not CTGF could be used as a potential biomarker for ascending aorta dilatation.
There are several limitations associated with this study. First, FEA techniques do not include the effects of blood flow. However, the effects of wall shear stress have been shown to be several orders of magnitude lower than static stresses calculated using pressure-deformation analyses.43 Secondly, static boundary conditions were assumed, so the systolic motion of the heart was not captured in this model. Studying the effects on ascending aortic wall stress through the cardiac cycle represents an area of future research. Thirdly, material properties derived from the strain testing of excised abdominal aortas were used in this work; studies have shown that the arterial wall in the ascending aorta has twice the breaking strength of AAA specimens, but detailed elastic properties of the ascending aorta are unavailable.44,45 Finally, a uniform wall thickness was assumed in these models, even though pathological specimens show significant variation in aortic thickness. Unfortunately, no validated techniques for extraction of local wall thickness from imaging data are available, particularly in the proximal ascending aorta where systolic motion artefact confounds any attempts to accurately determine aortic thickness. Further multi-scale studies linking micro- and macro-structure of the aortic wall with the mechanical forces are needed to fully evaluate the mechanism regulating the VSMC-matrix complex and, overall the aortic tissue homoeostasis. However, the delineation of the cellular and extracellular regulators of aortic dilation, such as CTGF and its molecular pathway could help in the surveillance of patients with aortic wall pathologies and in optimizing medical therapy as well as the timing of surgical intervention.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This project was supported by the ‘Harrison Memorial Fund’ of the University of Pennsylvania School Of Medicine (G.F.), by the Society of Vascular Surgery Clinical Research Seed Grant (G.F. and B.M.J.) and by a generous gift from Mr and Mrs Edward Rubin (G.F. and J.B.G.).
References
- 1.Cozijnsen L, Braam RL, Waalewijn RA, Schepens MAAM, Loeys BL, van Oosterhout MFM, et al. What is new in dilatation of the ascending aorta?: review of current literature and practical advice for the cardiologist. Circulation. 2011;123:924–928. doi: 10.1161/CIRCULATIONAHA.110.949131. [DOI] [PubMed] [Google Scholar]
- 2.Parish LM, Gorman JH, III, Kahn S, Plappert T, St John-Sutton MG, Bavaria JE, et al. Aortic size in acute type A dissection: implications for preventive ascending aortic replacement. Eur J Cardio-Thorac Surg. 2009;35:941–946. doi: 10.1016/j.ejcts.2008.12.047. [DOI] [PubMed] [Google Scholar]
- 3.Parolari A, Tremoli E, Songia P, Pilozzi A, Di Bartolomeo R, Alamanni F, et al. Biological features of thoracic aortic diseases. Where are we now, where are we heading to: established and emerging biomarkers and molecular pathways. Eur J Cardiothorac Surg. 2013;44:9–23. doi: 10.1093/ejcts/ezs647. [DOI] [PubMed] [Google Scholar]
- 4.Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88:520–529. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elefteriades JA. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg. 2002;74:S1877–S1880. doi: 10.1016/S0003-4975(02)04147-4. [DOI] [PubMed] [Google Scholar]
- 6.Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, et al. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400–1405. doi: 10.1016/j.athoracsur.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 7.Cohn LH, Rizzo RJ, Adams DH, Aranki SF, Couper GS, Beckel N, et al. Reduced mortality and morbidity for ascending aortic aneurysm resection regardless of cause. Ann Thorac Surg. 1996;62:463–468. doi: 10.1016/0003-4975(96)00280-9. [DOI] [PubMed] [Google Scholar]
- 8.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, et al. American College of Cardiology Foundation, Guidelines AHATFOP; Surgery AAFT, Radiology ACO, Association AS, Anesthesiologists SOC, Interventions SFCAA, Radiology SOI, Surgeons SOT, Medicine SFV, Imaging NASFC. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. JAC. 2010;55:e27–e129. doi: 10.1016/j.jacc.2010.02.015. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 9.Schmid F-X, Bielenberg K, Schneider A, Haussler A, Keyser A, Birnbaum D. Ascending aortic aneurysm associated with bicuspid and tricuspid aortic valve: involvement and clinical relevance of smooth muscle cell apoptosis and expression of cell death-initiating proteins. Eur J Cardiothorac Surg. 2003;23:537–543. doi: 10.1016/S1010-7940(02)00833-3. [DOI] [PubMed] [Google Scholar]
- 10.LeMaire SA, Wang X, Wilks JA, Carter SA, Wen S, Won T, et al. Matrix metalloproteinases in ascending aortic aneurysms: bicuspid versus trileaflet aortic valves. J Surg Res. 2005;123:40–48. doi: 10.1016/j.jss.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 11.Gaudino M, Anselmi A, Morelli M, Pragliola C, Tsiopoulos V, Glieca F, et al. Aortic expansion rate in patients With dilated post-stenotic ascending aorta submitted only to aortic valve replacement. JAC. 2011;58:581–584. doi: 10.1016/j.jacc.2011.03.040. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 12.LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8:103–113. doi: 10.1038/nrcardio.2010.187. Nature Publishing Group. [DOI] [PubMed] [Google Scholar]
- 13.Friedman T, Mani A, Elefteriades JA. Bicuspid aortic valve: clinical approach and scientific review of a common clinical entity. Expert Rev Cardiovasc Ther. 2008;6:235–248. doi: 10.1586/14779072.6.2.235. [DOI] [PubMed] [Google Scholar]
- 14.Poullis MP, Warwick R, Oo A, Poole RJ. Ascending aortic curvature as an independent risk factor for type A dissection, and ascending aortic aneurysm formation: a mathematical model. Eur J Cardiothorac Surg. 2008;33:995–1001. doi: 10.1016/j.ejcts.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 15.Khanafer K, Berguer R. Fluid-structure interaction analysis of turbulent pulsatile flow within a layered aortic wall as related to aortic dissection. J Biomech. 2009;42:2642–2648. doi: 10.1016/j.jbiomech.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 16.Nathan DP, Xu C, Gorman JH, Fairman RM, Bavaria JE, Gorman RC, et al. Pathogenesis of acute aortic dissection: a finite element stress analysis. Ann Thorac Surg. 2011;91:458–463. doi: 10.1016/j.athoracsur.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 17.Nathan DP, Xu C, Plappert T, Desjardins B, Gorman JH, Bavaria JE, et al. Increased ascending aortic wall stress in patients with bicuspid aortic valves. Ann Thorac Surg. 2011;92:1384–1389. doi: 10.1016/j.athoracsur.2011.04.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillippi JA, Klyachko EA, Kenny JP, Eskay MA, Gorman RC, Gleason TG. Basal and oxidative stress-induced expression of metallothionein is decreased in ascending aortic aneurysms of bicuspid aortic valve patients. Circulation. 2009;119:2498–2506. doi: 10.1161/CIRCULATIONAHA.108.770776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiellaro-Rafferty K, Weiss D, Joseph G, Wan W, Gleason RL, Taylor WR. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am J Physiol Heart Circ Physiol. 2011:H355–H362. doi: 10.1152/ajpheart.00040.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96:280–291. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 22.Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev. 2010;16:467–491. doi: 10.1089/ten.teb.2009.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Zhang J, Fu W, Guo D, Jiang J, Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. YMVA. 2012:1–14. doi: 10.1016/j.jvs.2012.05.084. [DOI] [PubMed] [Google Scholar]
- 24.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J-H, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524. doi: 10.1161/CIRCRESAHA.110.234237. [DOI] [PubMed] [Google Scholar]
- 26.Humphrey JD, Holzapfel GA. Mechanics, mechanobiology, and modeling of human abdominal aorta and aneurysms. J Biomech. 2012;45:805–814. doi: 10.1016/j.jbiomech.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parmacek MS. Myocardin: dominant driver of the smooth muscle cell contractile phenotype. Arterioscler Thromb Vasc Biol. 2008;28:1416–1417. doi: 10.1161/ATVBAHA.108.168930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, LeMaire SA, Chen L, Shen YH, Gan Y, Bartsch H, et al. Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation. 2006;114:I200–I205. doi: 10.1161/CIRCULATIONAHA.106.643940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE −/−mice. Clin Sci. 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP, Howatt DA, Subramanian V, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor-/- mice. Circ Res. 2011;108:574–581. doi: 10.1161/CIRCRESAHA.110.222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milewicz DM, Guo D-C, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genom Human Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 32.Ikonomidis JS, Ruddy JM, Benton SM, Arroyo J, Brinsa TA, Stroud RE, et al. Aortic dilatation with bicuspid aortic valves: cusp fusion correlates to matrix metalloproteinases and inhibitors. Ann Thorac Surg. 2012;93:457–463. doi: 10.1016/j.athoracsur.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies RR, Kaple RK, Mandapati D, Gallo A, Botta DM, Jr, Elefteriades JA, et al. Natural history of ascending aortic aneurysms in the setting of an unreplaced bicuspid aortic valve. Ann Thorac Surg. 2007;83:1338–1344. doi: 10.1016/j.athoracsur.2006.10.074. [DOI] [PubMed] [Google Scholar]
- 34.Li Z-Y, Sadat U, U-King-Im J, Tang TY, Bowden DJ, Hayes PD, et al. Association between aneurysm shoulder stress and abdominal aortic aneurysm expansion: a longitudinal follow-up study. Circulation. 2010;122:1815–1822. doi: 10.1161/CIRCULATIONAHA.110.939819. [DOI] [PubMed] [Google Scholar]
- 35.Gallo A, Saad A, Ali R, Dardik A, Tellides G, Geirsson A. Circulating interferon-γ-inducible Cys-X-Cys chemokine receptor 3 ligands are elevated in humans with aortic aneurysms and Cys-X-Cys chemokine receptor 3 is necessary for aneurysm formation in mice. J Thorac Cardiovasc Surg. 2012;143:704–710. doi: 10.1016/j.jtcvs.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ejiri J, Inoue N, Tsukube T, Munezane T, Hino Y, Kobayashi S, et al. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: protective role of statin and angiotensin II type 1 receptor blocker. Cardiovasc Res. 2003;59:988–996. doi: 10.1016/S0008-6363(03)00523-6. [DOI] [PubMed] [Google Scholar]
- 37.Bruemmer D, Daugherty A, Lu H, Rateri DL. Relevance of angiotensin II-induced aortic pathologies in mice to human aortic aneurysms. Ann N Y Acad Sci. 2011;1245:7–10. doi: 10.1111/j.1749-6632.2011.06332.x. [DOI] [PubMed] [Google Scholar]
- 38.Haskett D, Speicher E, Fouts M, Larson D, Azhar M, Utzinger U, et al. The effects of angiotensin II on the coupled microstructural and biomechanical response of C57BL/6 mouse aorta. J Biomech. 2012;45:772–779. doi: 10.1016/j.jbiomech.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 39.Patel R, Cardneau JD, Colles SM, Graham LM. Synthetic smooth muscle cell phenotype is associated with increased nicotinamide adenine dinucleotide phosphate oxidase activity: effect on collagen secretion. YMVA. 2006;43:364–371. doi: 10.1016/j.jvs.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 40.Branchetti E, Sainger R, Poggio P, Grau JB, Patterson-Fortin J, Bavaria JE, et al. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol. 2013;33:e66–e74. doi: 10.1161/ATVBAHA.112.300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batinić-Haberle I, Spasojević I, Tse HM, Tovmasyan A, Rajic Z, St Clair DK, et al. Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids. 2012;42:95–113. doi: 10.1007/s00726-010-0603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miriyala S, Spasojević I, Tovmasyan A, Salvemini D, Vujaskovic Z, St Clair D, et al. Manganese superoxide dismutase, MnSOD and its mimics. Biochim Biophys Acta. 2012;1822:794–814. doi: 10.1016/j.bbadis.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strecker C, Harloff A, Wallis W, Markl M. Flow-sensitive 4D MRI of the thoracic aorta: comparison of image quality, quantitative flow, and wall parameters at 1.5T and 3T. J Magn Reson Imaging. 2012;36:1097–1103. doi: 10.1002/jmri.23735. [DOI] [PubMed] [Google Scholar]
- 44.Vorp DA, Schiro BJ, Ehrlich MP, Juvonen TS, Ergin MA, Griffith BP. Effect of aneurysm on the tensile strength and biomechanical behavior of the ascending thoracic aorta. Ann Thorac Surg. 2003;75:1210–1214. doi: 10.1016/S0003-4975(02)04711-2. [DOI] [PubMed] [Google Scholar]
- 45.Vorp DA, Raghavan ML, Muluk SC, Makaroun MS, Steed DL, Shapiro R, et al. Wall strength and stiffness of aneurysmal and nonaneurysmal abdominal aorta. Ann N Y Acad Sci. 1996;800:274–276. doi: 10.1111/j.1749-6632.1996.tb33330.x. [DOI] [PubMed] [Google Scholar]