

Abstract
The incidence of Clostridium difficile infection (CDI) and associated mortality have increased rapidly worldwide in recent years. Therefore, it is critical to develop new therapies for CDI. In this study, we generated a novel, potently neutralizing, tetravalent, and bispecific antibody composed of 2 heavy-chain-only VH (VHH) binding domains against both TcdA and TcdB (designated “ABA”) that reverses fulminant CDI in mice infected with an epidemic 027 strain after a single injection of the antibody. We demonstrated that ABA bound to both toxins simultaneously and displayed a significantly enhanced neutralizing activity both in vitro and in vivo. Additionally, ABA was able to broadly neutralize toxins from clinical C. difficile isolates that express both TcdA and TcdB but failed to neutralize the toxin from TcdA−TcdB+ C. difficile strains. This study thus provides a rationale for the development of multivalent VHHs that target both toxins and are broadly neutralizing for treating severe CDI.
Keywords: Clostridium difficile, toxins, antibody, VHH, immunotherapy
Clostridium difficile is the most common cause of nosocomial antibiotic-associated diarrhea and is the etiologic agent of pseudomembranous colitis [1]. C. difficile infection (CDI) is primarily caused by 2 large exotoxins, TcdA and TcdB. It is estimated that >500 000 cases of CDI occur annually in the United States, with the yearly mortality rate ranging from 3% to 17%, depending on the strains. The incidence of CDI-associated mortality among patients is increasing rapidly because of the emergence of hypervirulent and antibiotic-resistant strains [2], and systemic complications are the major cause of death in C. difficile–associated disease [3].
Primary treatment for CDI involves the use of the antibiotics metronidazole and/or vancomycin. However, neither antibiotic is entirely effective, as high rates of recurrence occur despite initial successful treatment with these antibiotics [4]. The incidence of recurrence is estimated to be 20%–35%, after which there is an even greater probability (as high as 50%) of additional recurrences [5, 6]. Fidaxomicin, newly approved by the Food and Drug Administration, shows an improved efficacy over vancomycin at lowering the relapse rate [7]. Several experimental therapies are currently under development, including novel antibiotics [7, 8], probiotic and fecal transplant therapies [9, 10], novel vaccines [11, 12], and antibody-based therapies [13–16].
Camelidae species, including camels, llamas, and alpacas, produce both conventional immunoglobulin G (IgG) antibodies containing a heavy chain and light chain (IgG1) and unconventional IgGs (IgG2 and IgG3) that contain only a heavy chain (HCAbs) [17]. HCAbs bind antigens only through the variable domain of the heavy chain, thus allowing easy cloning of the DNA encoding this binding domain, which is known as a single-domain antibody or heavy-chain-only VH (VHH) [17]. Several groups have used VHHs for treating toxin-mediated diseases [18, 19]. Recently Hussack et al reported the generation of anti-Tcd VHHs from llamas that target the receptor-binding domains of the 2 toxins, some of which possessed toxin-neutralizing activity [20, 21]. Here, we report the isolation of panels of VHHs that recognize and neutralize either TcdA or TcdB. We further constructed fusion proteins consisting of multiple-antitoxin VHHs, which neutralize both TcdA and TcdB and alleviate fulminant disease symptoms in mice infected with C. difficile.
MATERIALS AND METHODS
Immunogens and Immunization
Two alpacas were immunized with purified full-length recombinant TcdA or TcdB (aTcdA and aTcdB) containing point mutations that inactivate the glucosyltransferase activity of these toxins [12, 22], as described previously [23, 24]. All animals were handled and cared for according to institutional animal care and use committee guidelines and in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animals were immunized subcutaneously up to 5 times at intervals of ≥3 weeks with 50–100 µg of aTcdA or aTcdB with alum adjuvant (with CpG in primary immunization). Blood samples were collected before each immunization for IgG titer determination. Five days following the final boost, peripheral blood lymphocytes (PBLs) were harvested as the source of VHH genetic material.
Construction of VHH Libraries and Screening
Two VHH phage display libraries were generated to obtain VHHs recognizing TcdA and TcdB. The libraries were produced from PBLs obtained from a aTcdA- or aTcdB-immune alpaca (described above), using methods previously described [25, 26]. Panning for VHH-displayed phage was done as described previously [25, 26] or by pull-down methods, using biotinylated TcdA or TcdB. For pull-down selection, the recombinant TcdA and TcdB [22] proteins were biotinylated using the Pierce EZ-Link NHS-PEG4 Biotin kit (Pierce Biotechnology, Rockford, IL) per the manufacturer's instructions. VHH clones were sequenced, and those with distinctly homologous complementarity-determining region sequences were considered to be clonally related; only the VHH in each group that had the most potent toxin-neutralizing activity or that produced the strongest enzyme-linked immunosorbent assay (ELISA) signal was pursued further. Six unique TcdA-binding VHHs and 11 unique TcdB-binding VHHs were identified. Selected VHH coding sequences were cloned into the pET32b expression vector (Novagen) for cytosolic expression of VHHs fused to thioredoxin in Escherichia coli Rosetta-gami 2 (DE3)pLacI cells (Novagen).
Generation of the VHH Heterotetramer AH3/E3/E3/AA6ABA (ABA)
VHHs having the most potent neutralizing activity and the highest binding affinity to distinct, nonoverlapping epitopes targeting each toxins were chosen for inclusion within a multimeric, multivalent antibody. For TcdA, VHHs AH3 and AA6 were selected for their potent neutralizing activity. For TcdB, 2 copies of the E3 VHH were selected, because E3 is a potent TcdB-neutralizing VHH targeting the well-conserved glucosyltransferase domain with particularly high affinity. To generate ABA, the coding sequences of individual VHHs were amplified and fused under the cytomegalovirus promoter of a pSEC91 plasmid. DNA encoding a flexible linker sequence ([G3S]4) was installed between each of the 4 VHH-coding sequences. Both an immunoglobulin κ-chain leader (for protein secretion) and a His(6)-tag (for purification) were added to the N-terminus of the tetramer. The insert was sequenced to ensure that the proper sequence was obtained, and the final construct was transfected into HEK293 cells. ABA purified from cell culture supernatants of ABA-secreting stable 293 clones displayed a single dominant band during sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) after GelBlue (Pierce) staining. The purified ABA showed no toxicity to mice after intravenously injection of doses up to 10 mg/kg.
ELISAs
Microplates were coated with 0.5 µg/mL recombinant TcdA or 0.5 µg/mL TcdB [22] overnight at 4°C and incubated with 50 µL bacterial supernatants or purified VHHs. After washes, horseradish peroxidase (HRP)–conjugated anti-E-tag antibody was added to plates, followed by analysis by a standard ELISA. For competition ELISA, serial dilutions of VHHs were mixed with serial dilutions of ABA before adding to plates coated with TcdA or TcdB. After incubation and washes, the binding of monomer VHHs was measured by adding a biotinylated anti-thioredoxin VHH generated by us, followed by HRP-conjugated streptavidin. To determine whether ABA is capable of binding the 2 toxins simultaneously, plates were coated with TcdA or TcdB before adding serial dilutions of ABA. After washes, serial dilutions of TcdB or TcdA, respectively, were added to the wells. After extensive washing, mouse monoclonal antibodies against TcdB or TcdA (List Biological Laboratories, Campbell, CA), respectively, were added to the wells before the addition of HRP-conjugated antimouse antibodies for detection.
In Vitro Neutralizing Assays
Mouse colonic epithelial CT26 cells and African green monkey kidney Vero cells (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/mL penicillin G, and 40 µg/mL streptomycin sulfate. Subconfluent CT26 or Vero cells (2.0 × 104 cells/well) were seeded in 96-well plates for 24 hours before the addition of toxin and VHH agents. Serially diluted VHHs and toxins were premixed using toxin at a concentration of 0.2 ng/mL for TcdB or 10 ng/mL for TcdA and then added to each well. In some experiments, 10-µL bacterial supernatants from 11 C. difficile strains were mixed with ABA (10 µg/mL) before addition to the Vero cell monolayer. This panel of strains was kindly provided by Dr Trevor Lawley and represent an assortment of genetically and geographically diverse clinical isolates [27, 28], Bacterial supernatant added without ABA acted as a control. After incubation for 24 hours, cells were observed under a phase-contrast microscope, and the percentage of cells that were rounded was assessed.
Systemic Challenge
Six-week-old female CD1 mice (Charles River Labs) were maintained in a pathogen-free animal biosafety level 2 facility. All mice used in the experiments were housed in groups of 5 per cage under the same conditions. Food, water, bedding, and cages were autoclaved. Mice (5 per group) were administered VHH monomers or ABA by intraperitoneal injection 1 hour before intraperitoneal challenge of a mixture of TcdA and TcdB (25 ng/mouse of each toxin). Mice were monitored hourly for signs of illness, including hunched posture, ruffled coat, and rapid breathing. Animals that became moribund were euthanized.
CDI Challenge
C57BL/6 mice were orally administered 105 C. difficile spores from the UK1 (BI/NAP1/027) strain after receiving antibiotic treatment, as previously described [12]. Mice (10 per group) were administered with ABA by intraperitoneal injection 24 hours after spore challenge. Mouse weights were measured daily, and the development of disease symptoms was monitored twice daily. Animals that became moribund or lost >20% of their body weight were euthanized.
Statistical Analysis
Data were analyzed by Kaplan–Meier survival analysis with a log-rank test of significance, analysis of variance, and 1-way analysis of variance, followed by Bonferroni posttests, using the Prism statistic software program. Results are expressed as mean ± standard error of mean.
RESULTS
Identification and Characterization of Anti-Tcd VHHs
Six anti-TcdA VHHs and 11 anti-TcdB VHHs were identified and expressed in E. coli and purified using nickel affinity chromatography (Supplementary Figure 1A). The domain location of the binding epitopes of each VHH in their targeted toxin (Figure 1A) was determined using Western blotting and ELISA of the VHHs against recombinant toxin fragments [29] or toxin chimeras [30]. The relative affinities of the VHHs for the different toxins was assessed by ELISA (Figure 1B), and the KDs for the toxins were determined by surface plasmon resonance (SPR) analysis (Supplementary Table 1). We further evaluated the ability of the VHHs to neutralize the cytotoxic activities of TcdA or TcdB on cultured cells. Four VHHs against TcdA (Figure 2A) and 3 VHHs against TcdB (Figure 2B) had strong neutralizing activity, capable of neutralizing toxin-mediated cytopathic effects at nanomolar concentrations.
Figure 1.

Binding of specific heavy-chain-only antibody (VHH) to TcdA and TcdB. A, Diagram illustrates specific binding of individual VHHs to the glucosyltransferase domain (GTD), cysteine protease domain (CPD), translocation domain (TD), or receptor-binding domain (RBD) in TcdA or TcdB. B and C, Enzyme-linked immunosorbent assay analysis of serially diluted anti-TcdA (B) or anti-TcdB (C) VHHs in microtiter plates coated with 0.5 µg/mL of TcdA or TcdB, respectively.
Figure 2.
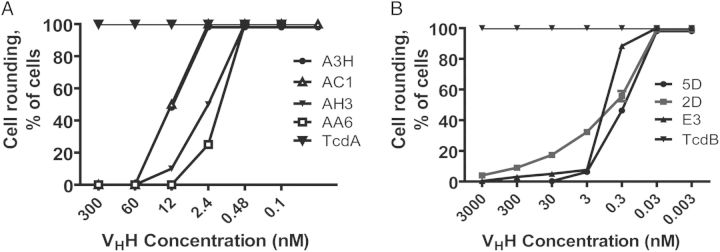
Neutralizing activities of anti-Tcd heavy-chain-only antibodies (VHHs). CT26 cells were exposed to either 10 ng/mL TcdA (A) or 0.2 ng/mL TcdB (B) premixed with a serial dilution of each anti-TcdA or anti-TcdB VHH, respectively, and then added to cells for 24 hours. Morphological changes in cells were observed under a phase-contrast microscope, and the percentage of cell rounding was assessed.
Generation of a Multivalent VHH Agent That Neutralizes Both TcdA and TcdB
Since it has been found necessary to neutralize both TcdA and TcdB to achieve optimal protection against CDI [15, 30], we generated ABA (Figure 3A). The selection of these VHHs was based on their high toxin affinities, their potent toxin neutralizing activities, and their binding to more well-conserved regions on these toxins. Purified ABA from stably transfected HEK293 cells displayed a 66-kDa band on SDS-PAGE (Supplementary Figure 1B). ABA successfully competed for the Tcd binding by its individual component VHHs, AH3, AA6, and E3, in a dose-dependent manner but failed to block AC1 binding to TcdA or B12 binding to TcdB (Figure 3B), thus indicating that each of the component VHHs in ABA remained functional. Additionally, in an ELISA, ABA demonstrated the capacity to bind to both toxins at the same time in a dose-dependent fashion, regardless of whether TcdA or TcdB was bound to the plate (Figure 4A and 4B). Thus, ABA is able to bind TcdA and TcdB simultaneously and is thus bispecific to both toxins. This finding was confirmed by SPR binding experiments in which ABA was coupled to a surface onto which TcdA and TcdB were titrated (Supplementary Figure 2). The maximal response value of the ABA surface was twice as high for both TcdA titrations as for the TcdB titration, suggesting that ABA is trispecific to 3 distinctive epitopes, with AH3 and AA6 subunits binding simultaneously to TcdA molecules along with E3 subunits binding to TcdB.
Figure 3.

Generation of bispecific ABA against TcdA and TcdB. A, Diagram of the ABA fusion protein. AH3/E3/E3/AA6 heavy-chain-only antibody (VHH) subunits were separated by a flexible linker sequence (FS). A His(6)-tag and E-tag were genetically fused at the N-terminal and C-terminal, respectively, of the ABA molecule. B, Checkerboard Enzyme-linked immunosorbent assay analysis of ABA competition with individual components. Serially diluted ABA (ng/mL) was mixed with different serially diluted thioredoxin/VHH fusion proteins targeting TcdA (AH3, AA6, or AC1) or TcdB (E3 or B12) and added to microplates coated with TcdA and TcdB, respectively. Biotinylated anti-thioredoxin VHH was used for detection.
Figure 4.
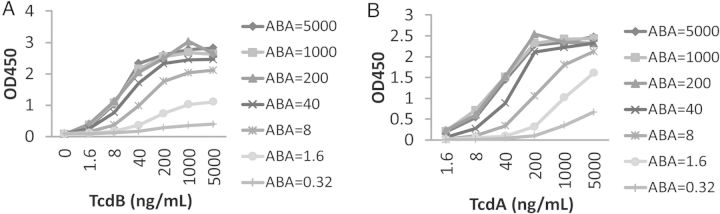
ABA binds to TcdA and TcdB simultaneously. Serially diluted ABA (ng/mL) was added to microplate wells coated with TcdA (A) or TcdB (B). After incubation and washes, serially diluted TcdB (A) or TcdA (B) were added to the plates. The binding of toxins was determined by detecting TcdB (A) or TcdA (B), respectively, using toxin-specific mouse monoclonal antibodies.
ABA Demonstrates Strong Neutralizing Activity
We next determined the neutralizing activity of ABA to prevent toxin-mediated cytopathic effects on cultured cells. ABA had substantially enhanced neutralizing activity against TcdA-induced cytopathic effects on CT26 cells, with a concentration of half-maximal activity (ED50) of around 10 pM, compared with the individual VHHs, which had ED50 values of around 3 nM (Figure 5A). The neutralizing activity of ABA against TcdB was comparable to that of E3 (Figure 5B). Further experiments demonstrated that ABA was capable of preventing the rounding of Vero cells induced by culture supernatants from 11 genetically and geographically diverse clinical C. difficile isolates that express both functional TcdA and TcdB (Table 1) [27, 28]. Interesting, ABA failed to neutralize TcdB in the culture supernatants from 2 TcdA−TcdB+ strains (ribotype 017), suggesting that the glucosyltransferase domains (GTDs) from these strains are significantly different from those from the tested TcdA+TcdB+ strains.
Figure 5.

In vitro and in vivo neutralization of TcdA and TcdB due to ABA. A and B, CT26 cells were exposed to serially diluted E3, AH3, AA6, or ABA together with either 10 ng/mL TcdA (A) or 0.2 ng/mL TcdB (B) for 24 hours. Morphological changes in cells were observed under a phase-contrast microscope, and the percentage of cell rounding was assessed. C, For in vivo neutralization, mice were injected intraperitoneally with the indicated doses of ABA or a mixture of the individual heavy-chain-only antibody (VHH) components, followed 1 hour later by intraperitoneal inoculation with a mixture of TcdA and TcdB (25 ng each/mouse). Overall mouse survival was analyzed by Kaplan–Meier survival curves. Abbreviation: PBS, phosphate-buffered saline.
Table 1.
ABA Neutralization Against Toxins Secreted From Different Clostridium difficile Strains
| Strain | Ribotype | REA Type | PFGE Type | Place/Date of Isolation/Source | ABA Neutralization |
|---|---|---|---|---|---|
| R20291 | 027 | BI | NAP1 | London/2006/human | Yes |
| CD196 | 027 | BI | NAP1 | France/1985/human | Yes |
| 630 | 012 | R | Zurich/1982/human | Yes | |
| M120 | 078 | BK | NAP7,8,9 | UK/2007/human | Yes |
| BI-9 | 001 | J | NAP2 | Gerding collection | Yes |
| Liv024 | 001 | J | NAP2 | Liverpool/2009/human | Yes |
| Liv022 | 106 | DH | NAP11 | Liverpool/2009/human | Yes |
| TL178 | 002 | G | NAP6 | Belfast/2009/human | Yes |
| TL176 | 014 | Y | NAP4 | Cambridge, UK/2009/human | Yes |
| TL174 | 015 | … | … | Cambridge, UK/2009/human | Yes |
| CD305 | 023 | … | … | London/2008/human | Yes |
| CF5 | 017 | CF | NAP9 | Belgium/1995/human | No |
| M68 | 017 | CF | NAP9 | Dublin/2006/human | No |
ABA was mixed with bacterial culture supernatants from 13 C. difficile strains and added to Vero cell monolayers. Cell rounding was observed under a light microscope. All supernatants alone caused cell rounding, whereas no cell rounding was observed when supernatants were mixed with ABA, except for CF5 and M68 strains.
Abbreviations: PFGE, pulsed-field gel electrophoresis; REA, restriction endonuclease analysis.
We further assessed the in vivo neutralizing activity of ABA by evaluating its capacity to protect mice from systemic toxicity. Mice were completely protected against a lethal challenge of mixed TcdA and TcdB (25 ng of each per mouse) after ABA treatment at concentrations as low as 3.2 µg/kg, whereas the individual component VHH combination failed to show complete protection at a dose as high as 1 mg/kg (P = .009; Figure 5C). Thus, the in vivo neutralizing activity of ABA is at least 300-fold more potent than that of the mixture of the individual components.
ABA Treatment Reverses Fulminant Disease Symptoms in Mice With CDI
Since ABA exhibits potent neutralizing activity against both toxins, we evaluated its therapeutic potential against fulminant CDI, to which no effective treatment is available. After oral challenge of antibiotic-treated mice with C. difficile spores from the NAP1/027 strain, a single dose of ABA (1 mg/kg) or vehicle control was parenterally administered to mice. At the time of ABA treatment, a majority of mice had developed severe diarrhea and were experiencing weight loss (Figure 6A). Treatment with ABA at a dose of 1 mg/kg significantly prevented the severe weight loss associated with CDI (Figure 6A). More importantly, mice treated with ABA were significantly protected from mortality induced by C. difficile challenge, compared with the phosphate-buffered saline control (Figure 6B). None of the mice in the 1 mg/kg or 40 μg/kg groups died, and only 1 mouse in the 200 μg/kg group died, whereas 60% of the mice in the vehicle control group died (Figure 6B).
Figure 6.

Therapeutic efficacy of ABA against Clostridium difficile infection in mice. C57BL/6 mice were pretreated with antibiotics before being challenged with UK1 spores. Mice were injected intraperitoneally with 1 mg/kg of ABA 24 hours after infection. Mouse weight change (A) and survival (B) were monitored and are plotted. The experiments were repeated twice with similar results (10 mice/experiment). Error bars denote standard errors of the mean. Abbreviation: PBS, phosphate-buffered saline.
DISCUSSION
The incidence of CDI-associated mortality has increased rapidly in recent years [3], while treatment options against severe and fulminant CDI remain limited [2]. In this study, we generated a novel bispecific VHH antibody that is capable of neutralizing both TcdA and TcdB and reversing fulminant disease symptoms in mice with CDI. Our study thus provides evidence for the usefulness and efficacy of bispecific VHH antibodies in the treatment of fulminant CDI in patients.
The glucosyltransferase-deficient holotoxins aTcdA and aTcdB [12] were used to immunize alpacas to induce potent B-cell responses. These atoxic holotoxins both remain soluble and appear to maintain native conformations and thus should induce antibody responses that are similar to those of wild-type toxins. As expected, the majority of neutralizing anti-TcdA VHHs recognize the C-terminal combined repetitive oligopeptides (CROPs), which has been established as the immunodominant domain of TcdA [12, 31]. However, a recent study has shown that a truncated TcdA protein, without the CROPs, can cause cytotoxicity in cells [32]. Our 2 most potent neutralizing anti-TcdA VHHs recognize the N-terminal GTD and the central translocation domain (TD), indicating that the toxins can also be effectively neutralized by blocking glucosyltransferase or the membrane insertion/cytosolic release of TcdA.
Unlike the anti-TcdA VHHs, all neutralizing anti-TcdB VHHs recognize the N-terminal GTD; none of the 5 VHHs that bind to the CROPs of TcdB demonstrated neutralizing activity, despite their high binding affinities. This result is in line with our recent finding that the major neutralizing epitopes of TcdB are largely located in the N-terminal portion of the toxin rather than in the C-terminal CROP region [12]. The CROP region of TcdB has been the traditional immunogen choice for vaccine development and generation of neutralizing antibodies [33–35]. This notion now warrants revision because of convincing data that the N-terminal portion of TcdB is the most effective domain as an immunogen for vaccines and immunotherapies against CDI. Further support for this concept is found in recent studies in which TcdB CROPs failed to induce protective titers of neutralizing antibodies [34, 35], while the GTD fragment of TcdB led to the generation of highly protective neutralizing antibody responses [36].
Because various C. difficile strains expressing different toxin isoforms are isolated from patients with CDI, it is important for therapeutic antibodies to target conserved toxin epitopes, thus conferring broad protection across clinically relevant isolates. Recent reports have shown that TcdA is relatively well conserved between historical and epidemic strains, whereas TcdB shows a significant degree of variability [37, 38]. The sequence variability in TcdB from TcdA+TcdB+ strains is mainly found within the C-terminal CROPs and the adjacent region (88% identity between historical and epidemic strains), whereas the N-terminal GTD is more conserved and shows 96% amino acid sequence identity [37]. Unlike the anti-TcdB monoclonal antibody undergoing clinical trials [14], which binds to the CROP region [33], E3 binds to the GTD of TcdB. In addition, ABA consists of 2 distinct VHHs, AH3 and AA6, that bind to the GTD and TD of TcdA, respectively. In fact, ABA was able to neutralize toxins a panel of genotypically diverse TcdA+TcdB+ clinical isolates collected from different geographic locations [27, 28], including some BI/NAP1/027 strains that are responsible for recent outbreaks of CDI, thus demonstrating the broad efficacy range of this agent against those strains producing both TcdA and TcdB.
Although the majority of pathogenic C. difficile produce both TcdA and TcdB, some clinical isolates produce only TcdB [39]. The TcdA−TcdB+ strain infections occur in sporadic form in Europe and North America but are more frequent in East Asian countries [40]. Sequence analysis of the TcdB gene from TcdA−TcdB+ strains identified multiple point mutations in GTD regions, with only 84% homology with that of laboratory strain VPI 10463 [39]. Since E3 binds to the GTD of TcdB and neutralizes its activity, we examined the ability of ABA to neutralizing TcdB from 2 TcdA−TcdB+ strains. Our data demonstrated that ABA failed to neutralize TcdB from these 2 TcdA−TcdB+ NAP9/CF/017 strains (Table 1), supporting the previous finding that the glucosyltransferase of TcdB from the TcdA−TcdB+ strains is significantly different from that of the VPI 10463 strain [39, 41]. Although ABA is able to broadly neutralizing toxins from TcdA+TcdB+ C. difficile strains, it is unable to neutralize TcdB from the tested TcdA−TcdB+ ribotype 017 strains and, thus, is unlikely to be therapeutically effective against CDI caused by these strains. Therefore, further improvement via the introduction of VHHs that are able to also neutralize TcdB from the 017 strains is desirable.
CDI causes a wide spectrum of clinical symptoms and outcomes, including mild diarrhea, fulminant disease, and death [2, 42]. A single dose of ABA was able to significantly protect mice against weight loss and fulminant CDI after C. difficile spore challenge. We have previously reported that toxins are released into the bloodstream of animals experimentally infected with C. difficile, a feature associated with severe and fulminant CDI in several animal models [43–45]. The potent neutralizing activity of ABA may allow for the rapid neutralization of circulating toxins liberated from the intestines of mice with fulminant CDI. In fact, a single injection of ABA of only 3.2 µg/kg provided full protection against lethal systemic toxin challenge in mice.
In this study, we effectively used a 1-mg/kg dose of ABA to treat CDI in mice, compared with the 50-mg/kg and 10-mg/kg doses of human monoclonal antibodies that have been used in hamsters [33] and patients [14], respectively, with CDI. The serum half-life of ABA in its current form is likely to be short because of its smaller size and its lack of an Fc domain. Thus, either multiple doses of the antibody in its current form or further modifications to increase bioavailability may be necessary to treat chronic infections. Multiple doses of VHHs may induce an anti-VHH antibody response and reduce the efficacy of subsequent treatments should patients develop multiple recurrent CDIs. Although VHHs are not particularly immunogenic, their potential immunogenicity can be further reduced via the humanization of the VHH scaffold [46]. In addition, a number of approaches are now available to improve the serum half-life of VHHs, such as genetic fusion with albumin-binding domains [47, 48] or immunoglobulins [49], as well as PEGylation [50], thereby enhancing their therapeutic potential.
In summary, we report here that a novel bispecific VHH antibody is able to rapidly alleviate fulminant CDI in mice. Our study thus demonstrates the feasibility of designing multivalent and bispecific VHH antibodies against both toxins with significant enhanced therapeutic efficacy to reduce the morbidity and mortality associated with this debilitating disease.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Disclaimer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by National Institute of Allergy and Infectious Diseases and National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (awards R01AI088748, R01DK084509, and R56AI99458 to H. F. and contract N01-AI-30050 and award U54 AI057159 to C. B. S.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Cloud J, Kelly CP. Update on Clostridium difficile associated disease. Curr Opin Gastroenterol. 2007;23:4–9. doi: 10.1097/MOG.0b013e32801184ac. [DOI] [PubMed] [Google Scholar]
- 2.Kelly CP, LaMont JT. Clostridium difficile--more difficult than ever. N Engl J Med. 2008;359:1932–40. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 3.Siemann M, Koch-Dorfler M, Rabenhorst G. Clostridium difficile-associated diseases. The clinical courses of 18 fatal cases. Intensive Care Med. 2000;26:416–21. doi: 10.1007/s001340051175. [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Fang H, Weintraub A, Nord CE. Distinct ribotypes and rates of antimicrobial drug resistance in Clostridium difficile from Shanghai and Stockholm. Clin Microbiol Infect. 2009;15:1170–3. doi: 10.1111/j.1469-0691.2009.02992.x. [DOI] [PubMed] [Google Scholar]
- 5.Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis. 2007;45:302–7. doi: 10.1086/519265. [DOI] [PubMed] [Google Scholar]
- 6.Johnson S. Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcomes. J Infect. 2009;58:403–10. doi: 10.1016/j.jinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Louie TJ, Miller MA, Mullane KM, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–31. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 8.Butler MM, Shinabarger DL, Citron DM, et al. MBX-500, a hybrid antibiotic with in vitro and in vivo efficacy against toxigenic Clostridium difficile. Antimicrob Agents Chemother. 2012;56:4786–92. doi: 10.1128/AAC.00508-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis. 2011;53:994–1002. doi: 10.1093/cid/cir632. [DOI] [PubMed] [Google Scholar]
- 10.Brandt LJ, Aroniadis OC, Mellow M, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol. 2012;107:1079–87. doi: 10.1038/ajg.2012.60. [DOI] [PubMed] [Google Scholar]
- 11.Tian JH, Fuhrmann SR, Kluepfel-Stahl S, Carman RJ, Ellingsworth L, Flyer DC. A novel fusion protein containing the receptor binding domains of C. difficile toxin A and toxin B elicits protective immunity against lethal toxin and spore challenge in preclinical efficacy models. Vaccine. 2012;30:4249–58. doi: 10.1016/j.vaccine.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Sun X, Zhang Y, et al. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun. 2012;80:2678–88. doi: 10.1128/IAI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly CP, Pothoulakis C, Vavva F, et al. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrob Agents Chemother. 1996;40:373–9. doi: 10.1128/aac.40.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowy I, Molrine DC, Leav BA, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 2010;362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 15.Roberts A, McGlashan J, Al-Abdulla I, et al. Development and evaluation of an ovine antibody-based platform for treatment of Clostridium difficile infection. Infect Immun. 2012;80:875–82. doi: 10.1128/IAI.05684-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marozsan AJ, Ma D, Nagashima KA, et al. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J Infect Dis. 2012;206:706–13. doi: 10.1093/infdis/jis416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wesolowski J, Alzogaray V, Reyelt J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198:157–74. doi: 10.1007/s00430-009-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams H, Brummelhuis W, Maassen B, et al. Specific immuno capturing of the staphylococcal superantigen toxic-shock syndrome toxin-1 in plasma. Biotechnol Bioeng. 2009;104:143–51. doi: 10.1002/bit.22365. [DOI] [PubMed] [Google Scholar]
- 19.Tremblay JM, Mukherjee J, Leysath CE, et al. A single VHH-based toxin neutralizing agent and an effector antibody protects mice against challenge with Shiga toxins 1 and 2. Infect Immun. 2013;81:4592–603. doi: 10.1128/IAI.01033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussack G, Arbabi-Ghahroudi M, Mackenzie CR, Tanha J. Isolation and characterization of Clostridium difficile toxin-specific single-domain antibodies. Methods Mol Biol. 2012;911:211–39. doi: 10.1007/978-1-61779-968-6_14. [DOI] [PubMed] [Google Scholar]
- 21.Hussack G, Arbabi-Ghahroudi M, van Faassen H, et al. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J Biol Chem. 2011;286:8961–76. doi: 10.1074/jbc.M110.198754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang G, Zhou B, Wang J, et al. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol. 2008;8:192. doi: 10.1186/1471-2180-8-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maass DR, Sepulveda J, Pernthaner A, Shoemaker CB. Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHHs) J Immunol Methods. 2007;324:13–25. doi: 10.1016/j.jim.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tremblay JM, Kuo CL, Abeijon C, et al. Camelid single domain antibodies (VHHs) as neuronal cell intrabody binding agents and inhibitors of Clostridium botulinum neurotoxin (BoNT) proteases. Toxicon. 2010;56:990–8. doi: 10.1016/j.toxicon.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukherjee J, Tremblay JM, Leysath CE, et al. A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PLoS One. 2012;7:e29941. doi: 10.1371/journal.pone.0029941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vance DJ TJ, Mantis NJ, Shoemaker CB. Stepwise Engineering of Heterodimeric Single Domain Camelid VHH Antibodies that Passively Protect Mice from Ricin Toxin. J Biol Chem. 2013;288:36538–47. doi: 10.1074/jbc.M113.519207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stabler RA, Dawson LF, Valiente E, et al. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS One. 2012;7:e31559. doi: 10.1371/journal.pone.0031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He M, Sebaihia M, Lawley TD, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107:7527–32. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He X, Sun X, Wang J, et al. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Infect Immun. 2009;77:2294–303. doi: 10.1128/IAI.01577-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Sun X, Zhang Y, et al. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun. 2012;80:2678–88. doi: 10.1128/IAI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castagliuolo I, Sardina M, Brun P, et al. Clostridium difficile toxin A carboxyl-terminus peptide lacking ADP-ribosyltransferase activity acts as a mucosal adjuvant. Infect Immun. 2004;72:2827–36. doi: 10.1128/IAI.72.5.2827-2836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olling A, Goy S, Hoffmann F, Tatge H, Just I, Gerhard R. The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile toxin A. PLoS One. 2011;6:e17623. doi: 10.1371/journal.pone.0017623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Babcock GJ, Broering TJ, Hernandez HJ, et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun. 2006;74:6339–47. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hussack G, Arbabi-Ghahroudi M, van Faassen H, et al. Neutralization of clostridium difficile toxin A with single-domain antibodies targeting the cell-receptor binding domain. J Biol Chem. 2011;286:8961–76. doi: 10.1074/jbc.M110.198754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Permpoonpattana P, Hong HA, Phetcharaburanin J, et al. Immunisation with Bacillus spores expressing toxin A peptide repeats protects against infection with toxin A+ B+ strains of Clostridium difficile. Infect Immun. 2011;79:2295–302. doi: 10.1128/IAI.00130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leuzzi R, Spencer J, Buckley A, et al. Protective efficacy induced by recombinant Clostridium difficile toxin fragments. Infect Immun. 2013;81:2851–60. doi: 10.1128/IAI.01341-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lanis JM, Barua S, Ballard JD. Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog. 2010;6:e1001061. doi: 10.1371/journal.ppat.1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stabler RA, He M, Dawson L, et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambol SP, Merrigan MM, Lyerly D, Gerding DN, Johnson S. Toxin gene analysis of a variant strain of Clostridium difficile that causes human clinical disease. Infect Immun. 2000;68:5480–7. doi: 10.1128/iai.68.10.5480-5487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Pai H, Seo MR, Kang JO. Clinical and microbiologic characteristics of tcdA-negative variant Clostridium difficile infections. BMC Infect Dis. 2012;12:109. doi: 10.1186/1471-2334-12-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huelsenbeck J, Dreger S, Gerhard R, Barth H, Just I, Genth H. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect Immun. 2007;75:801–9. doi: 10.1128/IAI.01705-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–36. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 43.Steele J, Feng H, Parry N, Tzipori S. Piglet models of acute or chronic Clostridium difficile illness. J Infect Dis. 2010;201:428–34. doi: 10.1086/649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He X, Wang J, Steele J, et al. An ultrasensitive rapid immunocytotoxicity assay for detecting Clostridium difficile toxins. J Microbiol Methods. 2009b;78:97–100. doi: 10.1016/j.mimet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steele J, Chen K, Sun X, et al. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis. 2012;205:384–91. doi: 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284:3273–84. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 47.Tijink BM, Laeremans T, Budde M, et al. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther. 2008;7:2288–97. doi: 10.1158/1535-7163.MCT-07-2384. [DOI] [PubMed] [Google Scholar]
- 48.Coppieters K, Dreier T, Silence K, et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006;54:1856–66. doi: 10.1002/art.21827. [DOI] [PubMed] [Google Scholar]
- 49.Harmsen MM, Van Solt CB, Fijten HP, Van Setten MC. Prolonged in vivo residence times of llama single-domain antibody fragments in pigs by binding to porcine immunoglobulins. Vaccine. 2005;23:4926–34. doi: 10.1016/j.vaccine.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 50.Harmsen MM, van Solt CB, Fijten HP, et al. Passive immunization of guinea pigs with llama single-domain antibody fragments against foot-and-mouth disease. Vet Microbiol. 2007;120:193–206. doi: 10.1016/j.vetmic.2006.10.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.