

Abstract
Introduction
N-succinimidyl 4-guanidinomethyl-3-[*I]iodobenzoate ([*I]SGMIB) has shown promise for the radioiodination of monoclonal antibodies (mAbs) and other proteins that undergo extensive internalization after receptor binding, enhancing tumor targeting compared to direct electrophilic radioiodination. However, radiochemical yields for [131I]SGMIB synthesis are low, which we hypothesize is due to steric hindrance from the Boc-protected guanidinomethyl group ortho to the tin moiety. To overcome this, we developed the isomeric compound, N-succinimidyl 3-guanidinomethyl-5-[131I]iodobenzoate (iso-[131I]SGMIB) wherein this bulky group was moved from ortho to meta position.
Methods
Boc2-iso-SGMIB standard and its tin precursor, N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate (Boc2-iso-SGMTB), were synthesized using two disparate routes, and iso-[*I]SGMIB synthesized from the tin precursor. Two HER2-targeted vectors — trastuzumab (Tras) and a nanobody 5F7 (Nb) — were labeled using iso-[*I]SGMIB and [*I]SGMIB. Paired-label internalization assays in vitro with both proteins, and biodistribution in vivo with trastuzumab, labeled using the two isomeric prosthetic agents were performed.
Results
When the reactions were performed under identical conditions, radioiodination yields for the synthesis of Boc2-iso-[131I]SGMIB were significantly higher than those for Boc2-[131I]SGMIB (70.7 ± 2.0% vs 56.5 ± 5.5%). With both Nb and trastuzumab, conjugation efficiency also was higher with iso-[131I]SGMIB than with [131I]SGMIB (Nb, 33.1 ± 7.1% vs 28.9 ± 13.0%; Tras, 45.1 ± 4.5% vs 34.8 ± 10.3%); however, the differences were not statistically significant. Internalization assays performed on BT474 cells with 5F7 Nb indicated similar residualizing capacity over 6 h; however, at 24 h, radioactivity retained intracellularly for iso-[131I]SGMIB-Nb was lower than for [125I]SGMIB-Nb (46.4 ± 1.3% vs 56.5 ± 2.5%); similar results were obtained using Tras. Likewise, a paired-label biodistribution of Tras labeled using iso-[125I]SGMIB and [131I]SGMIB indicated an up to 22% tumor uptake advantage at later time points for [131I]SGMIB-Tras.
Conclusion
Given the higher labeling efficiency obtained with iso-SGMIB, this residualizing agent might be of value for use with shorter half-life radiohalogens.
Keywords: Residualizing label, Radioiodination, Internalization, Monoclonal Antibody
1. Introduction
Targeted radiotherapy using monoclonal antibodies (mAbs) specifically reactive with internalizing receptors that are either uniquely or over expressed on cancer cells has been widely investigated [1]. To circumvent the loss of radioactivity from tumor cells after intracellular degradation of the radioiodinated mAbs [2, 3], a variety of residualizing labeling approaches have been developed including the use of iodotyramine-cellobiose conjugates [4-6], and d-amino acid peptide-DTPA conjugates [7], and peptides containing charged D-amino acids [8, 9].
Our group has been pursuing the development of an alternative approach for labeling internalizing mAbs that seeks to enhance retention of radiohalogens in cancer cells while attempting to avoid concomitantly increased normal tissue retention. The design hypothesis has been to utilize acylation agents that will result in the generation of labeled catabolites that would be charged at lysosomal pH and therefore will not be able to cross the lysosomal and cell membranes [10-14]. The most successful residualizing agent developed to date based on this premise is the guanidine-moiety-containing acylation agent, N-succinimidyl 4-guanidinomethyl-3-[131I]iodobenzoate ([131I]SGMIB) [15, 16], which likely reflects the fact that guanidine has a pKa of ∼13, and should remain exclusively positively charged at lysosomal pH. Indeed, when the anti-epidermal growth factor receptor variant III-reactive mAb L8A4 was labeled with [131I]SGMIB, significantly higher intracellularly retained radioactivity in tumor cells in vitro, compared to that from the directly labeled mAb was seen even at 24 h [15]. Moreover, an up to fivefold tumor targeting advantage was demonstrated for radioiodination of this and other internalizing vectors with SGMIB prosthetic group [17-19].
When we designed the SGMIB reagent, we opted to use the 1,3,4- over 1,3,5-isomer simply due to the ready availability of the starting material for the synthesis of both its standard and tin precursor. However, the yields for the synthesis of Boc2-[131I]SGMIB from its tin precursor were only 60-65%, significantly lower than that obtained with similar compounds [10, 20]. Hypothesizing that the lower radiochemical yield might be due to steric hindrance imparted by the bulky Boc2-guanidinomethyl group present at the ortho position of the tin moiety in the precursor, in the current study, we have developed an isomeric agent, N-succinimidyl 3-guanidinomethyl-5-[131I]iodobenzoate (iso-[131I]SGMIB) wherein the guanidinomethyl moiety has been moved to the 3-position to give a 1,3,5-substitution. Direct comparisons were made between Boc2-iso-[131I]SGMIB and Boc2-[131I]SGMIB with regard to radiochemical yield and the efficiency of conjugating iso-[131I]SGMIB and [131I]SGMIB to two HER2-targeting proteins — trastuzumab (Tras) and 5F7 nanobody. Cellular retention of radioactivity after internalization of two biomolecules, each labeled using the two isomeric prosthetic agents, was evaluated in vitro on HER2-expressing BT474 breast carcinoma cells. Finally, a paired-label biodistribution of trastuzumab radioiodinated using iso-[125I]SGMIB and [131I]SGMIB was performed in mice with subcutaneous BT474M1 breast carcinoma xenografts.
2. Materials and methods
2.1. General
Chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO) unless noted otherwise. Sodium [125I]iodide and sodium [131I]iodide in 0.1 N NaOH with specific activities of 2200 Ci/mmol and 1200 Ci/mmol respectively, were purchased from Perkin-Elmer Life and Analytical Sciences (Boston, MA). The Boc-protected tin precursor of 1,3,4-SGMIB and radioiodinated SGMIB were synthesized as reported previously [15, 16]. Aluminum-backed sheets (Silica gel 60 F254) used for analytical TLC and silica gel 60 for normal-phase column chromatography were obtained from EM Science (Gibbstown, NJ). In some cases, chromatography was also performed with the Biotage Isolera chromatography system (Charlotte, NC) using their pre-packed columns. Preparative thick layer chromatography was used for small-scale purification with plates obtained from Whatman (Clifton, NJ) or EM Science. High pressure liquid chromatography (HPLC) was performed using a Beckman Gold HPLC system equipped with a Model 126 programmable solvent module, a Model 166 NM variable wavelength detector, a Model 170 radioisotope detector and a Beckman System Gold remote interface module SS420X; data were acquired using the 32 Karat® software (Beckman Coulter, Inc., Brea, CA). Recently, the gamma detector in this system was replaced with a ScanRam RadioTLC scanner/HPLC detector combination (LabLogic; Brandon, Fl) and later radio HPLC analyses were performed with that detector. A 4.6 × 250 mm Partisil silica column (10 μm; Alltech, Deerfield, IL) was used for normal phase HPLC. PD-10 desalting columns for gel filtration were purchased from GE Healthcare (Piscataway, NJ). Instant thin layer chromatography (ITLC) was performed using silica gel impregnated glass fiber sheets (Pall Corporation, East Hills, NY) eluted with PBS, pH 7.4. Developed sheets were analyzed for radioactivity either using the TLC scanner described above or cutting the sheet into small strips and counting them in an automated gamma counter (LKB 1282, Wallac, Finland or Perkin Elmer Wizard II, Shelton, CT). Proton NMR spectra were obtained on a Varian 400 MHz NMR spectrometer (Palo Alto, CA); chemical shifts are reported in δ units using the residual solvent peak as reference. Mass spectra were recorded using an Agilent LC/MSD Trap for electrospray ionization (ESI) LC/MS or an Agilent LCMS-TOF with DART, a high resolution mass spectrometer used for ESI, DART and LC-MS.
2.2. Nb, mAb, cells, and culture conditions
Trastuzumab (Tras; Herceptin®) was purchased from Genentech (San Francisco, CA) and the anti-HER2 Nb [17, 18], 5F7 was a gift from Ablynx (Ghent, Belgium). All reagents used for cell studies were obtained from Invitrogen (Grand Island, NY). BT474 human breast carcinoma cells were cultured in DMEM/F12 medium containing 10% fetal calf serum (FCS), streptomycin (100 μg/mL), and penicillin (100 IU/mL) (Sigma Aldrich, MO). Cells were cultured at 37°C in a humidified incubator under 5% CO2 with media changed every two days. When about 80% confluent, cells were sub-cultured by trypsinization (0.05 % Trypsin- EDTA).
2.3. Synthesis of Boc-protected iodo standard and tin precursor-Method 1
2.3.1. 3-Amino-5-(methoxycarbonyl)benzoic acid (1)
A suspension of 10% Pd/C (0.35 g, 3.33 mmol) in ethanol (2 mL) was carefully added to a solution of 3-(methoxycarbonyl)-5-nitrobenzoic acid (2.5 g, 11.1 mmol) in ethanol (5 mL) in an argon-purged flask. Argon was displaced by hydrogen by repeated evacuation and hydrogen flushes and the mixture was stirred at 20°C for 5 h under a hydrogen atmosphere (balloon). After reduction reaction was completed, the reaction mixture was carefully filtered through a bed of Celite in a fritted funnel. Ethanol was evaporated from the filtrate, and the crude product chromatographed (40:60 ethyl acetate:hexanes) to yield 2.03 g (94%) of white solid: 1H NMR (CD3OD): δ 3.88 (s, 3H), 7.51 (d, 2H), 7.89 (s, 1H). MS (ESI), m/z: (positive mode) 196.1 (M+H)+; (negative mode) 194.1 (M-H)-.
2.3.2. 3-Iodo-5-(methoxycarbonyl)benzoic acid (2)
Aqueous hydriodic acid (57 wt %, 0.85 mL) was added to a solution of 3-amino-5-(methoxycarbonyl)benzoic acid (1; 1.96 g, 10.10 mmol) in THF (3 mL) at 0°C, and the mixture stirred for 5 min. A solution of sodium nitrite in water (2.08 g, 30.14 mmol; 3 mL) was added drop wise to the above mixture, and the stirring continued for another 40 min at 0 °C. A solution of potassium iodide (5.00 g, 30.12 mmol) in 3 mL of water was added to the resultant diazonium intermediate, and the mixture stirred for 2 h at 20°C. The reaction mixture was diluted with 300 mL of water and extracted with EtOAc (3 × 150 mL). The combined organic extract was washed with brine, dried with Na2SO4, and concentrated under reduced pressure. The crude product was purified by chromatography (1:1 ethyl acetate:hexanes) to yield 0.2 g (26%) of white solid: 1H NMR (CDCl3): δ 3.86 (s, 3H), 7.84 (s, 1H), 7.88 (s, 1H), 8.18 (s, 1H). MS (ESI), m/z: (positive mode) 329 (M+Na)+; (negative mode) 305 (M-H)-.
2.3.3. Methyl 3-(hydroxymethyl)-5-iodobenzoate (3)
Borane dimethyl sulfide complex (0.20 g; 2.63 mmol) was added drop wise to a solution of 3-iodo-5-(methoxycarbonyl)benzoic acid (2; 0.70 g, 2.29 mmol) in anhydrous chloroform (10 mL), and the mixture refluxed for 3 h. The mixture was partitioned between 0.1 M potassium carbonate and ethyl acetate, and the pooled ethyl acetate extract was washed with brine, and dried with Na2SO4. The crude product was purified by chromatography using 3:7 ethyl acetate:hexanes to yield 0.44 g (78%) of an oil: 1H NMR (CDCl3): δ 3.87 (s, 3H), 4.65 (s, 2H), 7.86 (s, 1H), 7.90 (s, 1H), 8.21 (s, 1H). GCMS m/z: 292 (M+H)+.
2.3.4. Methyl 3-iodo-5-((tosyloxy)methyl)benzoate (4)
A solution of tosyl chloride (0.39 g, 2.05 mmol) in 1 mL dichloromethane was added drop wise to a mixture of methyl 3-(hydroxymethyl)-5-iodobenzoate (3; 0.40 g, 1.37 mmol), triethylamine (0.21 g, 2.10 mmol), and DMAP (33 mg, 0.28 mmol) in dichloromethane (5 mL) at 0°C. The reaction mixture was stirred at 0°C for 30 min and the temperature gradually increased to 15°C over 2 h. Volatiles were removed and the product was isolated by chromatography using 3:7 ethyl acetate:hexanes to yield 0.49 g (80%) of an oil: 1H NMR (CDCl3): δ 2.33 (s, 3H), 3.79 (s, 3H), 4.93 (s, 2H), 7.21 (s, 1H), 7.23 (s, 1H), 7.60 (s, 1H), 7.64 (s, 1H), 7.66 (s, 1H), 7.74 (s, 1H), 8.15 (s, 1H). LCMS (ESI), m/z: 469.2 (M+Na)+. HRMS (DART) Calcd for C16H19INO5S (M+NH4)+: 464.0029. Found: 464.0015 ± 0.0001 (n=4).
2.3.5. Methyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (5)
Potassium tert-butoxide (1M, 1.65 mL, 1.65 mmol) in THF was taken in a dry flask and THF was evaporated with a stream of argon. N1, N2-bis(tert-butoxycarbonyl)guanidine (0.43 g, 1.65 mmol) was added to the flask followed by DMF (2 mL). Methyl 3-iodo-5-((tosyloxy)methyl)benzoate (4; 0.49 g, 1.10 mmol) in DMF (2 mL) was added to the above and the mixture stirred at 20°C for 30 min. The reaction mixture was worked up by partitioning between ethyl acetate and water, and the crude compound was purified by chromatography using 3:7 ethyl acetate:hexanes to yield 0.30 g (51%) of an oil: 1H NMR (CDCl3): δ 1.4 (s, 9H), 1.49 (s, 9H), 3.89 (s, 3H), 5.11 (s, 2H), 7.94 (s, 2H), 8.23 (s, 1H). LCMS (ESI), m/z: 534.3 (M+H)+. HRMS (DART) Calcd for C20H29IN3O6 (M+H) + : 534.1101. Found: 534.1093 ± 0.0006 (n=4).
2.3.6. 3-((1,2-Bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoic acid (6)
A solution of NaOH (544 mg, 13.6 mmol) in water (3 mL) was added to methyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (6; 290 mg, 0.54 mmol) in 10 mL of 2:1 dioxane:water and the mixture stirred at 20°C for 6 h. Dioxane was evaporated using a rotary evaporator under aspirator vacuum, and the residual material partitioned between ethyl acetate and sodium acetate buffer, pH 5.5. Ethyl acetate was evaporated from dried organic layer, and the crude material was subjected to column chromatography using 100% ethyl acetate to afford 17.9 mg (6%) of an oil: 1H NMR (CDCl3): δ 1.38 (s, 9H), 1.48 (s, 9H), 5.11 (s, 2H), 7.97 (s, 2H), 8.26 (s, 1H). LCMS (ESI), m/z: (positive mode) 520 (M+H)+, (negative mode) 518 (M-H)-. HRMS (DART) Calcd for C19H27IN3O6 (M+H)+ : 520.0945. Found: 520.0942 ± 0.0004 (n=4).
2.3.7. N-Succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (7)
EDC (7.88 mg, 0.04 mmol) was added to a mixture of 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoic acid (6; 17.8 mg, 0.03 mmol) and N-hydroxysuccinimide (4.73 mg, 0.04 mmol) in dry dichloromethane (5 mL), and the mixture was stirred at 20°C for 2 h. The precipitated urea derivative was filtered through a fritted funnel, washed with ethyl acetate, and the filtrate was concentrated. Preparative TLC purification using 1:1 ethyl acetate:hexanes afforded 12.8 mg (60%) of a white solid: 1H NMR (CDCl3): δ 1.42 (s, 9H), 1.48 (s,9H), 2.88 (s,4H), 5.15 (s, 2H), 8.03 (s, 1H), 8.11 (s, 1H), 8.32 (s, 1H). LCMS (ESI), m/z: 617.3 (M+H)+. HRMS (DART) Calcd for C23H30IN4O8 (M+H)+ : 617.1108. Found: 617.1100 ± 0.0004 (n=4).
2.3.8. Methyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate (8)
A mixture of methyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (5; 116 mg, 0.22 mmol), hexamethylditin (0.71 g, 2.18 mmol), and bis(triphenylphosphine)palladium(II) chloride (14 mg, 0.05 mmol) in 3 mL of dioxane was stirred at 80°C for 15 min. The cooled reaction mixture was filtered through a bed of Celite in a fritted funnel, and dioxane was evaporated from the filtrate. The crude material was subjected to preparative TLC using 3:7 ethyl acetae:hexanes to yield 80.1 mg (52%) of an oil: 1H NMR (CDCl3): δ 0.22 (s, 9H [119Sn-H d]), 1.31 (s, 9H), 1.41 (s, 9H), 3.82 (s, 4H), 5.09 (s, 2H), 7.66 (s, 1H), 7.84 (s,1H), 7.99 (s, 1H). LCMS (ESI), m/z: (negative mode) 569.1 (M-H)-. HRMS (DART) Calcd for C23H38N3O6Sn (M+H)+: 572.1783. Found: 572.1780 ± 0.0007 (n=4).
2.3.9. 3-((1,2-Bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoic acid (9)
A solution of NaOH (140 mg, 3.51 mmol) in 2 mL of water was added to a solution of methyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate (8; 80 mg, 1.4 mmol) in 4 mL of dioxane, and the heterogeneous mixture was stirred at 20°C for 6 h. Dioxane was evaporated, and the residual material partitioned between ethyl acetate and sodium acetate buffer, pH 5.5. Ethyl acetate was evaporated and the crude product purified by preparative TLC using 100% ethyl acetate to afford 28.5 mg (37%) of a pale yellow oil: 1H NMR (CDCl3): δ 0.31 (s, 9H [119Sn-H d]), 1.41 (s, 9H), 1.50 (s, 9H), 5.19 (s, 2H), 7.80 (s, 1H), 7.98 (s,1H), 8.09 (s, 1H). LCMS (ESI), m/z: (positive mode) 557 (M+H)+; (negative mode) 555 (M-H)-. HRMS (DART) Calcd for C22H36N3O6Sn (M+H)+: 558.16. Found: 558.1626 ± 0.0002 (n=4).
2.3.10. N-Succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)-benzoate (10)
EDC (11.58 mg, 0.06 mmol) was added to 28 mg (0.05 mmol) of 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoic acid (9) in 3 mL anhydrous dichloromethane followed by N-hydroxysuccinimide (6.95 mg, 0.06 mmol). The mixture was stirred at 20°C for 2 h. Dichloromethane was evaporated using a rotary evaporator and the resultant crude mixture was purified by preparative TLC using 1:1 ethyl aceatate:hexanes to afford 18.8 mg (57%) of a colorless oil: 1H NMR (CDCl3): δ 0.30 (s, 9H [119Sn-H d]), 1.4 (s, 9H), 1.47 (s, 9H), 2.88 (s, 4H), 5.16 (s, 2H), 7.87 (s, 1H), 7.98 (s,1H), 8.09 (s, 1H). LCMS (ESI), m/z: 654.3 (M+H)+. HRMS (DART) Calcd for C26H39N4O8Sn (M+H)+: 655.1790. Found: 655.1794 ± 0.0004 (n=4).
2.4. Synthesis of Boc-protected iodo standard and tin precursor-Method 2
2.4.1. 5-Iodoisophthalic acid (11)
A mixture of dimethyl 5-iodoisophthalate (Matrix Scientific, Columbia, SC; 6.00 g, 18.75 mmol) and lithium hydroxide (3.15 g, 132 mmol) were taken in 100 mL of 1:1 water:dioxane and the mixture stirred at 20°C for 2 h. Dioxane was evaporated, and the residual material was treated with 1M HCl to adjust the pH to 4. The resultant precipitate was extracted with ethyl acetate, the ethyl acetate layer dried with MgSO4, and the volatiles were evaporated to give a white powder. The solid was crystallized with isopropyl alcohol to afford 5.32 g, (97%) as a white powder. 1H NMR (CD3OD) 8.55 (d, 2H), 8.60 (d, 1H). LCMS (ESI) m/z: (negative mode) 291 (M-H)-.
2.4.2. 3-Iodo-5-((2-(trimethylsilyl)ethoxy)carbonyl)benzoic acid (12)
A mixture of 5-iodoisophthalic acid (11; 5.60 g, 19.18 mmol), EDC (4.41 g, 23.01 mmol), and DMAP (0.23 g, 1.92 mmol) in 50 mL of DMF was stirred at 20°C until the reaction mixture became homogeneous. The esterification reaction was initiated by the gradual addition of 2-(trimethylsilyl) ethanol (2.27 g, 19.18 mmol) over a period of 20 min, and the reaction was allowed to proceed for another 16 h at 20°C. The crude mixture was partitioned between water and ethyl acetate. The pooled ethyl acetate extract was dried over MgSO4, filtered, and ethyl acetate from filtrate evaporated. The residue was chromatographed using 4:1 hexanes:ethyl acetate to afford 1.9 g (25.3%) of a white solid: 1H NMR (CDCl3): δ 0.07 (s, 9H) 1.12 (m, 2H), 4.43 (m, 2H), 8.58 (s, 2H), 8.67 (s, 1H). LCMS (ESI), m/z: 393.3(M+H)+. HRMS (DART; negative mode) Calcd for C13H16IO4Si (M-H)-: 390.9863. Found: 390.9867 ± 0.0003 (n=4).
2.4.3. 2-(Trimethylsilyl)ethyl 3-(hydroxymethyl)-5-iodobenzoate (13)
A 1M solution of borane dimethyl sulfide in DCM (6.5 mL, 6.5 mmol) was gradually added to a solution of 3-iodo-5-((2 (trimethylsilyl)ethoxy)carbonyl)benzoic acid (12; 1.700 g, 4.33 mmol) in 75 mL of anhydrous THF at -20°C. The reaction mixture was gradually warmed to 20°C, stirred under argon atmosphere overnight, poured over ice, and partitioned between ethyl acetate and water. The pooled ethyl acetate solution was dried over MgSO4, filtered, and the filtrate concentrated by a rotary evaporator. The crude product was chromatographed using 5:1 hexanes:ethyl acetate to give 1.2 g (73.2%) of an oil: 1H NMR (CDCl3): δ 0.06 (s, 9H), 1.11 (m, 2H), 4.39 (m, 2H), 4.68 (s, 2H), 7.9 (s, 1H), 7.94 (s, 1H), 8.25 (s, 1H). LCMS (ESI), m/z: 379.3 (M+H)+. HRMS (DART) Calcd for C13H19INaO3Si (M+Na)+ : 401.0046. Found: 401.0036 ± 0.0001 (n=4).
2.4.4. 2-(Trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (14)
Methanesulfonyl chloride (0.27 mL, 3.47 mmol) was added to a solution of 2-(trimethylsilyl)ethyl 3-(hydroxymethyl)-5-iodobenzoate (13; 1.20 g, 3.17 mmol) in 35 mL of dichloromethane, and the resultant solution cooled to 0-5°C. Triethylamine (0.49 mL, 3.49 mmol) was added to the above. The reaction mixture was allowed to gradually warm to 20°C, stirred for 1 h, and partitioned between water and ethyl acetate. The pooled organic layers were dried over MgSO4, filtered, and the filtrate concentrated to dryness to give a low melting solid. A 1M solution of potassium tert-butoxide in THF (4.76 mL, 4.76 mmol) was added to 1,3-bis(tert-butoxycarbonyl)guanidine (1.23 g, 4.76 mmol) in 35 mL of DMF and the mixture stirred at 20°C for 30 min. The resultant solution of 1,3-bis(tert-butoxycarbonyl)guanidine potassium salt was added to the crude mesylate prepared above, and the reaction mixture stirred at 20°C for 17 h. The reaction mixture was partitioned between water and ethyl acetate, and the pooled ethyl acetate layer dried with MgSO4, filtered, and concentrated. The crude compound was purified via chromatography using 5:1 hexanes:ethyl acetate to afford 1.1 g (50.9%) of a solid: 1H NMR (CDCl3): δ 0.05 (s, 9H), 1.10 (m, 2H), 1.34 (s, 9H), 1.43 (s, 9H), 4.37 (m, 2H), 5.11 (s, 2H), 7.91 (s, 2H), 8.23 (s, 1H). LCMS (ESI), m/z: 620.2 (M+H)+. HRMS (DART) Calcd for C24H39IN3O6Si (M+H)+ : 620.1653. Found: 620.1662 ± 0.0001 (n=4).
2.4.5. N-Succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (7)
Tetrabutyl ammonium fluoride in THF (1M; 0.45 mL, 0.45 mmol) was added to a solution of 2-(trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (14; 210 mg, 0.34 mmol) in 25 mL of THF at 0-5°C under argon, and the mixture stirred at 20°C overnight. THF was evaporated, and the residual material partitioned between ethyl acetate and water. The combined ethyl acetate extract was dried with MgSO4, and concentrated. N-hydroxysuccinimide, (65 mg, 0.56 mmol), DMAP (11 mg, 0.09 mmol), and EDC (108 mg, 0.56 mmol) were added to a solution of the above crude intermediate in 25 mL of ethyl acetate and the mixture stirred at 20°C overnight. Water was added and the layers separated. The organic layer was washed once with NaHCO3, dried with MgSO4, and concentrated. The crude material was purified by chromatography using 2:1 hexanes:ethyl acetate to afford 119 mg (57%) of a white solid: 1H NMR (CDCl3): δ 1.45 (s, 9H), 1.50 (s, 9H), 2.91 (s, 4H), 5.14 (s, 2H), 8.07 (s, 1H), 8.13 (s, 1H), 8.35 (s, 1H). LCMS(ESI), m/z: 617.3 (M+H)+. HRMS (DART) Calcd for C23H30IN4O8 (M+H)+ : 617.1108. Found: 617.1100 ± 0.0004 (n=4).
2.4.6. 2-(Trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl) benzoate (15)
A mixture of 2-(trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (14; 1.00 g, 1.61 mmol), hexamethylditin (2.64 g, 8.07 mmol), and bis(triphenylphosphine)palladium(II) chloride (1.13 g, 1.61 mmol) in 40 mL dioxane was heated at 100°C for 1 h, and then concentrated on rotary evaporator. The crude product was mixed with ether, and insolubles filtered through a bed of Celite. The residue, obtained from the evaporation of ether from the filtrate, was purified via chromatography using 5:1 hexanes:ethyl acetate to afford 0.44 g (42%) of an oil: δ 0.06 (s, 9H), 0.28 (s, 9H [119Sn-H d]), 1.10 (m,2H), 1.38 (s, 9H), 1.47 (s, 9H), 4.39 (m, 2H), 5.12 (s, 2H), 7.7 (s, 1H), 7.88 (s, 1H), 8 (s, 1H). LCMS (ESI), m/z: 658.2 (M+H)+. HRMS (DART) Calcd for C27H48N3O6SiSn (M+H)+ : 658.2334. Found: 658.2352 ± 0.0002 (n=4).
2.4.7. N-Succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl) benzoate (10)
Tetrabutyl ammonium fluoride in THF (1M; 0.168 mL, 0.168 mmol) was added to a solution of 2-(trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate (15; 0.1 g, 0.15 mmol) in 20 mL of THF at 0-5°C. The solution was stirred overnight under argon. THF was evaporated, and the residual material was partitioned between ethyl acetate and water. The pooled ethyl acetate fractions were dried over MgSO4, filtered, and ethyl acetate evaporated from the filtrate. N-hydroxysuccinimide (26.3 mg, 0.23 mmol), DMAP (4.65 mg, 0.04 mmol), and EDC (43.8 mg, 0.23 mmol) were added to the above crude intermediate in 25 mL of dry ethyl acetate, and the mixture stirred overnight at 20°C. Sodium bicarbonate solution was added and the layers separated. The aqueous layer was further extracted with ethyl acetate and the combined ethyl acetate layers were washed with brine, dried with MgSO4, and filtered. Ethyl acetate was evaporated from the filtrate and the crude material was purified by preparative TLC using 2:1 hexanes:ethyl acetate to yield 42 mg (42.2%) of a colorless oil: 1H NMR (CDCl3): δ 0.30 (s, 9H [119Sn-H d]), 1.40 (s, 9H), 1.47 (s, 9H), 2.89 (s, 4H), 5.16 (s, 2H), 7.87 (s, 1H), 7.99 (s, 1H), 8.09 (s, 1H). LCMS(ESI), m/z: 654.3 (M+H)+. HRMS (FAB+) Calcd for C26H39N4O8Sn (M+H)+: 655.1790. Found: 655.1794 ± 0.0004 (n=4).
2.5. Radiochemistry
2.5.1. N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-[131I]iodobenzoate ([131I]7)
Solutions of HOAc in CHCl3 {3% (v/v), 2 μL}, tert-butyl hydroperoxide (TBHP) in CHCl3 {30% (w/v), 5 μL) — prepared by mixing 70% (w/v) aqueous solution of TBHP with CHCl3 and drying twice with Na2SO4, and 10 in CHCl3 (15 μL) were added to a vial containing 0.5 - 4 mCi of 131I in 2-4 μL of 0.1 N NaOH. The mixture was stirred at 20°C for various periods of time and injected onto a normal phase HPLC column that was eluted isocratically with 25/75 (v/v) ethyl acetate/hexanes containing 0.2% (v/v) HOAc at a flow rate of 1mL/min. The HPLC fractions containing the product [131I]7 (tR = 28 min) were isolated and the solvents evaporated to dryness. Radiochemical yield of Boc2-iso-[131I]SGMIB was studied as a function of precursor amount using a 1 h reaction time, and as a function of reaction time using 50 μg of precursor. Radiochemical yields for the synthesis of Boc2-[131I]SGMIB and Boc2-iso-[131I]SGMIB were compared by conducting labeling reactions under identical conditions using 50 μg of the respective precursor and a reaction time of 30 min. For some studies Boc2-SGMIB was also labeled using 125I.
2.5.2. N-Succinimidyl 3-guanidinomethyl-5-[131I]iodobenzoate (iso-[131I]SGMIB)
Solvent was evaporated from the HPLC fractions containing [131I]7 using an argon stream, and the residual radioactivity was transferred to a half-dram vial with small volumes of ethyl acetate and then evaporated to dryness. Trifluoroacetic acid (100 μL) was added to the above vial, the vial vortexed, and left at 20°C for 10 min. Trifluoroacetic acid was evaporated, and to insure its complete removal, 3 × 50 μL of ethyl acetate was added, and evaporated each time.
2.5.3. Radiolabeling of trastuzumab and 5F7 Nb using [*I]SGMIB or iso-[*I]SGMIB
A solution of protein in 0.1 M borate buffer, pH 8.5 (50 μL, 2 mg/mL) was added to about 1-2 mCi of the dry prosthetic agent in a half-dram vial, and the mixture kept at 20°C for 20 min. Labeled protein was isolated by gel filtration though a PD-10 column (GE Healthcare, Piscataway, NJ) that was eluted with PBS, pH 7.4. The conjugation efficiency was in the range of 24.5% – 40.0%, 10.1% - 42.4%, 38.4% - 42.2%, and 27.5% - 44.2% for iso-[131I]SGMIB-Nb, [131I]SGMIB-Nb, iso-[131I]SGMIB-Tras, and [131I]SGMIB-Tras, respectively.
2.6. Evaluation of protein-associated radioactivity and immunoreactivity
Protein-associated radioactivity was determined by trichloroacetic acid (TCA) precipitation, ITLC, and SDS-PAGE. TCA precipitability was determined in a paired-label format by incubating about 5 ng each of Tras (or Nb) labeled with [125I]SGMIB or iso-[131I]SGMIB, 800 μL of 2% bovine serum albumin (BSA), and 100 μL of 20% TCA at 20°C for 15 min. Protein-associated radioactivity was also evaluated by ITLC using PBS, pH 7.4 as the eluent. Under these conditions, the intact protein remains at the origin (Rf = 0) and both acylation agents elute with an Rf value of 0.7 – 0.8. The integrity of labeled proteins was further assessed by SDS-PAGE under nonreducing conditions and subsequent phosphor imaging as previously described for Nb [18]. The immunoreactivity of the labeled proteins was determined by the Lindmo assay using magnetic beads coated with the extracellular domain of HER2 or as control for nonspecific binding, with BSA [8, 18]. These assays were performed in a paired-label format for each HER2-targeted protein by incubating their radiolabeled SGMIB and iso-SGMIB conjugates (about 5 ng each) with three doubling concentrations of both positive and negative beads, and immunoreactive fraction determined as previously described [21].
2.7. Internalization Assays
The ability of the two prosthetic groups for trapping radioiodine within HER2-expressing cells after internalization was determined by paired-label assays using BT474 breast carcinoma cells. Cells were plated at a density of 8 × 105 cells per well in 3 mL medium in 6-well plates. After overnight incubation at 37°C, cells were incubated at 4°C for 30 min. Medium was replaced with fresh medium containing labeled protein pair {5 nM each; Tras (or Nb) [125/131I]SGMIB and iso-[125/131I]SGMIB conjugates} and the cells were incubated at 4°C for 1 h. Parallel experiments also were performed with a 100-fold molar excess of Tras to determine nonspecific uptake. The fraction of radioiodine activity initially bound to the cells that was present on the cell surface, intracellular compartment and leaked into cell culture supernatant as a function of time (1-24 h) was determined as described previously [17, 18]. The fraction of radioactivity in the cell culture supernatants that remained protein-associated was determined by TCA. Triplicate samples were used for each time point, and in the case of labeled Tras, the entire experiment was repeated twice.
2.8. Biodistribution in mice bearing BT474M1 xenografts
Animals experiments were performed following the guidelines established by the Duke University Institutional Animal Care and Use Committee. BT474M1 xenografts were established as reported before [17]. and the studies were initiated when the tumor sizes were about 300-500 mm3. Groups of five animals were injected via the tail vein with about 5 μCi each of iso-[125I]SGMIB-Tras (2.5 μg; 17.2 pmol) and [131I]SGMIB-Tras (1.8 μg; 12.4 pmol). Animals were killed by an overdose of isofluorane at 4, 12, 24 and 48 h, and tumor and other tissues of interest along with blood and urine were collected. Blot-dried solid tissues, blood and urine were weighed and counted for radioiodine activity along with injection standards in an automated gamma counter using the 125I/131I dual-label program. From these, percentages of injected dose- per organ and per gram of tissue were computed. The statistical significance of differences between the uptake of two radioisotopes in each tissue was calculated using the Microsoft Excel paired Student t test; the difference was considered to be significant for p values less than 0.05.
3. Results and discussion
The guanidine-substituted acylation agent, SGMIB, has excelled as a residualizing labeling method for use with internalizing mAbs and their fragments. Higher tumor targeting in vitro and in vivo has been observed when mAbs, their fragments and peptides were radiohalogenated using this template compared to the same biomolecule radioiodinated by the direct electrophilic approach [15, 17-19, 22-24]. Moreover, when SGMIB was utilized to radioiodinate the HER2-targeted Nb, tumor uptake and retention was more than two fold higher than those observed previously with radionuclide/labeling method/Nb combination [17]. Unfortunately, potential clinical translation of the SGMIB method has been impeded by relatively low radiolabeling yield for the synthesis of the intermediate, Boc2-SGMIB, which is about 65% at best. Hypothesizing that low yields might be due to the presence of the relatively bulky Boc2-guanidinomethyl group at the ortho position of the tin moiety in the precursor, we designed an isomeric molecule wherein the Boc2-guanidinomethyl group was moved from the ortho to the meta position.
Two approaches were evaluated for the synthesis of both the Boc2-iso-SGMIB standard and the corresponding tin precursor. The first involved the intermediate compound 3 (Scheme 1). Our efforts to synthesize this intermediate following a reported method [25] were not fruitful. In the published method, the starting material, 3-(methoxycarbonyl)-5-nitrobenzoic acid was subjected to the following sequence of reactions: borane dimethyl sulfide (BMS)-mediated reduction of acid to alcohol, hydrogenation of nitro to amino, and finally conversion of amino to iodo. Unfortunately, the yield for hydrogenation was only 38%. Assuming that this might be due to poisoning of the catalyst by residual sulfur-containing compound(s) generated from BMS, we changed the reaction sequence to hydrogenation, amino to iodo conversion and finally, acid to alcohol transformation. Although hydrogenation yield (94%) improved considerably, due to the modest yield for the Sandmeyer reaction, the overall yield for the three steps, 19%, was substantially lower than the 53% yield reported by Takano et al. [25]. The alcohol function in intermediate 3 was converted to a tosyloxy group, which was displaced with the Boc2-guanidine by adaptation of a protocol reported [26] to render compound 5. In the transesterification of 5 to 7, the isolated yield for the acid intermediate 6, was only 6% presumably due to the susceptibility of Boc groups to the acid-base work up conditions. Esterification of 6 under typical conditions yielded the standard of Boc2-iso-SGMIB in 60% yield for that step.
Scheme 1.
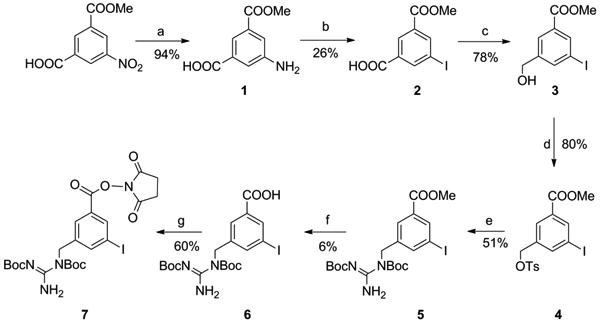
a) H2, Pd/C b) NaNO2, HI c) BMS d) TsCI, TEA, DMAP e) Bis-Boc2-guanidine, Potassium tert-butoxide f) NaOH g) A/-hydroxysuccinimide, EDC.
Scheme 2 shows the method used for the synthesis of N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate 10, the tin precursor for the synthesis of iso-[*I]SGMIB, starting from the intermediate 5. Compound 5 was subjected to the Stille reaction by treatment with hexamethyl ditin and a palladium catalyst to yield 8. Alkaline hydrolysis of 8 yielded 9, and the acid was re-esterified with N-hydroxysuccinimide using EDC to yield the desired tin precursor 10.
Scheme 2.
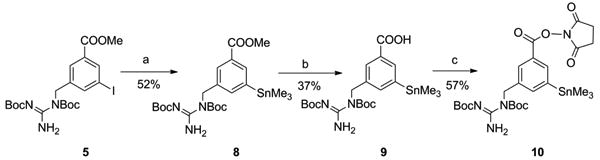
a) Hexamethylditin, (Ph3P)2PdCl2 b) NaOH c) N-hydroxysuccinimide, EDC
Due to the difficulty encountered in the conversion of 5 to 6, and as a result, the poor cumulative yield obtained with the synthetic approach described above, we pursued an alternative strategy for the synthesis of 7 and 10 starting from dimethyl 5-iodoisophthalate (Scheme 3). Instead of the methyl ester employed in Scheme 1, a trimethylsilylethyl ester intermediate as used for the original SGMIB, was sought. Typically, trimethylsilylethyl protecting groups are removed by treatment with tetrabutyl ammonium fluoride (TBAF). Due to the solubility of tetrabutyl ammonium salts in organic solvents, acidification can be avoided and the salt can be used directly for re-esterification with N-hydroxysuccinimide. Dimethyl 5-iodoisophthalate was hydrolyzed as reported [27], and the resultant 5-iodoisophthalic acid was converted to its mono trimethylsilylethyl ester 12. Formation of the diester was unavoidable resulting in only modest yields for the desired monoester (25%). Reduction of the remaining free acid group in 12 rendered alcohol 13, which was converted to the corresponding mesylate. The crude mesylate intermediate was subjected to guanidinylation, yielding compound 14. Stannylation of 14 under Stille conditions gave compound 15. Both 14 and 15 were subjected to ester deprotection with TBAF and re-esterification to obtain the respective target compounds 7 and 10. The overall yield for the last two conversions was higher for the iodo (57%) than for the tin derivative (42%). This is most likely due to the lability of tin moiety for TBAF [28]. NMR and mass spectral data for all new compounds reported herein are consistent with their structures.
Scheme 3.
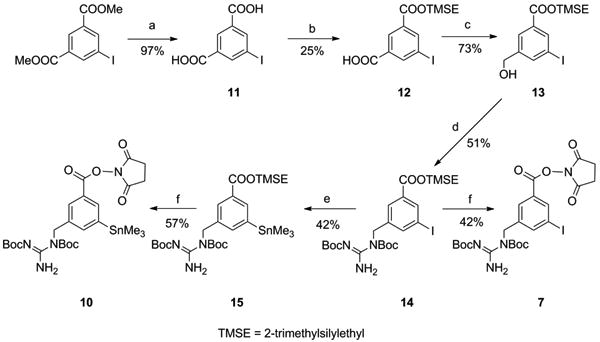
a) LiOH b) 2-Trimethylsilylethyl alcohol, EDC, DMAP c) BMS d) i) MsCI, TEA ii) Boc2-guanidine, Pot. tert-butoxide e) Hexamethylditin, (Ph3)2PdCl2 f) i) Tetrabutyl ammonium fluoride ii) N-hydroxysuccinimide, EDC, DMAP
To facilitate comparison, radioiodination of 10 was conducted using the same oxidizing agent and solvent that was used previously with the original SGMIB reagent (Scheme 4). Radiochemical yield for the synthesis of [131I]7 (Boc2-iso-[131I]SGMIB) was studied as a function of precursor amount and reaction time. Radiochemical yields, determined by HPLC, increased with increasing precursor amount when the reaction was conducted for 1 h (Figure 1A). The yields were 53.5 ± 5.9% (n=3), 61.0 ± 3.6% (n=9), 64.0 ± 2.7% (n=10), and 70.7 ± 2.0% (n=7) when 25 μg, 50 μg, 100 μg and 200 μg, of tin precursor was used, respectively. Except for 100 μg versus 50 μg, all the differences were statistically significant (p < 0.05). Although the mass amounts of radioiodide were substantially sub-stoichiometric, these results suggest that radioiodination yields for this reaction are dependent on precursor amount. Dependence of radioiodination yields on precursor amounts and the use of large molar excess of precursors relative to iodide are not uncommon [29, 30]. The labeling yields also increased with increasing time when a constant amount 50 μg of precursor was used (Figure 1B); radiochemical yields of 29.1 ± 3.7% (n=3), 50.3 ± 1.7 (n=3), 57.9 ± 5.3 (n=3), and 61.0 ± 4.0% (n = 9) were obtained when the reaction was performed for 5, 15, 30 and 60 min, respectively. Only the difference in yields between 30 and 60 min was not statistically significant. Taken together, although slightly higher yields were obtained when 200 μg of precursor was used, reasonable yields could be obtained using 50 μg precursor and a reaction time of 30 min.
Scheme 4.
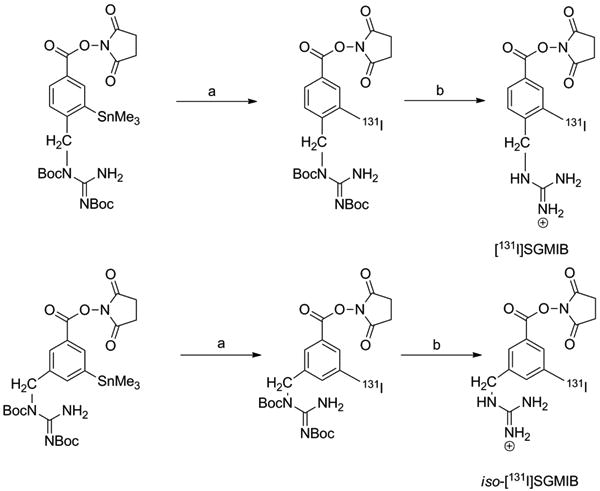
a) [131I]lodide, tert-butyl hydroperoxide, HOAc, CHCI3, 1 h b)TFA
Figure 1.
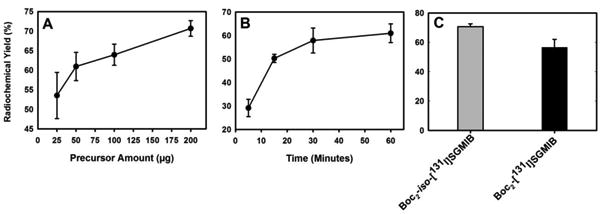
A) Radiochemical yields for the synthesis of Boc2-iso-[131I]SGMIB as a function of precursor amount (1h reaction time, n = 3-10). B) Radiochemical yields for the synthesis of Boc2-iso-[131I]SGMIB as a function of reaction time (50 μg precursor amount, n = 3-9). (C) Radioiodination yields: Boc2-iso-[131I]SGMIB versus Boc2-[131I]SGMIB (200 μg precursor, 1 h reaction time).
Next, to investigate whether, as hypothesized, better radioiodination yields will be obtained with the 1,3,5-isomer, reactions were performed under identical conditions using 200 μg of tin precursor of both 1,3,4- and 1,3,5-isomers and a 1-h reaction time. As shown in Figure 1C, the radiochemical yields for the synthesis of Boc2-iso-[131I]SGMIB (70.7 ± 2.0%; n=7) was about 25% higher (p < 0.05) than that obtained for Boc2-[131I]SGMIB (56.5 ± 5.5% n =3), consistent with steric hindrance playing a role.
As was done in the original SGMIB procedure, Boc groups from Boc2-iso-[*I]SGMIB were removed by TFA treatment before running the protein conjugation reactions. The efficiency of conjugation of iso-[131I]SGMIB to trastuzumab and 5F7 Nb was compared to that of [131I]SGMIB under identical conditions of pH, protein concentration, and temperature. Although yields for the conjugation of iso-[131I]SGMIB to Nb (33.1 ± 7.1%; n = 6) were higher than those obtained with [131I]SGMIB (28.9 ± 13.0%; n =6), the difference was not statistically significant. Likewise, with trastuzumab, iso-[131I]SGMIB demonstrated some advantage with respect to conjugation efficiency {45.1 ± 4.5% (n= 4) versus 34.8 ± 10.3% (n = 5) for [131I]SGMIB}; however, again, the difference was not statistically significant. These results are somewhat unexpected because from a steric effect perspective, if one isomer could have an advantage for this conjugation, it would be the 1,3,4-isomer. The two isomers differ with respect to the position of the guanidinomethyl moiety, which is further away from the reaction center carbonyl group in the 1,3,4-isomer, offering less steric hindrance. On the other hand, assuming the net effect of the guanidinomethyl group is electron-withdrawing, the electron density at the carbonyl carbon will be less in the 1,3,5-isomer compared to 1,3,4-isomer, making the conjugation more facile for the 1,3,5-isomer. When the two protein labeling steps — radioiodination and conjugation — are considered, based on the above results, iso-SGMIB will have a 40% and 60% advantage in available labeled product for 5F7 Nb and trastuzumab, respectively. This could be of particular practical significance when labeling with short-lived and/or expensive radiohalogens such as 123I 124I, and 211At.
The characteristics of 5F7 Nb and trastuzumab labeled using iso-[*I]SGMIB were compared in paired-label format to those of the same protein labeled using [*I]SGMIB. A TCA precipitation assay indicated that 97.4 ± 0.7% and 98.4 ± 0.3% of the radioactivity was associated with Nb labeled using iso-[131I]SGMIB and [125I]SGMIB, respectively; with trastuzumab, these values were 98.0 ± 0.1% and 98.5 ± 0.3%, respectively. ITLC analysis indicated that more than 97.5% of radioiodine activity was protein-associated for all four labeled conjugates. Labeled proteins were also analyzed by SDS-PAGE under non-reducing conditions. Quantitative phosphor imaging analysis indicated that more than 98% of the radioactivity was associated with the intact protein in each case. To determine whether labeling trastuzumab and 5F7 Nb with iso-[131I]SGMIB compromised binding to HER2, immunoreactive fractions were determined by Lindmo assay using magnetic beads coated with HER2 extracellular domain. The immunoreactive fraction calculated for iso-[131]SGMIB-Nb and [125I]SGMIB-Nb were 81.8 ± 1.4% and 84.5 ± 0.8%, respectively; corresponding values obtained for trastuzumab were 75.4 ± 1.4% and 74.1 ± 0.6%, respectively. Taken together, these results suggest that as expected, there are no isomer-dependent effects on quality control characteristics, with trastuzumab or the Nb.
We next evaluated whether changing the position of the guanidinomethyl group in the SGMIB template affected the residualizing ability of proteins labeled by this approach. Using paired-label assays on BT474 cells in vitro, the internalization and cell processing of iso-[131I]SGMIB-Tras and [125I]SGMIB-Tras were compared; iso-[125I]SGMIB-Nb and [131I]SGMIB-Nb also were likewise compared. In all cases, conjugate uptake was HER2-mediated as demonstrated by the fact that it was reduced to less than 1% of control values when the cells were co-incubated with 100-fold molar excess of unlabeled trastuzumab. With both proteins, the percentage of initially bound radioactivity that was cell-associated and that internalized was almost identical for the two isomer conjugates up to 6 h (Figure 2 and 3); however, the percentage of initially-bound radioactivity that was both cell-associated and internalized from iso-[125I]SGMIB-Nb at 24 h was only about 82-83% of those seen for [131I]SGMIB-Nb (HER2-specific intracellular radioactivity: 46.4 ± 1.3% versus 56.5 ± 2.5%; p < 0.05) (Figure 2B). As is the case with the Nb, the internalized radioactivity from iso-[131I]SGMIB-Tras at 24 h (Figure 3B) was considerably less than that from [125I]SGMIB-Tras (specific % initially bound: 29.7 ± 2.3% versus 45.9 ± 5.5%; p < 0.05). The percentage of initially bound radioactivity that was present in the intracellular compartment was higher for trastuzumab than with Nb for both labeling methods at early time points whereas at 24 h, the opposite behavior was observed. The first observation likely reflects the ability of the divalent trastuzumab molecule to form HER2 dimers on the cell surface, while the second might reflect differences in intracellular catabolism and/or receptor recycling between the two HER2-targeted entities.
Figure 2.
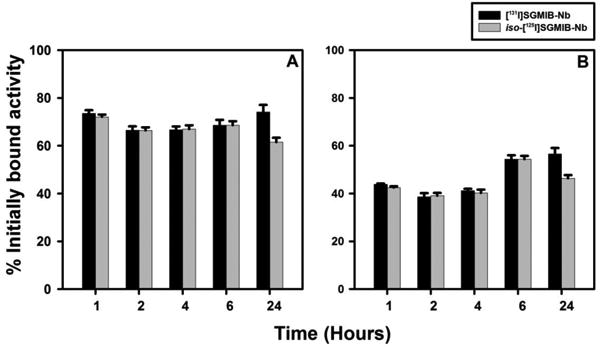
Paired-label in vitro internalization of iso-[125I]SGMIB-Nb (gray) and [131I]SGMIB-Nb (black) by BT474 cells. Cells were allowed to take up the radiotracers at 4°C for an hour, brought up to 37°C and processed at 1 h, 2 h, 4 h, 6 h, and 24 h as described in the text. Specific (in the absence of trastuzumab minus in the presence) total cell-associated radioactivity (cell surface-bound + internalized) (A) and intracellularly trapped activity (B).
Figure 3.
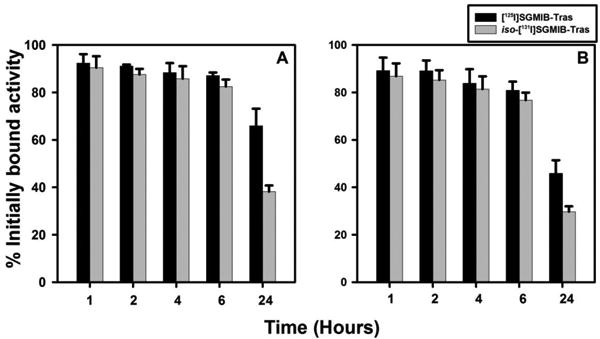
Paired-label in vitro internalization of iso-[131I]SGMIB-Tras (gray) and [125I]SGMIB-Tras (black) by BT474 cells. Cells were allowed to take up the radiotracers at 4°C for an hour, brought up to 37°C and processed at 1 h, 2 h, 4 h, 6 h, and 24 h as described in the text. Specific (in the absence of trastuzumab minus in the presence) total cell-associated radioactivity (cell surface-bound + internalized) (A) and intracellularly trapped activity (B).
The fraction of radioactivity present in the cell culture supernatants, as expected, exhibited complementary behavior to the trends observed for internalized radioactivity. The increased fraction of radioactivity in cell culture supernatant with time could be due to several processes including dissociation, recycling and/or generation of labeled catabolites. To investigate the later, the fraction of radioactivity in cell culture supernatants that remained protein-associated radioactivity was determined by TCA precipitation in the trastuzumab experiment. In concordance with the cell uptake data, the fraction of radioactivity that was protein-associated in cell culture supernatants was nearly identical for both trastuzumab conjugates through the 6-h time point. However, at 24 h, only 42.4 ± 1.7% of the radioactivity was protein-associated in the case of iso-[131I]SGMIB-Tras compared to 50.4 ± 1.9% for [125I]SGMIB-Tras suggesting either a higher rate of catabolism and/or cellular escape of low molecular weight catabolites for the iso-SGMIB conjugate.
We next evaluated the tissue distribution of the two radioiodinated trastuzumab conjugates in order to determine whether differences in residualizing ability observed in vitro were predictive of in vivo behavior. The experiment was performed in SCID mice bearing subcutaneous breast carcinoma xenografts established using BT474M1 cells, a clone of the original BT474 cell line, but more tumorigenic [31]. The uptake of radioiodine activity in tumor and normal tissues from the paired-label experiment are shown in Table 1. With the exception of large intestines at later time points, radioiodine levels from co-administered iso-[125I]SGMIB-Tras and [131I]SGMIB-Tras were quite similar in normal tissues at all time points. Tumor uptake of both agents increased up to 24 h but dropped at 48 h. Consistent with the in vitro results, the tumor uptake of iso-[125I]SGMIB-Tras was similar (97%) to that for [131I]SGMIB-Tras at 4 h but the difference increased as a function of time such that the uptake of iso-[125I]SGMIB-Tras was 9%, 16%, and 22% lower than that of [131I]SGMIB-Tras at 12 h, 24 h, and 48 h, respectively. The uptake in thyroid, an indicator of deiodination, was minimal and virtually identical for both tracers throughout the observation period (Figure 4), suggesting that the lower residualizing capacity of the iso-[125I]SGMIB-Tras conjugate cannot be explained by a greater susceptibility to deiodination.
Table 1.
Paired-label uptake of iodine radioactivity in various tissues after intravenous administration of trastuzumab labeled using [131I]SGMIB and iso[125I]SGMIB in SCID mice bearing BT474M1 xenografts.
| Tissue | %Injected Dose/grama | |||||||
|---|---|---|---|---|---|---|---|---|
| 4 h | 12 h | 24 h | 48 h | |||||
| SGMIB | iso-SGMIB | SGMIB | iso-SGMIB | SGMIB | iso-SGMIB | SGMIB | iso-SGMIB | |
| Liver | 19.77 ± 7.12 | 18.78 ± 6.62 | 12.59 ± 2.48 | 11.99 ± 2.39 | 7.01 ± 1.14 | 6.69 ± 1.10 | 5.24 ± 1.77 | 5.80 ± 1.83b |
| Spleen | 36.71 ± 14.50 | 37.80 ± 14.85 | 20.44 ± 5.91 | 20.69 ± 5.86b | 13.14 ± 3.77 | 13.76 ± 3.41b | 5.58 ± 1.75 | 5.80 ± 1.83 |
| Lung | 12.90 ± 3.77 | 13.71 ± 4.16 | 10.71 ± 3.97 | 11.29 ± 4.09 | 5.18 ± 0.91 | 5.40 ± 0.96 | 3.76 ± 0.99 | 4.01 ± 1.01 |
| Heart | 9.90 ± 3.73 | 10.30 ± 3.65 | 7.52 ± 2.04 | 7.85 ± 1.99b | 3.40 ± 0.52 | 3.52 ± 0.53 | 2.12 ± 0.66 | 2.22 ± 0.73b |
| Kidney | 11.16 ± 3.55 | 11.38 ± 3.70 | 7.85 ± 2.28 | 8.04 ± 2.32 | 3.84 ± 0.33 | 3.85 ± 0.35b | 2.63 ± 0.71 | 2.72 ± 0.71 |
| Stomach | 2.99 ± 1.39 | 3.14 ± 1.47b | 1.79 ± 0.62 | 1.83 ± 0.63 | 1.37 ± 0.41 | 1.42 ± 0.45b | 0.63 ± 0.33 | 0.72 ± 0.38 |
| Sm. Intestine | 5.22 ± 2.07 | 5.37 ± 2.16 | 2.73 ± 0.78 | 3.13 ± 0.82 | 1.63 ± 0.19 | 1.93 ± 0.25 | 1.05 ± 0.28 | 1.41 ± 0.44 |
| Intestine | 2.83 ± 0.64 | 2.66 ± 0.67 | 2.88 ± 0.90 | 3.66 ± 0.93 | 2.11 ± 0.26 | 3.29 ± 0.68 | 1.11 ± 0.23 | 1.93 ± 0.43 |
| Muscle | 1.92 ± 0.77 | 1.99 ± 0.79b | 2.16 ± 0.85 | 2.31 ± 0.88 | 1.47 ± 0.15 | 1.53 ± 0.17 | 0.89 ± 0.17 | 0.94 ± 0.17 |
| Blood | 27.80 ± 12.18 | 28.18 ± 12.53b | 19.93 ± 6.93 | 20.09 ± 6.63b | 9.05 ± 0.87 | 9.13 ± 0.96b | 5.35 ± 1.45 | 5.63 ± 1.59 |
| Bone | 3.72 ± 1.14 | 3.75 ± 1.04b | 2.68 ± 0.79 | 2.71 ± 0.86b | 1.66 ± 0.57 | 1.67 ± 0.56b | 1.12 ± 0.35 | 1.10 ± 0.33b |
| Brain | 1.11 ± 0.32 | 1.17 ± 0.36 | 0.84 ± 0.34 | 0.87 ± 0.35 | 0.33 ± 0.05 | 0.34 ± 0.06b | 0.20 ± 0.04 | 0.21 ± 0.04b |
| Tumor | 10.97 ± 4.34 | 10.65 ± 4.10 | 16.82 ± 5.20 | 15.26 ± 4.60 | 17.89 ± 3.24 | 15.08 ± 2.41 | 12.83 ± 2.22 | 10.06 ± 1.43 |
Mean ± SD (n = 5).
Difference between the uptake of two isotopes not statistically significant.
Figure 4.
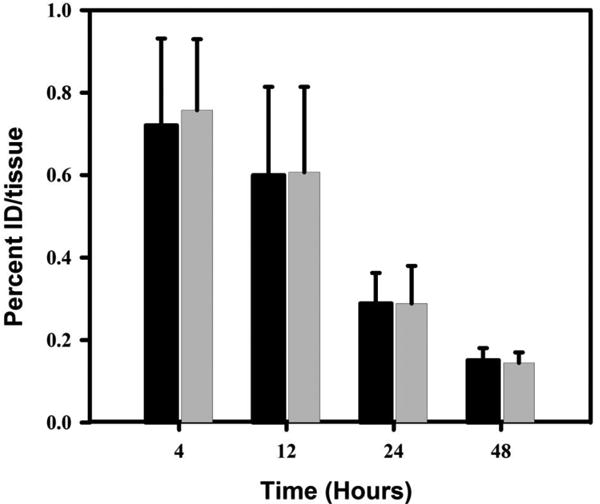
Uptake of radioiodine in thyroid after administration of iso-[125I]SGMIB-Tras and [131I]SGMIB-Tras in SCID mice bearing BT474M1 xenografts.
The results from these in vitro and in vivo experiments suggest that at later time points iso-SGMIB is not as effective as a residualizing agent compared with the original SGMIB prosthetic group. Although the reason for this behavior is not known at this time, one possibility is that iso-SGMIB structure is more susceptible to deiodination in biological milieu. By following the argument described (vide supra), the electron density at the iodine-bearing carbon should be less in the 1,3,4-isomer than in 1,3,5-isomer and thus iso-SGMIB should be less susceptible to deiodination by nucleophilic displacement. However, as noted above, the thyroid uptake values for both prosthetic groups were not significantly different.
Differences in biological characteristics of antibodies and their fragments labeled with isomeric prosthetic agents are not unprecedented. When antibodies were labeled with radiometals using stereoisomeric forms of a DTPA derivative, significant differences in the uptake in tumor and other tissues have been observed, which was attributed to the differential stability of the isomeric chelating agent-metal complexes [32, 33]. Moreover, we have labeled an anti-tenascin antibody 81C6 and a F(ab')2 fragment of OC 125, an antibody reactive to ovarian cancers using two isomeric forms of N-succinimidyl [*I]iodobenzoate (SIB) [34]. Deiodination in vivo, and uptake in thyroid and stomach as a result, were consistently lower for the meta-isomer when the biodistribution of these labeled proteins or their potential catabolites was performed. While the position of iodine in the isomeric SIB agents was different with respect to the carboxylate/carbamide moiety, it was in the meta-position in both SGMIB and iso-SGMIB. At any rate, as pointed out above, the thyroid uptake data clearly indicate that possible differential deiodination was not an issue. As noted before, differential catabolism and/or recycling, perhaps reflecting modification of different lysine residues on the proteins by the two isomers might be the reason for their different biological behavior. Determination of the position of labeling by mass spectrometry along with peptide mapping using trypsin digestion of protein[35] conjugated with unlabeled SGMIB derivatives, and/or labeled catabolites may throw light on this issue.
4. Conclusions
In summary, we have developed a method for the synthesis of iso-SGMIB, an analogue of the original residualizing agent SGMIB. Compared to SGMIB, 1.4- to 1.6-fold higher overall yields can be obtained using this agent for the labeling of internalizing mAbs and fragments. While both 5F7 Nb and trastuzumab could be radioiodinated using iso-[*I]SGMIB with the preservation of immunoreactivity, in vitro internalization assays using HER2-expressing BT474 breast cancer cells indicated that the residualizing ability of iso-SGMIB was not as prolonged as that observed with SGMIB. The tumor uptake obtained from biodistribution in vivo further corroborated this finding. Given the higher labeling efficiency obtained with iso-SGMIB, this residualizing agent might be of value for use with the shorter half-life radiohalogens 123I and 211At. In addition, further studies are warranted to investigate why the residualizing ability of the two isomers differed at later time points.
Acknowledgments
This work was supported by Grants CA32324 and CA154291 from the National Institutes of Health. The technical assistance of Xiao-Guang, M.D., with the animal studies is greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bast RC, Zalutsky MR, Kreitman RJ, Frankel AE. Monoclonal Serotherapy. 8th. Hamilton, Ontario: B.C. Decker; 2010. [Google Scholar]
- 2.Geissler F, Anderson SK, Venkatesan P, Press O. Intracellular catabolism of radiolabeled anti-mu antibodies by malignant B-cells. Cancer Res. 1992;52:2907–15. [PubMed] [Google Scholar]
- 3.Press OW, Shan D, Howell-Clark J, Eary J, Appelbaum FR, Matthews D, et al. Comparative metabolism and retention of iodine-125, yttrium-90, and indium-111 radioimmunoconjugates by cancer cells. Cancer Res. 1996;56:2123–9. [PubMed] [Google Scholar]
- 4.Ali SA, Eary JF, Warren SD, Badger CC, Krohn KA. Synthesis and radioiodination of tyramine cellobiose for labeling monoclonal antibodies. Int J Rad Appl Instrum B. 1988;15:557–61. doi: 10.1016/s0969-8051(88)80015-5. [DOI] [PubMed] [Google Scholar]
- 5.Moerlein SM, Dalal KB, Ebbe SN, Yano Y, Budinger TF. Residualizing and non-residualizing analogues of low-density lipoprotein as iodine-123 radiopharmaceuticals for imaging LDL catabolism. Int J Rad Appl Instrum B. 1988;15:141–9. doi: 10.1016/0883-2897(88)90080-3. [DOI] [PubMed] [Google Scholar]
- 6.Schilling U, Friedrich EA, Sinn H, Schrenk HH, Clorius JH, Maier-Borst W. Design of compounds having enhanced tumour uptake, using serum albumin as a carrier--Part II. In vivo studies. Int J Rad Appl Instrum B. 1992;19:685–95. doi: 10.1016/0883-2897(92)90103-6. [DOI] [PubMed] [Google Scholar]
- 7.Govindan SV, Mattes MJ, Stein R, McBride BJ, Karacay H, Goldenberg DM, et al. Labeling of monoclonal antibodies with diethylenetriaminepentaacetic acid-appended radioiodinated peptides containing D-amino acids. Bioconjugate Chem. 1999;10:231–40. doi: 10.1021/bc980075g. [DOI] [PubMed] [Google Scholar]
- 8.Foulon CF, Reist CJ, Bigner DD, Zalutsky MR. Radioiodination via D-amino acid peptide enhances cellular retention and tumor xenograft targeting of an internalizing anti-epidermal growth factor receptor variant III monoclonal antibody. Cancer Res. 2000;60:4453–60. [PubMed] [Google Scholar]
- 9.Vaidyanathan G, Alston KL, Bigner DD, Zalutsky MR. Nε -(3-[*I]Iodobenzoyl)-Lys5-Nα- maleimido-Gly1-GEEEK ([*I]IB-Mal-D-GEEEK): a radioiodinated prosthetic group containing negatively charged D-glutamates for labeling internalizing monoclonal antibodies. Bioconjugate Chem. 2006;17:1085–92. doi: 10.1021/bc0600766. [DOI] [PubMed] [Google Scholar]
- 10.Garg S, Garg PK, Zalutsky MR. N-succinimidyl 5-(trialkylstannyl)-3-pyridinecarboxylates: a new class of reagents for protein radioiodination. Bioconjugate Chem. 1991;2:50–6. doi: 10.1021/bc00007a009. [DOI] [PubMed] [Google Scholar]
- 11.Garg S, Garg PK, Zhao XG, Friedman HS, Bigner DD, Zalutsky MR. Radioiodination of a monoclonal antibody using N-succinimidyl 5-iodo-3-pyridinecarboxylate. Nucl Med Biol. 1993;20:835–42. doi: 10.1016/0969-8051(93)90149-o. [DOI] [PubMed] [Google Scholar]
- 12.Reist CJ, Garg PK, Alston KL, Bigner DD, Zalutsky MR. Radioiodination of internalizing monoclonal antibodies using N-succinimidyl 5-iodo-3-pyridinecarboxylate. Cancer Res. 1996;56:4970–7. [PubMed] [Google Scholar]
- 13.Shankar S, Vaidyanathan G, Affleck D, Welsh PC, Zalutsky MR. N-succinimidyl 3- [131I]iodo-4-phosphonomethylbenzoate ([131I]SIPMB), a negatively charged substituent-bearing acylation agent for the radioiodination of peptides and mAbs. Bioconjugate Chem. 2003;14:331–41. doi: 10.1021/bc025636p. [DOI] [PubMed] [Google Scholar]
- 14.Vaidyanathan G, White BJ, Affleck DJ, Zhao XG, Welsh PC, McDougald D, et al. SIB-DOTA: a trifunctional prosthetic group potentially amenable for multi-modal labeling that enhances tumor uptake of internalizing monoclonal antibodies. Bioorg Med Chem. 2012;20:6929–39. doi: 10.1016/j.bmc.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaidyanathan G, Affleck DJ, Li J, Welsh P, Zalutsky MR. A polar substituent-containing acylation agent for the radioiodination of internalizing monoclonal antibodies: N-succinimidyl 4-guanidinomethyl-3-[131I]iodobenzoate ([131I]SGMIB) Bioconjugate Chem. 2001;12:428–38. doi: 10.1021/bc0001490. [DOI] [PubMed] [Google Scholar]
- 16.Vaidyanathan G, Zalutsky MR. Synthesis of N-succinimidyl 4-guanidinomethyl-3-[*I]iodobenzoate: a radio-iodination agent for labeling internalizing proteins and peptides. Nat Protoc. 2007;2:282–6. doi: 10.1038/nprot.2007.20. [DOI] [PubMed] [Google Scholar]
- 17.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Improved tumor targeting of Anti-HER2 nanobody through N-succinimidyl 4-guanidinomethyl-3-Iodobenzoate radiolabeling. J Nucl Med. 2014;55:650–6. doi: 10.2967/jnumed.113.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl Med Biol. 2013;40:52–9. doi: 10.1016/j.nucmedbio.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaidyanathan G, Jestin E, Olafsen T, Wu AM, Zalutsky MR. Evaluation of an anti-p185(HER2) (scFv-C(H)2-C(H)3)2 fragment following radioiodination using two different residualizing labels: SGMIB and IB-Mal-D-GEEEK. Nucl Med Biol. 2009;36:671–80. doi: 10.1016/j.nucmedbio.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg PK, Archer GE, Jr, Bigner DD, Zalutsky MR. Synthesis of radioiodinated N-succinimidyl iodobenzoate: optimization for use in antibody labelling. Int J Rad Appl Instrum A. 1989;40:485–90. doi: 10.1016/0883-2889(89)90131-7. [DOI] [PubMed] [Google Scholar]
- 21.Lindmo T, Boven E, Cuttitta F, Fedorko J, Bunn PA., Jr Determination of the immunoreactive fraction of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J Immunol Methods. 1984;72:77–89. doi: 10.1016/0022-1759(84)90435-6. [DOI] [PubMed] [Google Scholar]
- 22.Vaidyanathan G, Affleck DJ, Bigner DD, Zalutsky MR. Improved xenograft targeting of tumor-specific anti-epidermal growth factor receptor variant III antibody labeled using N-succinimidyl 4-guanidinomethyl-3-iodobenzoate. Nucl Med Biol. 2002;29:1–11. doi: 10.1016/s0969-8051(01)00277-3. [DOI] [PubMed] [Google Scholar]
- 23.Vaidyanathan G, Affleck DJ, Bigner DD, Zalutsky MR. N-succinimidyl 3-[211At]astato-4-guanidinomethylbenzoate: an acylation agent for labeling internalizing antibodies with alpha-particle emitting 211At. Nucl Med Biol. 2003;30:351–9. doi: 10.1016/s0969-8051(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 24.Vaidyanathan G, Boskovitz A, Shankar S, Zalutsky MR. Radioiodine and 211At-labeled guanidinomethyl halobenzoyl octreotate conjugates: potential peptide radiotherapeutics for somatostatin receptor-positive cancers. Peptides. 2004;25:2087–97. doi: 10.1016/j.peptides.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 25.Takano Y, Koizumi M, Takarada R, Kamimura MT, Czerminski R, Koike T. Computer-aided design of a factor Xa inhibitor by using MCSS functionality maps and a CAVEAT linker search. J Mol Graph Model. 2003;22:105–14. doi: 10.1016/s1093-3263(03)00140-2. [DOI] [PubMed] [Google Scholar]
- 26.Vaidyanathan G, Zalutsky MR. A new route to guanidines from bromoalkanes. J Org Chem. 1997;62:4867–9. [Google Scholar]
- 27.Vaidyanathan G, White BJ, Affleck DJ, Zhao XG, Welsh PC, McDougald D, et al. SIB-DOTA: a trifunctional prosthetic group potentially amenable for multi-modal labeling that enhances tumor uptake of internalizing monoclonal antibodies. Bioorg Med Chem. 2012;20:6929–39. doi: 10.1016/j.bmc.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim SJ, Adiyaman Y, Saha G, Powell WS, Rokach J. A photoaffinity probe for 5-hydroxyeicosanoid dehydrogenase suitable for radioiodination. Tetrahedron Lett. 2001;42:4445–8. [Google Scholar]
- 29.Rossouw DD, Macheli L. Large-scale synthesis of no-carrier-added [I-123] mIBG, using two different stannylated precursors. J Labelled Compd Radiopharm. 2009;52:499–503. [Google Scholar]
- 30.Vaidyanathan G, Zalutsky MR. No-carrier-added synthesis of meta-[131I]iodobenzylguanidine. Appl Radiat Isot. 1993;44:621–8. doi: 10.1016/0969-8043(93)90179-e. [DOI] [PubMed] [Google Scholar]
- 31.Yu Z, Xia W, Wang HY, Wang SC, Pan Y, Kwong KY, et al. Antitumor activity of an Ets protein, PEA3, in breast cancer cell lines MDA-MB-361DYT2 and BT474M1. Mol Carcinog. 2006;45:667–75. doi: 10.1002/mc.20212. [DOI] [PubMed] [Google Scholar]
- 32.Roselli M, Milenic DE, Brechbiel MW, Mirzadeh S, Pippin CG, Gansow OA, et al. In vivo comparison of CHX-DTPA ligand isomers in athymic mice bearing carcinoma xenografts. Cancer Biother Radiopharm. 1999;14:209–20. doi: 10.1089/cbr.1999.14.209. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi H, Wu C, Yoo TM, Sun BF, Drumm D, Pastan I, et al. Evaluation of the in vivo biodistribution of yttrium-labeled isomers of CHX-DTPA-conjugated monoclonal antibodies. J Nucl Med. 1998;39:829–36. [PubMed] [Google Scholar]
- 34.Garg PK, Slade SK, Harrison CL, Zalutsky MR. Labeling proteins using aryl iodide acylation agents: influence of meta vs para substitution on in vivo stability. Int J Rad Appl Instrum B. 1989;16:669–73. doi: 10.1016/0883-2897(89)90136-0. [DOI] [PubMed] [Google Scholar]
- 35.Manikwar P, Zimmerman T, Blanco FJ, Williams TD, Siahaan TJ. Rapid identification of fluorochrome modification sites in proteins by LC ESI-Q-TOF mass spectrometry. Bioconjugate Chem. 2011;22:1330–6. doi: 10.1021/bc100560c. [DOI] [PMC free article] [PubMed] [Google Scholar]