

Abstract
A mild and concise approach for the construction of 3,4-dihydro-2H-pyran ring integrated into the A-ring of the natural product oridonin using an optimized inverse electron demand hetero-Diels-Alder (IED HDA) reaction is reported herein. A self-dimerization of the exocyclic enone installed in the A-ring through a homo-HDA reaction was identified to exclusively give a dimeric ent-kaurane diterpenoid with the spirochroman core. Moreover, the efficient cross-HDA cycloadditions of this enone with various vinyl ethers or vinyl sulfides, instead of its own homo-HDA dimerization, were achieved in regio- and stereoselective manners, thus providing the access to novel dihydropyran-fused diterpenoids as potential anticancer agents to overcome chemoresistance.
Introduction
ent-Kaurane diterpenoids, stemming from geranylgeranyl pyrophosphate (GGPP) catalyzed by cyclases, present an important class of tetracyclic diterpene natural products.1–3 As shown in Figure 1, their structures are characterized by a perhydrophenantrene scaffold (A-, B- and Crings) fused with a cyclopentane unit (D-ring) formed by a bridge of two carbons between C-8 and C-13.3 Many of them, especially the ent-kaurane with an exo-methylene cyclopentanone moiety in the D-ring, possess a broad spectrum of biological activities such as anticancer, antibacterial, antifungal, anti-HIV, anti-inflammatory and anti-tuberculosis.2,4.
Figure 1.

Chemical structures of oridonin (1), representative natural products and derivatives containing the pyran moiety
Oridonin (1), a naturally occurring 7,20-epoxy-ent-kaurane, was isolated from Chinese medicinal herb Isodon rubescens in the genus isodon, which are traditionally used in China and Japan for the treatment of various human diseases.5 In recent years, it has been drawing a rising attention for cancer biologists due to its remarkable growth inhibition and apoptosis-inducing activity in cancer therapy.6 Accumulating evidence has revealed that 1 exhibits anti-proliferative activity against various cancer cells through unique but versatile mechanisms including cell cycle arrest, apoptosis, and autophagy, in which a series of transcription factors, protein kinases as well as pro- and/or anti-apoptotic proteins are involved.7 While oridonin is a promising anticancer drug candidate with great potential for clinical development, it still suffers from moderate potency, limited aqueous solubility and poor bioavailability.7c,8 Consequently, the template of 1 becomes an attractive scaffold for chemists to pursue better oridonin-like compounds through structural modifications. Compared to extensive biological studies on 1, the pursuit of chemical transformations based on its scaffold remains sparse, probably due to synthetic challenges arising from its densely functionalized, stereochemistry-rich ent-kaurane frameworks including a chemically reactive α,β-unsaturated ketone functionality in the D-ring and a 6-hydroxyl-7-hemiacetal moiety in the B-ring. Previously, most of oridonin derivatives have been synthesized attending to coupling ester appendages to its hydroxyl groups, periodate oxidative cleavage of 6,7-diol into 6,7-seco-ent-kaurane-type derivatives, or splitting the D-ring to form 15,16-seco-kauranoids.9,10
As part of our ongoing anticancer drug discovery program inspired by structurally intriguing natural products, we have directed our effort in developing efficient synthetic methodologies based on the unique ent-kaurane scaffold of 1 to generate novel A-ring modified diterpenoids, while keeping key reactive pharmacophores intact with the aim to improve potency and drug-like properties.11 Recently, a protecting group-free synthetic method was successfully established by our group to concisely obtain a series of A-ring thiazole-fused derivatives with enhanced activity and improved solubility,11a indicating that A-ring modifications appear to be tolerable for yielding biologically interesting molecules and allow further construction of other heterocycles thereby increasing the structural diversity. With the representative molecules as examples shown in Figure 1, the pyran ring system is a common and important structural motif that is widespread in various anticancer natural products and their synthetic analogues such as bryostatin 1 (in Phase II clinical trial) and eribulin mesylate (in the clinic) derived from halichondrin B.12–14 All of these observations combine to clearly justify our chemical effort to explore efficient synthetic approaches that incorporate pyran ring systems into the oridonin template. To date, a number of useful methods are available to form dihydropyran units, including hetero-Diels-Alder cycloadditions, Prins cyclizations, olefin metathesis, Petasis-Ferrier rearrangement, intramolecular oxy-Michael addition and Maitland-Japp reaction.15–20 Among them, hetero-Diels-Alder reaction has emerged as one of the most powerful tools to build this cyclic system due to its highly regio- and stereoselective nature.15a Particularly, the inverse-electron-demand hetero-Diels-Alder (IED HDA) reactions of electron-poor enones with electron-rich alkene dienophiles have been well established as an efficient method for the stereospecific construction of 3,4-dihydro-2H-pyran analogues.15a,21 Nevertheless, to the best of our knowledge, applications of this type of HDA reaction based on the complex ent-kaurane scaffold of 1 have never been explored. In this article, we describe our development of lanthanide Lewis acid (LA)-catalyzed IED HDA reactions based on the exocyclic enone installed in the A-ring under mild conditions for successfully furnishing a series of novel 3,4-dihydro-2H-pyran-fused derivatives in a regio- and stereoselective manner, providing access to an expanded natural scaffold-based compound library as potential anti-cancer agents to overcome chemoresistance.
Results and Discussions
Figure 2 presents our retrosynthetic analysis for constructions of various 3,4-dihydro-2H-pyran-fused derivatives of 1. As a key step, IED HDA reaction of electron-rich vinyl dienophiles with the exocyclic α,β-unsaturated ketone functionality created in the A-ring would provide a direct route to the desired pyran ring. However, two crucial synthetic challenges arising from this strategy remain to be addressed: (1) How to selectively perform HDA reaction at the A-ring, rather than the enone system in the D-ring that is a key bioactivity center; and (2) How to achieve the desired cross-Diels-Alder cycloaddition instead of homo-Diels-Alder dimerization from the diversity perspective. Therefore, controlled regio- and stereoselective cross-HDA reactions at the A-ring of 1 are highly desired.
Figure 2.
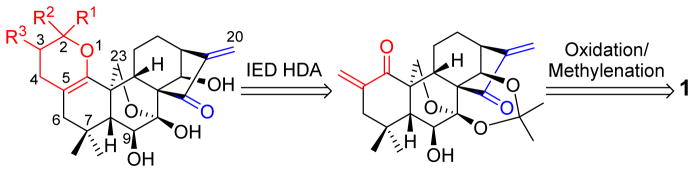
Retrosynthetic analysis for the construction of various substituted 2H-pyran-fused oridonin derivatives
Our synthetic effort commenced with 1, a naturally abundant and commercially available ent-kaurane diterpenoid (Scheme 1). Selective oxidation of 1 with Jones reagent readily provided the 1-ketone derivative 2 in 82% yield.22 The direct α-methylenation of 2 utilizing paraformaldehyde (PFA) and dimethylamine hydrochloride in refluxing 1,4-dioxane for 4 h, without any protection, smoothly afforded intermediate 3, in which the exocyclic enone system was successfully installed in the A-ring. However, 3 turned out to be unstable and was prone to undergo self-dimerization automatically. It was found that the purified 3 was allowed to stand at −20 °C or rt for 21 days to solely generate the spirochroman product 4 in 68% and 72% yield, respectively (Table 1). When the temperature was increased to 80 °C for 2 days, 4 was obtained in 80% yield.
Scheme 1.

Synthesis of Dimer 4 through HDA Type Self-Dimerization
Table 1.
Hetero-Diels-Alder Type Dimerization of 3 under Different Reaction Conditions
| Entry | Solvent | Temperature (°C) | Time (days) | Yield (%)a |
|---|---|---|---|---|
| 1 | None | −20 | 21 | 68 |
| 2 | None | rt | 21 | 72 |
| 3 | None | 80 | 2 | 80 |
| 4 | 1,4-dioxaneb | rt | 30 | 35 |
| 5 | 1,4-dioxaneb | 110 | 4 | 70 |
Isolated yields.
The concentration is 0.5 mol/L
In a solution of 1,4-dioxane (0.5 mol/L) at rt, the reaction proceeded more slowly than that without solvent; on the contrary, the reaction was accelerated to give 4 in 70% yield by refluxing the reaction solution at 110 °C for 4 days. Inspired by these findings, we attempted to access 4 directly from 2 through one-pot tandem α-methylenation/homo-HDA reactions. Thus, refluxing 2 with PFA and dimethylamine hydrochloride in 1,4-dioxane for 4 days readily furnished 4 in 65% yield. The structure of 4 was well characterized by spectroscopic data including 1H and 13C NMR, HRMS, HMQC and HMBC. According to previous reported self-HDA cycloaddition of α-alkylidene ketones,23 the formation of 4 from 3 is considered to be derived through A-ring-selective HDA reaction between the exocyclic enone of one molecule and the exomethylene of the other, in which the enone approached the exo-methylene from the less hindered α-face, but not the blocked β-face due to the bulky ent-kaurane ring system leading to a high facial selectivity. The stereochemistry of the spiro carbon C-2′ of 4 was thus tentatively assigned as R configuration, which was further confirmed through X-ray crystallographic analysis at a later stage by converting 4 into 5. Interestingly, the naturally occurring enone of oridonin was reported to presumably undergo dimerization into bisrubescensin C in vivo (Figure 1).12 Nevertheless, in our cycloaddition reactions, only the enone system in the A-ring was selectively involved, while the one in the D-ring was found intact. The high regioselectivity of this homo-HDA reaction on the enone system is likely ascribed to the less crowded steric environment of the heterodiene in the A-ring in comparison with that in the D-ring.
Since we failed to obtain the single crystal of compound 4 suitable for X-ray crystallographic analysis to determine the configurations of the chiral centers, 4 was further subjected to an epoxidation reaction by treatment with m-CPBA in CH2Cl2 at rt, exclusively leading to β-epoxide 5 in a good yield of 80%. In this case, the structure of 5 was unambiguously determined by X-ray crystallographic analysis (Scheme 2), which secured the stereochemistry of both 4 and 5. In this step, the reaction occurred preferentially at the 1-ene rather than two exo-methylenes in the D and D′ rings, respectively, and selectively formed 1,2-epoxide ring from the less sterically hindered β-face. Although some natural ent-kaurane dimers isolated from the genus isodon have been previously reported,24 homo-dimers 4 and 5 are the first examples of dimeric ent-kaurane diterpenoids with the intact enone functionality in the D-ring.
Scheme 2.
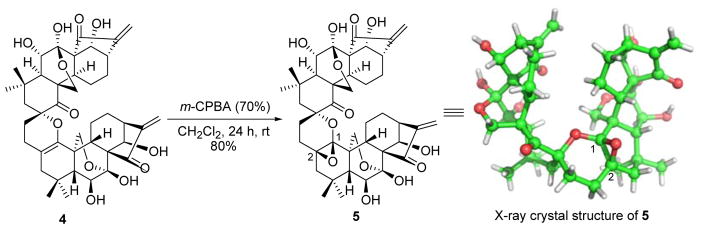
Regio- and Stereoselective Epoxidation of 4 Mediated by m-CPBA
While selectively achieving 4 generated excitement within our group, preventing the homo-dimerization of 3 during the development of A-ring-selective cross-HDA reactions with the aim to diversely construct pyran-fused derivatives was essential. Initially, 3 without any protection was chosen as the heterodiene to undergo Eu(fod)3-catalyzed cross-HDA reaction with n-butyl vinyl ether at rt for the purpose of atom-economy. To our disappointment, the reaction was very complex with a mixture of several side products including homo-dimer 4 as well as partially recovered 3. Accordingly, the acetonide protection of 7,14-dihydroxyl of 3 into 7 was deemed necessary to avoid potential side reactions.22 Moreover, introduction of the acetonide protecting group might also enhance the steric effect of the heterodiene in the D-ring leading to an improved regioselectivity for the A-ring. To reduce the chance of self-dimerization as much as possible, the protecting group was first installed to form 6 followed by α-methylenation to give the acetonide 7, instead of the direct protecting reaction of 3 (Scheme 3). With 7 in hand, we attempted to investigate its cross-HDA reaction with n-butyl vinyl ether. As shown in Table 2 (entries 1 and 2), when the reaction was performed in a solution of 1,4-dioxane or THF at rt using 10 mol% of Eu(fod)3 as the catalyst, only trace amount of the desired cycloadducts 8 and 9 were detected after 72 h; instead, dimer 10 was obtained in 24% and 21% yields, respectively, together with partially recovered 7. Increasing the reaction temperature to 80 °C predominantly led to dimer 10 in 62% yield along with cycloadducts 8 and 9 in total 15% isolated yield (Table 2, entry 3). Both 8 and 9 were fully characterized by spectroscopic data including 1H and 13C NMR, HRMS, HMBC, HMQC and NOESY, respectively, owing to their good separation by preparative TLC. The stereochemistry of these two isomers was also secured later by the X-ray crystallographic analysis of analogue 20.
Scheme 3.
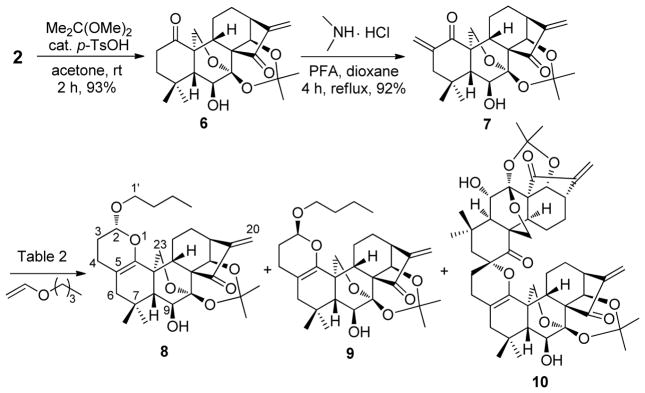
Cross-HDA Reaction of 7 with n-Butyl Vinyl Ether
Table 2.
Optimization of Cross-HDA Reaction Conditions for 8 and 9a
| Entry | Catalyst | Solvent | Temperature (°C) | Time (h) | dr (8/9)b | Yield (%)c | |
|---|---|---|---|---|---|---|---|
| 8 and 9 | 10 | ||||||
| 1 | Eu(fod)3 | 1,4-dioxaned | rt | 72 | —e | trace | 24 |
| 2 | Eu(fod)3 | THFd | rt | 72 | — | trace | 21 |
| 3 | Eu(fod)3 | 1,4-dioxaned | 80 | 72 | 45:55 | 15 | 62 |
| 4 | Eu(fod)3 | n-butyl vinyl ether | rt | 168 | 53:47 | 63 | 18 |
| 5 | None | n-butyl vinyl ether | rt | 72 | — | NRf | 14 |
| 6 | ZnCl2 | n-butyl vinyl ether | 32 | 72 | — | NRg | — |
| 7 | Yb(fod)3 | n-butyl vinyl ether | 32 | 72 | 10:90 | 68 | 14 |
| 8 | Cu(OTf)2 | n-butyl vinyl ether | 32 | 2 | — | decomposed | — |
| 9 | Ti(O-iPr)4 | n-butyl vinyl ether | 32 | 72 | — | 13 | 30 |
| 10 | (±)-BINOL | n-butyl vinyl ether | 32 | 72 | — | 8 | 28 |
7 (0.1 mmol), n-butyl vinyl ether (1 mL), and 10% mol of catalysts.
Determined by isolated yield.
Isolated yield.
7 (1 equiv), n-butyl vinyl ether (4 equiv) and solvents (1 mL).
Not determined.
No reaction.
Polymerization of vinyl ether was observed.
Since the undesired homo-dimerization of 7 was still predominant, we attempted to employ n-butyl vinyl ether as solvent to favor the desired cross-HDA reaction. To our delight, treatment of 7 with 10 mol % of Eu(fod)3 in a solution of n-butyl vinyl ether for 7 days provided 8 and 9 in total 63% yield with the diastereomeric ratio of 53:47 as major products over dimer 10 (18% yield). Moreover, no cycloadducts of the enone in the D-ring were found presumably due to the aforementioned hindered steric effects (Table 2, entry 4). In the absence of Eu(fod)3 (Table 2, entry 5), no desired cycloadducts 8 and 9 were observed, suggesting that lanthanide Lewis acid (LA) catalyst is a prerequisite for this HDA reaction due to its unique properties for coordination to the ketone functionality of the enone system in the A-ring, leading to its activation. Recently, important advances have been made in auxiliary-controlled IED HDA reactions to stereoselectively construct chiral pyran moieties.15a,25 7 with a stereochemistry-rich framework could be considered as a chiral auxiliary heterodiene, but the stereoselectivity in our case was very poor. Therefore, we continued our effort to optimize the reaction conditions by screening other Lewis acids and hydrogen bond donor catalysts. To our delight, it was found that 10 mol% of Yb(fod)3 at 32 °C offered an optimal result leading to 8 and 9 in total 68% yield with an enhanced diastereomeric ratio of 10:90 (Table 2, entry 7). Although their endo/exo selectivity can not be exactly determined due to the structural nature of these substrates, such IED HDA reactions catalyzed by Eu(fod)3 or Yb(fod)3 are likely endo-selective according to the relevant literature.25a–b,27 It is also reported that different coordination modes of Eu(fod)3 and Yb(fod)3 may account for their different facial selectivity in HDA reaction.26 As depicted in Figure 3, the high facial selectivity induced by Yb(fod)3 is probably ascribed to the different facial steric surrounding. The 7,20-epoxy blocks the α-face, and consequently, the dienophile approaches to the heterodiene mainly from the less hindered β-face in an endo-selective manner. In the case of more oxophilic Eu(fod)3, it is likely not only to activate the enone of the A-ring, but also coordinate to the 6-hydroxyl to form the bidentate complex from β-face, leading to the blockage of both α- and β-faces, which eventually results in the poor facial selectivity and longer reaction time. Other catalysts such as Ti(O-iPr)4 and (±)-BINOL also promote this reaction, but the yields are much lower (entries 9 and 10). In addition, no reaction or decomposition was observed when ZnCl2 or Cu(OTf)2 was employed as catalysts (entries 6 and 8).
Figure 3.
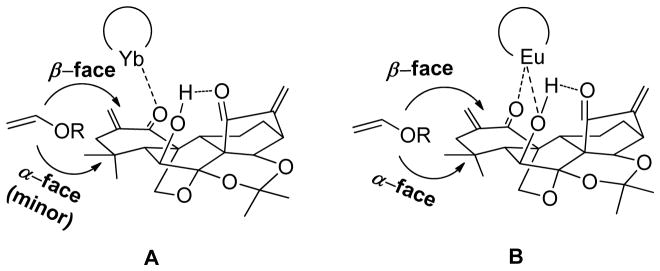
Plausible cross-HDA cycloaddition mode catalyzed by Yb(fod)3 (A) or Eu(fod)3 (B)
The optimized cross-HDA reaction condition was then applied for the synthesis of various substituted pyran-fused derivatives of 1 to explore the generality and scope. Several different vinyl ethers as well as vinyl sulfide were employed as the dienophiles to react with 7. To avoid self-dimerization during the reaction workup, intermediate 7 was directly used in the following HDA reaction without further purification. From the results summarized in Scheme 4, 10 mol% Yb(fod)3 was also found to be the effective catalyst and all reactions proceeded smoothly, affording the desired cycloadducts. In cases of ethyl vinyl ether, isobutyl vinyl ether, tert-butyl vinyl ether, 2-chloroethyl vinyl ether and allyl vinyl ether, the corresponding cycloadducts (compounds 11–20) were obtained in total 52–59% yields (2 steps) with roughly 10:90 ratios, generally similar to that of n-butyl vinyl ether. The steric effects of the substituents on vinyl ether did not show significant difference in terms of yields and diastereomeric ratios (Scheme 4, compounds 11–16). The high selectivity for the vinyl ether double bond versus the allyl double bond was also observed in the case of allyl vinyl ether (scheme 4, compounds 17–18). Furthermore, ethyl vinyl sulfide gave a slightly increased yield with completely controlled β-face selectivity to solely achieve compound 24 after shorter reaction time in comparison with ethyl vinyl ether. Different from others, the poor facial selectivity (dr = 55:45, α/β) was unexpectedly observed when 1,4-butanediol vinyl ether was used as the dienophile. Interestingly, exchange of lanthanide catalyst from Yb(fod)3 to Eu(fod)3 exclusively led to compound 21 in 70% yield with totally controlled α-face selectivity, and no diastereoisomer 22 was found. Although the exact explanation on the switchable facial selectivity in this HDA reaction is still unclear, the hydroxyl of the dienophile, at least in part, contributes to the high facial selectivity induced by Eu(fod)3 owning to its superior chelating ability.
Scheme 4.

Substrate Scope of One-Pot Cross-HDA Reactions with Various Vinyl Ether and Vinyl Sulfide
The terminal hydroxyl group of 21 could be considered as a starting point for further functional group transformations to generate structural diversity. As shown in Scheme 5, mesylation of 21 with MsCl in the presence of Et3N selectively produced intermediate 25 in 86% yield, which was followed by treatment with NaN3 to furnish a valuable azide 26 (63%) useful for building a potential compound library.
Scheme 5.
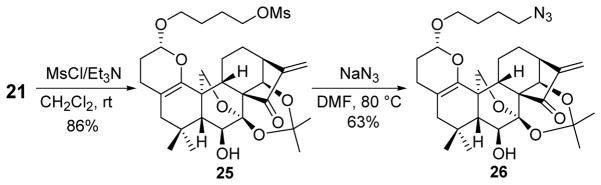
Further Functional Group Transformations of 21
To further explore the generality and scope of this reaction for the diversity of the ent-kaurane scaffold, 3,4-dihydro-2H-pyran was selected as the dienophile to undergo Yb(fod)3-catalyzed cross-HDA reaction with 7. (2R,3S)-27 was obtained in 35% yield as the main cycloadduct, along with trace amount of (2S,3R)-28, after a long reaction time (7 days) likely through an exo-selective HDA reaction (Scheme 6). In this case, 3,4-dihydro-2H-pyran approaches to the heterodiene from the β-face in an exo-selective manner to give 27 due to the enhanced steric effect of the cyclic vinyl ether with the ent-kaurane ring system.
Scheme 6.
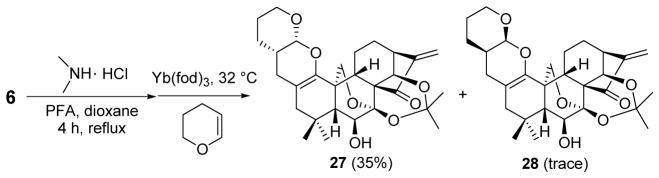
One-Pot Cross-HDA Reaction of 6 with 3,4-Dihydro-2H-Pyran
Considering that 3,4-dihydro-2H-pyran moieties are versatile synthetic building blocks for generation of various functionalized heterocycles, carbohydrates and natural products,28 we inevitably became interested in further chemical transformation based on this attractive scaffold. As shown in Scheme 7, treatment of compound 11 with 5% HCl (aq) for 45 min readily provided the deprotected derivative 29 in 78% yield, which was further hydrolyzed with 5% HCl (aq) for another 4 h to solely yield aldehyde 31 in 82% yield with high β-face selectivity. Acetonide deprotection of diastereoisomer 12 under the same condition gave the corresponding deprotected product 30, which was also prone to cleavage of the dihydropyran ring affording 31 in 60% yield (two steps). The structure of 31 was also well determined by 1H and 13C NMR, HRMS, HMBC, HMQC and NOSEY. 31 could be used as another common building block to extend the structural diversity.
Scheme 7.
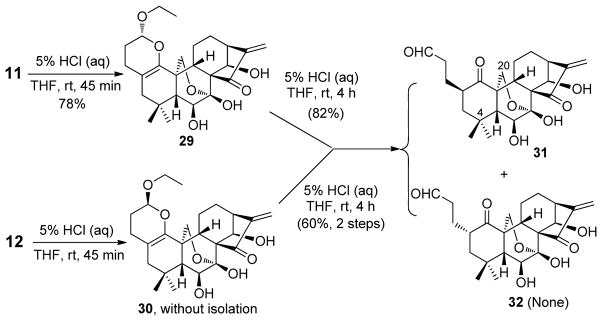
Hydrolysis Reactions of Compounds 11 and 12 by 5% HCl (aq)
The growth inhibitory effects of the newly generated pyran-fused diterpenoids were evaluated in four breast cancer cell lines MCF-7 (ER-positive), MDAMB-231 (ER-negative and triple-negative), MDA-MB-468 (ER-negative and triple-negative) as well as MCF-7/ADR (adriamycin-resistant) using MTT assays as described in the Experimental Section. The capability of these new molecules to inhibit the growth of cancer cells was summarized in Table 3 and compared with that of adriamycin and 1 spontaneously. Most of these new compounds not only exhibited significantly enhanced antiproliferative effects on aggressive breast cancer MDA-MB-468, and MDA-MB-231 cells relative to 1, but also displayed marked growth inhibitory activity against drug-resistant breast cancer MCF-7/ADR cells. Intriguingly, compound 12 demonstrated potencies in the submicromolar range with significantly improved capability to overcome chemoresistance.
Table 3.
Antiproliferative effects of pyran-fused diterpenoids against human breast cancer cells.a
| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | MDA-MB-468 | MCF-7/ADR | |
| 1 | 4.4 ± 1.4 | 28.0 ± 1.4 | 5.3 ± 1.4 | 34.8 ± 2.5 |
| Adriamycin | 0.67 ± 0.45 | 2.3 ± 0.22 | 0.65 ± 0.26 | >10b |
| 2 | 2.1 ± 0.35 | 2.5 ± 0.25 | 3.9 ± 2.2 | 9.0 ± 0.42 |
| 4 | 3.9 ± 0.17 | 7.8 ± 1.9 | 4.6 ± 0.18 | 12.7 ± 0.61 |
| 8 | 2.4 ± 1.5 | 2.2 ± 0.39 | 3.0 ± 0.67 | 3.8 ± 1.2 |
| 9 | 2.4 ± 1.9 | 3.7 ± 0.28 | 5.8 ± 1.9 | 6.8 ± 3.5 |
| 11 | 3.5 ± 2.2 | 6.1 ± 2.4 | 5.4 ± 0.98 | 4.7 ± 3.2 |
| 12 | 0.44 ± 0.27 | 0.54 ± 0.14 | 0.52 ± 0.18 | 1.6 ± 0.72 |
| 13 | 2.2 ± 1.3 | 1.8 ± 0.22 | 2.6 ± 0.08 | 4.4 ± 0.90 |
| 14 | 5.8 ± 3.8 | 8.2 ± 0.57 | 7.0 ± 1.0 | 10.3 ± 1.7 |
| 17 | 4.3 ± 1.8 | 7.1 ± 0.22 | 4.9 ± 0.85 | 4.3 ± 2.4 |
| 18 | 2.1 ± 0.98 | 3.3 ± 0.23 | 4.4 ± 1.5 | 3.2 ± 0.54 |
| 19 | 2.3 ± 1.0 | 3.3 ± 1.8 | 3.0 ± 1.0 | 3.1 ± 1.1 |
| 20 | 6.8 ± 3.4 | 7.2 ± 1.3 | 7.3 ± 0.23 | 8.7 ± 0.27 |
| 21 | 2.1 ± 1.1 | 3.2 ± 0.24 | 2.7 ± 0.14 | 4.5 ± 1.5 |
| 22 | 5.2 ± 2.9 | 5.9 ± 2.0 | 6.0 ± 0.29 | 5.3 ± 1.3 |
| 24 | 2.3 ± 1.4 | 2.9 ± 0.54 | 2.8 ± 0.42 | 3.8 ± 0.47 |
| 26 | 7.8 ± 3.2 | 7.6 ± 0.4 | 6.6 ± 0.77 | 8.6 ± 0.70 |
| 28 | 2.4 ± 0.78 | 2.5 ± 0.77 | 3.3 ± 0.58 | 2.9 ± 0.32 |
| 29 | 1.9 ± 0.97 | 3.3 ± 1.01 | 2.2 ± 0.68 | 3.2 ± 0.89 |
Breast cancer cell lines: MCF-7 (ER-positive), MDA-MB-231 and MDA-MB-468 (ER-negative and triple-negative, highly aggressive), MCF-7/ADR (adriamycin-resistant, adriamycin a.k.a. doxorubicin). Software: MasterPlex ReaderFit 2010, MiraiBio, Inc. Values are the mean ± SE of three independent experiments.
If a specific compound is given a value >10, it indicates that a specific IC50 cannot be calculated from the data points collected, meaning “no effect”.
Conclusions
In summary, we have developed a mild and concise method for the efficient construction of 3,4-dihydro-2H-pyran ring fused into the A-ring of oridonin using optimized cross-IED HDA reactions. It was found that 3 was prone to undergo self-dimerization through a homo-HDA reaction of the exocyclic enone in the A-ring leading to an unprecedented dimeric ent-kaurane diterpenoid 4 with the spirochroman core, the structure of which was later secured by X-ray crystallographic analysis of epoxide 5. Meanwhile, the desired lanthanide Lewis acid-catalyzed cross-HDA cycloadditions of 7 with various vinyl ethers or vinyl sulfides were achieved in regio- and stereoselective manners, instead of its homo-HDA dimerization, thus providing a series of diversely substituted dihydropyran-fused diterpenoids. Interestingly, lanthanide catalysts Yb(fod)3 and Eu(fod)3 displayed different facial selectivity during the cycloaddition probably owning to the facial discrimination by their different coordination modes to the unique scaffold of 1. Further functional group transformations based on our synthesized derivatives gave rise to additional versatile synthetic building blocks 26 and 31 for more advanced diversity. Intriguingly, these new molecules have demonstrated superior antiproliferative effects against various breast cancer cells including aggressive triple-negative breast cancer cells with great potential to overcome chemoresistance. Our success in the efficient synthesis of 3,4-dihydro-2H-pyran-fused diterpenoids allows for this important natural product scaffold to be evolved into new platforms for drug discovery and identification of novel targets and signaling pathways associated with cancer resistance.
EXPERIMENTAL SECTION
Synthesis of (2R,4aR,5S,6S,6aR,6a′R,7′S,8′S,8a′R,9S,11aS,11bS,11′S,13a′S,13b′S,14R,16′R)- 5,6,7′,8′,14,16′-hexahydroxy-4,4,6′,6′-tetramethyl-8,10′-dimethylene- 4,4a,5,6,6′,6a′,7′,8′,9,10,11,11a,11′,12′,13′,13a′-hexadecahydro-3′H-spiro[6,11b-(epoxymethano)-6a,9-methanocyclohepta[a]naphthalene-2,2′-8,13b-(epoxymethano)-8a,11-methanocyclohepta[3,4]benzo[1,2-h]chromene]-1,7,9′ (3H,4′H,5′H,8H,10′H)-trione (4)
A mixture of 222 (0.10 g, 0.27 mmol), dimethylammonium chloride (48 mg, 0.59 mmol), paraformaldehyde (17 mg) in 1,4-dioxane (3 mL) was refluxed for 4 h. The reaction mixture was then diluted with 5 mL of water and extracted with 15 mL of dichloromethane three times. The extract was washed with saturated NaHCO3 (aq) solution (5 mL) and brine (5 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 60% EtOAc in hexanes afforded the desired product 3 as a colorless gel (77 mg, 75%). 1H NMR (300 MHz, CDCl3/CD3OD = 10:1): δ 6.22 (s, 1H), 6.00 (d, 1H, J = 1.5 Hz), 5.62 (s, 1H), 5.29 (d, 1H, J = 1.5 Hz), 4.91 (s, 1H), 4.28 (d, 1H, J = 10.2 Hz), 4.01 (d, 1H, J = 9.9 Hz), 3.78 (d, 1H, J = 9.0 Hz), 3.02 (d, 1H, J = 9.3 Hz), 2.50 (m, 3H), 2.00 (m, 2H), 1.84 (d, 1H, J = 9.0 Hz), 1.60 (m, 1H), 1.28 (m, 1H), 1.22 (s, 3H), 0.99 (s, 3H). 13C NMR (75 MHz, CDCl3/CD3OD = 10:1): δ 206.4, 199.9, 150.9, 142.0, 125.4, 121.7, 98.1, 72.4, 72.0, 66.1, 61.4, 59.6, 49.5, 47.3, 46.2, 42.6, 32.6, 29.7, 29.4, 21.2, 18.9. HRMS Calcd for C21H26O6: [M + H]+ 375.1802; found 375.1811.
Heating 3 (77 mg, 0.20 mmol) neat at 80 °C for 2 days provided dimer 4 as a colorless solid (61 mg, 80%); [α]25D +100 (c 0.10, CH2Cl2 /MeOH = 10:1). 1H NMR (600 MHz, CDCl3/CD3OD = 14:1) δ 6.48 (s, 1H), 6.17 (s, 1H), 5.81 (s, 1H), 5.58 (s, 1H), 4.89 (s, 1H), 4.71 (s, 1H), 4.37 (d, 1H, J = 10.8 Hz), 4.03 (d, 1H, J = 10.8 Hz), 4.01 (d, 1H, J = 9.6 Hz), 3.97 (d, 1H, J = 9.6 Hz), 3.79 (d, 1H, J = 8.4 Hz), 3.75 (d, 1H, J = 8.4 Hz), 3.10 (d, 1H, J = 9.0 Hz), 2.95 (d, 1H, J = 9.6 Hz), 2.61 (dd, 1H, J = 13.8 Hz, 4.8 Hz), 2.55 (dt, 1H, J = 14.4 Hz, 8.4 Hz), 2.30 (m, 2H), 2.08 (m, 2H), 1.94 (m, 3H), 1.86 (m, 2H), 1.66 (m, 3H), 1.55 (m, 5H), 1.47 (m, 1H), 1.15 (s, 3H), 1.14 (s, 3H), 1.04 (s, 3H), 0.95 (s, 3H). 13C NMR (150 MHz, CDCl3/CD3OD = 14:1) δ 207.3, 206.8, 206.6, 151.6, 151.4, 142.0, 122.5, 121.6, 112.4, 98.4, 98.1, 80.4, 73.2, 72.7 (2C), 72.5, 65.7, 64.7, 62.3, 61.4, 60.6, 57.5, 53.8, 45.9, 43.4, 43.0, 42.0, 33.2, 32.3, 31.5, 30.8, 30.4, 30.0, 29.0, 25.2, 23.3, 20.9 (3C), 18.6. HRMS Calcd for C42H52O12: [M + H]+ 749.3532; found 749.3540.
Synthesis of (2R,6aR,7S,7aR,7a1R,10aR,11S,13aS,13bS)-2-butoxy-7-hydroxy-6,6,9,9-tetramethyl-16-methylene-2,3,4,5,6,6a,7,10a,11,12,13,13a-dodecahydro-7a,13b-(epoxymethano)-7a1,11-ethano[1,3]dioxino[4′,5′,6′:4,5]naphtho[2,1-h]chromen-17-one (8) and (2S,6aR,7S,7aR,7a1R,10aR,11S,13aS,13bS)-2-butoxy-7-hydroxy-6,6,9,9-tetramethyl-16-methylene-2,3,4,5,6,6a,7,10a,11,12,13,13a-dodecahydro-7a,13b-(epoxymethano)-7a1,11-ethano[1,3]dioxino[4′,5′,6′:4,5]naphtho[2,1-h]chromen-17-one (9)
A mixture of 6 (50 mg, 0.12 mmol), dimethylammonium chloride (21 mg, 0.26 mmol), and paraformaldehyde (8.0 mg) in 1,4-dioxane (2 mL) was refluxed for 4 h. The reaction mixture was then diluted with 3 mL of water and extracted with 10 mL of dichloromethane three times. The extract was washed with saturated NaHCO3 (aq) solution (5 mL) and brine (5 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using silica gel column; elution with 60% EtOAc in hexanes afforded the desired product 7 as a colorless gel (41 mg, 80%). 1H NMR (300 MHz, CDCl3): δ 6.17 (s, 1H), 6.03 (s, 1H), 5.59 (s, 1H), 5.27 (s, 1H), 5.20 (d, 1H, J = 12.0 Hz), 4.87 (s, 1H), 4.24 (d, 1H, J = 9.9 Hz), 4.01 (d, 1H, J = 9.9 Hz), 3.91 (dd, 1H, J = 12.0 Hz, 9.0 Hz), 3.07 (d, 1H, J = 9.0 Hz), 2.48 (m, 3H), 1.96 (m, 2H), 1.83 (d, 1H, J = 8.7 Hz), 1.68 (m, 1H), 1.65 (s, 3H), 1.45 (m, 1H), 1.43 (m, 1H), 1.35 (s, 3H), 1.25 (s, 3H), 1.00 (s, 3H).13C NMR (75 MHz, CDCl3) δ 204.6, 199.0, 150.4, 141.7, 125.5, 120.8, 101.3, 95.8, 71.6, 70.0, 65.9, 59.5, 55.8, 47.0 (2C), 46.6, 40.1, 32.8, 30.2, 30.1, 30.0, 25.3, 21.5, 19.0. HRMS Calcd for C24H30O6: [M + H]+ 415.2115; found 415.2109.
To a solution of 7 (41 mg, 0.10 mmol) in n-butyl vinyl ether (1 mL) was added Yb(OTf)3 (11 mg, 0.01 mmol) at rt. The resulting mixture was stirred at 32 °C for 72 h. The reaction mixture was then diluted with 3 mL of water and extracted with 10 mL of dichloromethane three times. The extract was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was further purified using preparative TLC developed by 15% EtOAc in hexanes to afford the desired product 8 (3.4 mg) and 9 (31.1 mg) as colorless amorphous gel in total 68% yield.
8: [α]25D –10 (c 0.10, CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 6.14 (s, 1H), 5.54 (s, 1H), 5.31 (d, 1H, J = 12.0 Hz), 4.89 (m, 1H), 4.85 (d, 1H, J = 1.2 Hz), 4.23 (dd, 1H, J = 9.6 Hz, 0.9 Hz), 4.00 (d, 1H, J = 9.6 Hz), 3.89 (dd, 1H, J = 12.0 Hz, 8.7 Hz), 3.53 (dt, 1H, J = 9.0 Hz, 6.6 Hz), 3.37 (dt, 1H, J = 9.6 Hz, 6.3 Hz), 3.03 (d, 1H, J = 9.3 Hz), 2.49 (m, 1H), 1.86 (m, 7H), 1.65 (s, 3H), 1.62 (m, 2H), 1.49 (m, 4H), 1.34 (s, 3H), 1.29 (m, 2H), 1.16 (s, 3H), 1.01 (s, 3H), 0.89 (t, 3H, J = 7.2 Hz). 13C NMR (75 MHz, CDCl3) δ 204.7, 150.9, 140.7, 119.9, 108.8, 100.9, 95.4 (2C), 72.0, 70.1, 67.4, 64.1, 59.0, 56.5, 50.1, 45.2, 40.6, 40.3, 32.9, 31.8, 30.8, 30.6, 30.1, 26.8, 25.3, 21.9, 20.9, 20.6, 19.5, 13.9. HRMS Calcd for C30H42O7: [M + H]+ 515.3003; found 515.2999.
9: [α]25D +118 (c 0.10, CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 6.14 (s, 1H), 5.54 (s, 1H), 5.34 (d, 1H, J = 11.7 Hz), 4.86 (d, 1H, J = 0.9 Hz), 4.58 (dd, 1H, J = 9.0 Hz, 1.5 Hz), 4.18 (d, 1H, J = 8.7 Hz), 3.99 (d, 1H, J = 9.6 Hz), 3.89 (dd, 1H, J = 12.0 Hz, 8.7 Hz), 3.77 (dt, 1H, J = 9.3 Hz, 6.3 Hz), 3.45 (dt, 1H, J = 9.3 Hz, 6.3 Hz), 3.03 (d, 1H, J = 9.0 Hz), 2.48 (m, 1H), 1.95 (m, 7H), 1.66 (s, 3H), 1.64 (m, 2H), 1.55 (m, 4H), 1.39 (m, 2H), 1.34 (s, 3H), 1.17 (s, 3H), 1.03 (s, 3H), 0.93 (t, 3H, J = 7.2 Hz). 13C NMR (75 MHz, CDCl3) δ 204.7, 150.9, 142.7, 120.0, 107.4, 100.9, 99.9, 95.4, 72.0, 70.1, 68.7, 63.9, 58.6, 56.5, 49.8, 44.8, 40.6, 40.3, 32.9, 31.7, 30.8 (2C), 30.1, 28.4, 25.9, 25.4, 21.3, 20.4, 19.3, 13.8. HRMS Calcd for C30H42O7: [M + H]+ 515.3003; found 515.2994.
Synthesis of (2R,6aR,7S,8S,8aR,11S,13aS,13bS,16R)-2-ethoxy-7,8,16-trihydroxy-6,6-dimethyl-10-methylene-3,4,5,6,6a,7,8,10,11,12,13,13a-dodecahydro-8,13b-(epoxymethano)-8a,11-methanocyclohepta[3,4]benzo[1,2-h]chromen-9(2H)-one (29) and 3-((2R,4aR,5S,6S,6aR,9S,11aS,11bS,14R)-5,6,14-trihydroxy-4,4-dimethyl-8-methylene-1,7-dioxododecahydro-1H-6,11b-(epoxymethano)-6a,9-methanocyclohepta[a]naphthalen-2-yl)propanal (31)
To a solution of 11 (10 mg, 0.02mmol) in THF (1.0 mL) was added 5% HCl aqueous solution (0.4 mL) at rt. The resulting mixture was stirred at rt for 45 min, and then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 2.5% methanol in dichloromethane to afford the desired product 29 (7.2 mg, 78%) as a colorless amorphous gel. [α]25D +12 (c 0.10, CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 6.18 (s, 1H), 5.87 (dd, 1H, J = 12.0 Hz, 4.2 Hz), 5.56 (s, 1H), 5.11 (m, 1H), 4.92 (s, 2H), 4.69 (s, 1H), 4.27 (dd, 1H, J = 9.6 Hz, 0.9 Hz), 3.99 (d, 1H, J = 9.9 Hz), 3.79 (dd, 1H, J = 12.0 Hz, 9.0 Hz), 3.59 (dq, 1H, J = 9.6 Hz, 6.9 Hz), 3.44 (dq, 1H, J = 9.6 Hz, 6.9 Hz), 3.02 (d, 1H, J = 9.9 Hz), 2.44 (m, 1H), 1.97 (m, 4H), 1.78 (m, 2H), 1.67 (m, 3H), 1.52 (m, 2H), 1.15 (t, 3H, J = 7.2 Hz), 1.13 (s, 3H), 1.00 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 206.6, 151.6, 140.5, 120.9, 109.7, 97.7, 95.2, 73.5, 72.0, 64.7, 63.0, 62.5, 58.5, 53.3, 45.1, 42.6, 41.4, 33.0, 30.4, 30.2, 26.9, 22.2, 20.7, 20.5, 15.1. HRMS Calcd for C25H34O7: [M + H]+ 447.2377; found 447.2380.
To a solution of 29 (5.0 mg, 0.01mmol) in THF (1.0 mL) was added 5% HCl aqueous solution (0.2 mL) at rt. The resulting mixture was stirred at rt for 4 h, and then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 3% methanol in dichloromethane to afford the desired product 31 (3.8 mg, 82%) as a colorless amorphous gel. [α]25D +172 (c 0.10, CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 9.71 (t, 1H, J = 1.5 Hz), 6.26 (s, 1H), 5.89 (d, 1H, J = 11.7 Hz), 5.65 (s, 1H), 5.41 (br s, 1H), 4.85 (s, 1H), 4.54 (br s, 1H), 4.34 (d, 1H, J = 10.2 Hz), 4.01 (dd, 1H, J = 10.5 Hz, 1.2 Hz), 3.78 (dd, 1H, J = 12.0 Hz, 9.0 Hz), 3.06 (d, 1H, J = 9.3 Hz), 2.59 (m, 1H), 2.43 (m, 3H), 2.13 (m, 3H), 1.88 (m, 2H), 1.65 (m, 1H), 1.55 (m, 1H), 1.28 (m, 1H), 1.18 (s, 3H), 0.98 (m, 1H), 0.92 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 212.9, 206.3, 201.7, 150.7, 122.3, 98.2, 73.3, 71.9, 64.5, 61.5, 59.0, 50.2, 48.9, 46.6, 42.6 (2C), 41.2, 32.7, 30.7, 29.4, 24.7, 21.9, 19.0. HRMS Calcd for C23H30O7: [M + H]+ 419.2064; found 419.2071.
To a solution of 12 (5.0 mg, 0.01mmol) in THF (1.0 mL) was added 5% HCl aqueous solution (0.3 mL) at rt. The resulting mixture was stirred at rt for 5 h, and then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 3% methanol in dichloromethane to afford the desired product 31 (2.5 mg, 60%) as a colorless amorphous gel.
Supplementary Material
Acknowledgments
This work was supported by grants P50 CA097007, P30 DA028821, R21 MH093844 (JZ) from the National Institutes of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from the Gulf Coast Consortia (GCC), a training fellowship from the Keck Center for Interdisciplinary Bioscience Training of the GCC (NIGMS grant T32 GM089657), Sealy and Smith Foundation grant (to the Sealy Center for Structural Biology and Molecular Biophysics), John Sealy Memorial Endowment Fund, and the Center for Addiction Research (CAR) at UTMB. We thank Drs. Lawrence C. Sowers and Jacob A. Theruvathu for the NMR spectroscopy assistance, Drs. Huiling Liu and Carol Nilsson for mass spectrometry assistance, as well as Dr. Yana Cen for helpful discussion.
Footnotes
Electronic Supplementary Information (ESI) available: Part of the detailed experimental procedures, copies of NMR spectra for all new compounds and X-ray CIF files for compounds 5 and 20. See DOI:10.1039/b000000x/
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Bruneton J. Pharmacognosy, Phytochemistry, Medicinal Plants. 3. Lavoisier; London: 1995. pp. 387–511. [Google Scholar]
- 2.Sun HD, Huang SX, Han QB. Nat Prod Rep. 2006;23:673–698. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- 3.García PA, de Oliveira AB, Batista R. Molecules. 2007;12:455–483. doi: 10.3390/12030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Li SH, Wang J, Niu XM, Shen YH, Zhang HJ, Sun HD, Li ML, Tian QE, Lu Y, Cao P, Zheng QT. Org Lett. 2004;6:4327–4330. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]; b) Ikezoe T, Yang Y, Bandobashi K, Saito T, Takemoto S, Machida H, Togitani K, Koeffler HP, Taguchi H. Mol Cancer Ther. 2005;4:578–586. doi: 10.1158/1535-7163.MCT-04-0277. [DOI] [PubMed] [Google Scholar]; c) Wang L, Zhao WL, Yan JS, Liu P, Sun HP, Zhou GB, Weng ZY, Wu WL, Weng XQ, Sun XJ, Chen Z, Sun HD, Chen SJ. Cell Death Differ. 2007;14:306–317. doi: 10.1038/sj.cdd.4401996. [DOI] [PubMed] [Google Scholar]; d) Ye YC, Wang HJ, Xu L, Liu WW, Liu BB, Tashiro SI, Onodera S, Ikejima T. Acta Pharmacol Sin. 2012;33:1055–1061. doi: 10.1038/aps.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yeh SH, Chang FR, Wu YC, Yang YL, Zhuo SK, Hwang TL. Planta Med. 2005;71:904–909. doi: 10.1055/s-2005-871234. [DOI] [PubMed] [Google Scholar]; f) Han QB, Lu Y, Wu L, He ZD, Qiao CF, Xu HX, Zheng QT, Sun HD. Tetrahedron Lett. 2005;46:5373–5375. [Google Scholar]; g) Bruno M, Rosselli S, Pibiri S, Piozzi F, Bondi ML, Simmonds MSJ. Phytochemistry. 2001;58:463–474. doi: 10.1016/s0031-9422(01)00252-7. [DOI] [PubMed] [Google Scholar]
- 5.a) Fujita E, Nagao Y, Node M, Kaneko K, Nakazawa S, Kuroda H. Experientia. 1976;32:203–206. doi: 10.1007/BF01937766. [DOI] [PubMed] [Google Scholar]; b) Fujita E, Nagao Y, Kaneko K, Nakazawa S, Kuroda H. Chem Pharm Bull. 1976;24:2118–2127. doi: 10.1248/cpb.24.2118. [DOI] [PubMed] [Google Scholar]; c) Wang RL. Chin J Cancer. 1984;8:50–52. [Google Scholar]; d) Yin F, Liang JY, Liu J. J Chin Pharm Univ. 2003;34:302–304. [Google Scholar]
- 6.a) Guan YZ, Wei TH. J Med Radiol Technol. 2005;236:43–45. [Google Scholar]; b) Zhou G, Kang H, Wang L, Gao L, Liu P, Xie J, Zhang F, Weng X, Shen Z, Chen J, Gu L, Yan M, Zhang D, Chen S, Wang Z, Chen Z. Blood. 2007;109:3441–3450. doi: 10.1182/blood-2006-06-032250. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bohanon FJ, Wang X, Ding C, Ding Y, Radhakrishnan GL, Rastellini C, Zhou J, Radhakrishnan RS. J Surg Res. 2014;190:55–63. doi: 10.1016/j.jss.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Li C, Wang E, Cheng Y, Bao J. Int J Biochem Cell Biol. 2011;43:701–704. doi: 10.1016/j.biocel.2011.01.020. [DOI] [PubMed] [Google Scholar]; b) Ikezoe T, Yang Y, Bandobashi K, Saito T, Takemoto S, Machida H, Togitani K, Koeffler HP, Taguchi H. Mol Cancer Ther. 2005;4:578–586. doi: 10.1158/1535-7163.MCT-04-0277. [DOI] [PubMed] [Google Scholar]; c) Hsieh TC, Wijeratne EK, Liang JY, Gunatilaka AL, Wu JM. Biochem Biophys Res Commun. 2005;337:224–231. doi: 10.1016/j.bbrc.2005.09.040. [DOI] [PubMed] [Google Scholar]; d) Jin S, Shen JN, Wang J, Huang G, Zhou JG. Cancer Biol Ther. 2007;6:261–268. doi: 10.4161/cbt.6.2.3621. [DOI] [PubMed] [Google Scholar]; e) Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera S, Ikejima T. Arch Biochem Biophys. 2009;490:70–75. doi: 10.1016/j.abb.2009.08.011. [DOI] [PubMed] [Google Scholar]; f) Cheng Y, Qiu F, Ye YC, Guo ZM, Tashiro S, Onodera S. FEBS J. 2009;276:1291–1306. doi: 10.1111/j.1742-4658.2008.06864.x. [DOI] [PubMed] [Google Scholar]; g) Hu HZ, Yang YB, Xu XD, Shen HW, Shu YM, Ren Z. Acta Pharmacol Sin. 2007;28:1819–1826. doi: 10.1111/j.1745-7254.2007.00667.x. [DOI] [PubMed] [Google Scholar]; h) Chen S, Gao J, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Int J Oncol. 2005;26:579–588. [PubMed] [Google Scholar]; i) Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, Wen JZ, Zhou H, Qu LH. Mol Cancer Ther. 2012;11:1155–1165. doi: 10.1158/1535-7163.MCT-12-0066. [DOI] [PubMed] [Google Scholar]
- 8.a) Xu W, Sun J, Zhang T, Ma B, Cui S, Chen D, He Z. Acta Pharmacol Sin. 2006;27:1642–1646. doi: 10.1111/j.1745-7254.2006.00440.x. [DOI] [PubMed] [Google Scholar]; b) Xu W, Sun J, Zhang T, Ma B, Chen D, He Z. J Shenyang Pharm Univ. 2007;4:220–222. [Google Scholar]
- 9.a) Xu J, Yang J, Ran Q, Wang L, Liu J, Wang Z, Wu X, Hua W, Yuan S, Zhang L, Shen M, Ding Y. Bioorg Med Chem Lett. 2008;18:4741–4744. doi: 10.1016/j.bmcl.2008.06.097. [DOI] [PubMed] [Google Scholar]; b) Wang L, Ran Q, Li D, Yao H, Zhang Y, Yuan S, Zhang L, Shen M, Xu J. Chin J Nat Med. 2011;9:194–198. [Google Scholar]; c) Wang L, Li D, Xu S, Cai H, Yao H, Zhang Y, Jiang J, Xu J. Eur J Med Chem. 2012;52:242–250. doi: 10.1016/j.ejmech.2012.03.024. [DOI] [PubMed] [Google Scholar]; d) Li D, Xu S, Cai H, Pei L, Zhang H, Wang L, Yao H, Wu X, Jiang J, Sun Y, Xu J. Eur J Med Chem. 2013;64:215–221. doi: 10.1016/j.ejmech.2013.04.012. [DOI] [PubMed] [Google Scholar]; e) Li D, Xu S, Cai H, Pei L, Wang L, Wu X, Yao H, Jiang J, Sun Y, Xu J. Chem Med Chem. 2013;8:812–818. doi: 10.1002/cmdc.201200559. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Zhang Y, Lu W, Nan F. Org Biomol Chem. 2011;9:4436–4439. doi: 10.1039/c1ob05611e. [DOI] [PubMed] [Google Scholar]
- 11.a) Ding C, Zhang Y, Chen H, Yang Z, Wild C, Chu L, Liu H, Shen Q, Zhou J. J Med Chem. 2013;56:5048–5058. doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ding C, Zhang Y, Chen H, Wild C, Wang T, White MA, Shen Q, Zhou J. Org Lett. 2013;15:3718–3721. doi: 10.1021/ol4015865. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ding C, Zhang Y, Chen H, Yang Z, Wild C, Ye N, Ester CD, Xiong A, White MA, Shen Q, Zhou J. J Med Chem. 2013;56:8814–8825. doi: 10.1021/jm401248x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bohanon FJ, Wang X, Ding C, Ding Y, Radhakrishnan GL, Rastellini C, Zhou J, Radhakrishnan RS. J Surg Res. 2014;190:55–63. doi: 10.1016/j.jss.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang SX, Xiao WL, Li LM, Li SH, Zhou Y, Ding LS, Lou LG, Sun HD. Org Lett. 2006;8:1157–1160. doi: 10.1021/ol0531379. [DOI] [PubMed] [Google Scholar]
- 13.Gitlitz BJ, Tsao-Wei DD, Groshen S, Davies A, Koczywas M, Belani CP, Argiris A, Ramalingam S, Vokes EE, Edelman M, Hoffman P, Ballas MS, Liu SV, Gandara DR. J Thorac Oncol. 2012;7:574–578. doi: 10.1097/JTO.0b013e31823f43ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ajani JA, Jiang Y, Faust J, Chang BB, Ho L, Yao JC, Rousey S, Dakhil S, Cherny RC, Craig C, Bleyer A. Invest New Drugs. 2006;24:353–357. doi: 10.1007/s10637-006-6452-1. [DOI] [PubMed] [Google Scholar]
- 15.a) Pellissier H. Tetrahedron. 2009;65:2839–2877. [Google Scholar]; b) Dossetter AG, Jamison TF, Jacobsen EN. Angew Chem, Int Ed. 1999;38:2398–2400. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; c) Danishefsky SJ, Selnick HG, Zelle RE, DeNinno MP. J Am Chem Soc. 1988;110:4368–4378. [Google Scholar]
- 16.a) Adams DR, Bhatnagar SP. Synthesis. 1977:661–672. [Google Scholar]; b) Jasti R, Rychnovsky SD. J Am Chem Soc. 2006;128:13640–13648. doi: 10.1021/ja064783l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wildemann H, Dunkelmann P, Muller M, Schmidt B. J Org Chem. 2003;68:799–804. doi: 10.1021/jo0264729. [DOI] [PubMed] [Google Scholar]
- 18.a) Smith AB, Fox RJ, Razler TM. Acc Chem Res. 2008;41:675–687. doi: 10.1021/ar700234r. [DOI] [PubMed] [Google Scholar]; b) Smith AB, Verhoest PR, Minbiole KP, Schelhaas M. J Am Chem Soc. 2001;123:4834–4836. doi: 10.1021/ja0105055. [DOI] [PubMed] [Google Scholar]
- 19.a) Kirkland TA, Colucci JL, Geraci S, Marx MA, Schneider M, Kaelin DE, Jr, Martin SF. J Am Chem Soc. 2001;123:12432–12433. doi: 10.1021/ja011867f. [DOI] [PubMed] [Google Scholar]; b) Pattenden G, Plowright AT. Tetrahedron Lett. 2000;41:983–986. [Google Scholar]
- 20.a) Mukaiyama T. Org React. 1982;28:203–331. [Google Scholar]; b) Clarke PA, Martin WHC, Hargreaves JM, Wilson C, Blake AJ. Chem Comm. 2005:1061–1063. doi: 10.1039/b416247a. [DOI] [PubMed] [Google Scholar]
- 21.a) Boger DL. In: Comprehensive Organic Synthesis. Trost BM, Flemming I, Paquette LA, editors. Vol. 5. Pergamon; Oxford: 1991. pp. 451–512. chap. 4.1. [Google Scholar]; b) Juhl K, Jørgensen KA. Angew Chem, Int Ed. 2003;42:1498–1501. doi: 10.1002/anie.200250652. [DOI] [PubMed] [Google Scholar]; c) Cao CL, Sun XL, Kang YB, Tang Y. Org Lett. 2007;9:4151–4154. doi: 10.1021/ol701669b. [DOI] [PubMed] [Google Scholar]; d) Gallier F, Hussain H, Martel A, Kirschning A, Dujardin G. Org Lett. 2009;11:3060–3063. doi: 10.1021/ol901065e. [DOI] [PubMed] [Google Scholar]
- 22.Zhou W, Cheng Y. Acta Chim Sinica. 1990;48:1185–1190. [Google Scholar]
- 23.a) Li C, Yu X, Lei X. Org Lett. 2010;12:4284–4287. doi: 10.1021/ol101705j. [DOI] [PubMed] [Google Scholar]; b) Uroos M, Hayes CJ. Org Lett. 2010;12:5294–5297. doi: 10.1021/ol102296t. [DOI] [PubMed] [Google Scholar]; c) Li C, Dian L, Zhang W, Lei X. J Am Chem Soc. 2012;134:12414–12417. doi: 10.1021/ja305464s. [DOI] [PubMed] [Google Scholar]
- 24.a) Shen XY, Isogai A, Furihata K, Sun HD, Suzuki A. Phytochemistry. 1994;35:725–729. [Google Scholar]; b) Na Z, Li SH, Xiang W, Zhao AH, Li CM, Sun HD. Chin J Chem. 2002;20:884–886. [Google Scholar]; c) Han QB, Lu Y, Zhang LL, Zheng QT, Sun HD. Tetrahedron Lett. 2004;45:2833–2837. [Google Scholar]
- 25.a) Gizecki P, Dhal R, Toupet L, Dujardin G. Org Lett. 2000;2:585–588. doi: 10.1021/ol991326j. [DOI] [PubMed] [Google Scholar]; b) Gallier F, Hussain H, Martel A, Kirschning A, Dujardin G. Org Lett. 2009;11:3060–3063. doi: 10.1021/ol901065e. [DOI] [PubMed] [Google Scholar]; c) Messer R, Schmitz A, Moesch L, Häner R. J Org Chem. 2004;69:8558–8560. doi: 10.1021/jo048351+. [DOI] [PubMed] [Google Scholar]; d) Johnson SC, Crasto C, Hecht SM. Chem Commun. 1998:1019–1020. [Google Scholar]
- 26.a) Cousins RPC, Ding WC, Pritchard RG, Stoodley RJ. Chem Commun. 1997:2171–2172. [Google Scholar]; b) Turov AV, Tkachuk AA, Khilya VP. Chemistry of Heterocyclic Compounds. 2004;40:986–991. [Google Scholar]; c) Turov AV, Bondarenko SP, Tkachuk AA, Khilya VP. Russ J Org Chem. 2005;41:47–53. [Google Scholar]
- 27.a) Wada E, Pei W, Yasuoka H, Chin U, Kanemasa S. Tetrahedron. 1996;5:1205–1220. [Google Scholar]; b) Wada E, Yasuoka H, Kanemasa S. Chem Lett. 1994:145–148. [Google Scholar]; c) Bogdanowicz-Szwed K, Palasz A. Monatsh Chem. 1997;128:1157–1172. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.