

Abstract
We previously demonstrated that blocking the hepatocyte growth factor (HGF) receptor, c-Met, using a HGF antagonist, NK4, inhibited arthritis in a rheumatoid arthritis (RA) model mice. In the present study, we investigated the role of c-Met signaling in synovial cell function. We demonstrated that synovial tissues from RA patients and MH7A cells, a human RA synovial cell line, expressed HGF and c-Met. HGF and c-Met expression in RA synovium was increased compared to osteoarthritis synovium suggesting increased c-Met signaling in RA synovial cells. The c-Met inhibitor, SU11274, inhibited ERK1/2 and AKT phosphorylation in HGF-stimulated MH7A cells. MEK and PI3K inhibitors suppressed production of matrix metalloproteinase-3 (MMP-3), vascular endothelial growth factor (VEGF) and prostaglandin E2 (PGE2) by MH7A cells, suggesting that c-Met-MEK-ERK and c-Met-PI3K-AKT pathways are involved positively regulating MH7A cell function. Although SU11274 suppressed MMP-3 and VEGF production it enhanced PGE2 production by MH7A cells suggesting that negative regulation by c-Met signaling, independent of the MEK-ERK and PI3K-AKT pathways, is involved in PGE2 production. Blocking c-Met signaling may be therapeutically useful to inhibit angiogenesis and cartilage and bone destruction by inhibiting VEGF and MMP-3 production, while enhancing PGE2 production in synovial cells in RA.
Keywords: c-Met, Hepatocyte growth factor, Rheumatoid arthritis, Synovial cell
Introduction
Rheumatoid arthritis (RA) is characterized by an aggressive synovial expansion due to angiogenesis and subsequent destruction of the underlying cartilage and bone (Szekanecz and Koch 2008, Scot et al. 2010, Szekanecz et al. 2010). Synovial cells produce inflammatory mediators, proteases, and angiogenic growth factors such as prostaglandin E2 (PGE2), matrix metalloproteinases (MMPs), and vascular endothelial growth factor (VEGF) (Pap et al. 2000).
MMPs are responsible for the destruction of cartilage and bone in RA joints. In particular, MMP-1 and MMP-3 are critical proteases in tissue degradation in RA (Walakovits et al. 1992, Green et al. 2003). PGE2 plays a major role in angiogenesis through the expression of VEGF in rheumatoid synovium (Ben-Av et al. 1995). Moreover, PGE2 has been shown to trigger bone resorption by osteoclasts (Mino et al. 1998) suggesting that the inflammation of RA is closely related to the production of PGE2 by synovial cells (Sano et al. 1992). TNF-a is a well-known cytokine that stimulates the production of MMPs and PGE2 through activation of intracellular signaling pathways including mitogen-activated protein kinase (MAPK) and activator protein-1 (AP-1) (Han et al. 2001, Reunanen et al. 2002, Martel-Pelletier et al. 2003, Akaogi et al. 2006).
New blood vessel formation is critically important for synovial expansion in RA. Angiogenic growth factors, such as VEGF and hepatocyte growth factor (HGF), enhance angiogenesis and induce synovial cell proliferation (Nagashima et al. 2001, Szekanecz et al. 2009, 2010). The HGF receptor, c-Met, appears to induce synovial cell proliferation in RA. We previously demonstrated that blocking c-Met signaling using a HGF antagonist, NK4, inhibited arthritis in a RA model of SKG mice (Tsunemi et al. 2013).
SU11274 is a prototypic anti-c-Met small molecule and an effective inhibitor of HGF-dependent signaling. SU11274 abrogates HGF-induced phosphorylation of c-Met and its downstream signal (Sattler et al. 2003). In the present study, we examined the effect of blocking c-Met signaling using NK4 or SU11274 on synovial cell function using a human synovial cell line in vitro.
Materials and methods
Cell line and cell culture
MH7A synovial cells isolated from intra-articular soft tissues of the knee joints of RA patients were obtained from Riken Cell Bank (Saitama, Japan). MH7A is a cell line established by transfecting with the SV40 T antigen (Miyazawa et al. 1988). MH7A cells were cultured in RPMI 1640 (Sigma, St. Louis, MO) containing 10% heat-inactivated fetal bovine serum (FBS) (Whittaker, Walkersville, MD), 100 units/ml of penicillin and 100 μg/ml of streptomycin (Invitrogen, San Diego, CA) at 37°C in an atmosphere of 5% CO2 in air.
Immunohistochemical analysis of HGF and c-Met expression in RA synovium
Synovial tissue specimens isolated from patients with RA and osteoarthritis (OA) at the time of arthroscopic biopsy or total joint replacement were stained as reported previously (Kitano et al. 2006). Briefly, synovial tissue specimens were preserved in 10% formalin, embedded in paraffin, and serially sectioned onto microscope slides at a thickness of 4 μm. Synovial tissues were stained with either anti-human c-Met antibody (1:200 dilution in PBS, rabbit IgG: Santa Cruz Biotechnology) or anti-human HGF antibody (1:200 dilution in PBS, rabbit IgG: Santa Cruz Biotechnology), washed, incubated with biotinylated goat anti-rabbit IgG (1:1000 dilution in PBS: Santa Cruz Biotechnology), washed, incubated with avidin-biotinylated horseradish peroxidase complex (ABC) and diaminobenzidine tetrahydrochloride ([DAB], Elite kit; Vector Laboratories, Inc., Burlingame, CA, USA), and counterstained with Mayer’s hematoxylin. All patients gave informed consent, and the Institutional Medical Ethics Committee approved the study protocol.
Western blot analysis
Western blot analysis was performed as reported previously (Dong et al. 2001). Briefly, MH7A cells (1 × 107 cells/well) were lysed in RIPA lysis buffer (Santa Cruz Biotechnology, CA), and protein content was determined using Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA) with bovine serum albumin as a standard. Each sample (20 μg) was resolved on 1% polyacrylamide gels under denaturing conditions and then transferred to 0.45-μm nitrocellulose membranes. After blocking overnight at 4°C with 5% nonfat milk in Tris-buffered saline-0.01% Tween 20 (Santa Cruz Biotechnology), membranes were incubated with primary antibodies against either phospho-extracellular signal-regulated kinase (ERK) 1/2, phospho-AKT, or β-actin (1:1000 dilution in PBS; Santa Cruz Biotechnology) after 12 h stimulation in the presence of HGF (10 ng/ml) with or without the c-Met inhibitor, SU11274 (Tocris Bioscience, UK). After washing the membranes with Tris buffered saline–0.05% Tween 20 (washing buffer), HRP-conjugated goat anti-rabbit secondary antibody (1:1000 dilution in PBS; Santa Cruz Biotechnology) was added, followed by incubation for 45 min. After further washing, the color was developed with luminol reagent (Santa Cruz Biotechnology), and HRP activity of the blots was analyzed using an LAS1000 imager. Luminol staining was done according to manufacturer’s instructions (Santa Cruz).
Measurement of PGE2, VEGF and MMP-3 production by MH7A cells
MH7A cells (2 × 104 cells/well) were plated in flat-bottomed 24-well microplates and cultured with or without either NK4 or c-Met inhibitor, SU11274 (Tocris Bioscience) in the presence or absence of 100 ng/ml TNF-α (Genzyme Pharmaceuticals, Cambridge, MA, USA). MH7A cells (2 × 104 cells/well) were also cultured with or without either the MEK inhibitor, PD98059 (Cell Signaling Technology, MA), or the PI3K inhibitor, LY294002 (Cell Signaling Technology), in the presence or absence of 100 ng/ml TNF-α. After 72 hours of incubation, PGE2, VEGF and MMP-3 production was determined by assaying supernatants with ELISAs using either anti-human PGE2, VEGF and MMP-3 monoclonal antibodies (Genzyme), respectively, according to the manufacturer's protocol.
Statistical analysis
Results are expressed as the mean ± SEM. The significance of the difference between the experimental results and control values was determined by a Student’s t-test. P values less than 0.05 were considered significant.
Results
HGF and c-Met expression in the synovium of RA patients
To examine whether c-Met signaling pathways are involved in the pathogenesis of RA, we examined the expression of HGF and c-Met in the synovium of OA and RA patients. Immunohistochemical analysis demonstrated that HGF and c-Met expression in synovial lining cells was higher in RA patients than in OA patients (Figure 1A). We also demonstrated that MH7A cells, a synovial cell line established from RA patients, expressed HGF and c-Met by Western blot analysis (Figure 1B). These results suggest that c-Met protein is increased in the synovial tissues of RA patients.
Figure 1.
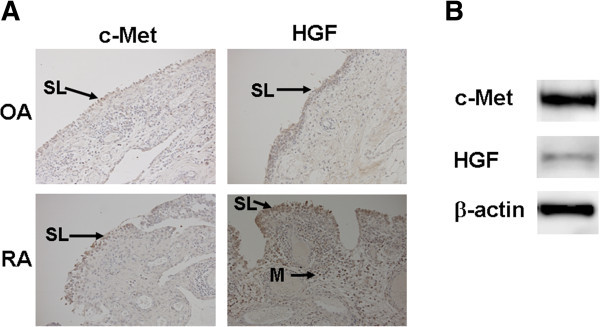
c-Met and HGF expression in synovial cells. A. Immunohistochemical analysis of joints. c-Met and HGF expression in synovial lining cells of RA patients was higher than in OA patients. HGF was expressed in infiltrating mononuclear cells (SL, synovial lining cell layer; M, mononuclear cells). Original magnification ×200. B. Western blot analysis of MH7A cells. c-Met and HGF were expressed in MH7A cells.
SU11274 inhibits ERK1/2 and AKT phosphorylation by MH7A cells
MEK/ERK and PI3-AKT signaling pathways can be activated by a variety of growth factors such as insulin and HGF (Dong et al. 2001). To determine whether ERK or AKT was downstream of c-Met signaling, we examined the effect of the SU11274 inhibitor on ERK1/2 and AKT phosphorylation in response to HGF treatment. Stimulation of MH7A cells with HGF led to a significant increase in phosphorylation of ERK1/2 and AKT. Conversely, although treatment with the c-Met inhibitor (0.25 nM) did not affect treatment with the c-Met inhibitor (2.5 nM) reduced HGF-induced ERK1/2 and AKT phosphorylation, indicating that minimal dose of the c-Met inhibitor to block HGF-induced p-ERK1/2 and p-AKT expression is 0.25 nM (Figure 2). These results suggest that c-Met-MEK-ERK and c-Met-PI3-AKT signaling pathways are utilized in MH7A cells.
Figure 2.
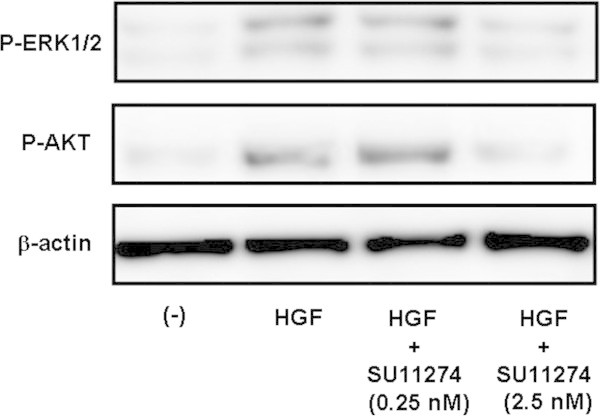
Effects of the SU11274 inhibitor on ERK1/2 and AKT phosphorylation in MH7A cells. MH7A cells were treated with HGF (10 ng/ml) with or without SU11274 (0.25 - 2.5 nM) and p-ERK1/2 and p-AKT expression was determined by Western blot analysis. We also determined β-actin expression as a control. Representative data of p-ERK1/2 and p-AKT expression by MH7A cells is shown.
Both MEK-ERK and PI3-AKT signaling pathways are involved in the production of MMP-3, VEGF and PGE2 by MH7A cells
To confirm the involvement of MEK-ERK and PI3-AKT signaling pathways in the production of MMP-3, VEGF and PGE2, we examined whether pretreatment with either MEK or PI3K inhibitors affected MMP-3, VEGF and PGE2 production. Both the MEK inhibitor, PD98059 and the PI3K inhibitor, LY294002, inhibited MMP-3, VEGF and PGE2 production (Figure 3). These results suggest that both MEK-ERK and PI3-AKT signaling pathways are involved in MMP-3, VEGF and PGE2 production.
Figure 3.
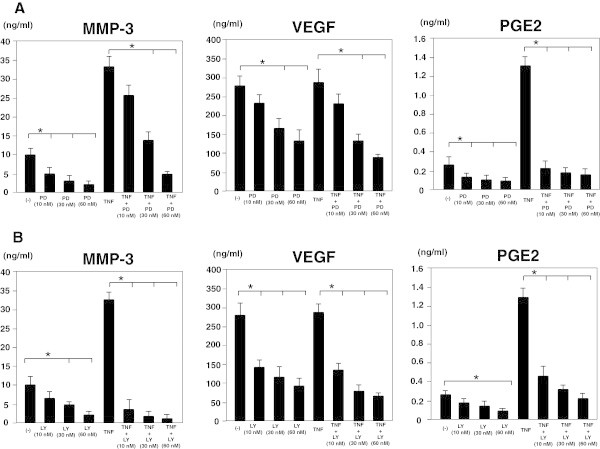
Effects of MEK or PI3K inhibitors on the production of MMP-3, VEGF and PGE2 by MH7A cells. A. MH7A cells (2 x 104 cells/well) were cultured with the MEK inhibitor, PD98059 (10 - 60 nM), in the presence or absence of 100 ng/ml TNF-α. After 72 hours of incubation, MMP-3, VEGF and PGE2 production was determined by analysis of supernatants using ELISA. B. MH7A cells (2 x 104 cells/well) were cultured with the PI3K inhibitor, LY294002 (10 - 60 nM), in the presence or absence of 100 ng/ml TNF-α. After 72 hours of incubation, MMP-3, VEGF and PGE2 production was determined by analysis of supernatants using ELISA. Data represent the mean ± SD of three independent experiments. *P < 0.01.
Blocking c-Met signaling using NK4 or SU11274 inhibits MMP-3 and VEGF, but enhances PGE2 production by MH7A cells
MH7A cells were treated with either NK4 or the c-Met inhibitor, SU11274, in the presence or absence of TNF-α for 72 hours and the production of MMP-3, VEGF and PGE2 was determined by ELISA. TNF-α significantly enhanced MMP-3 and PGE2, but not VEGF production by MH7A cells. Both NK4 and SU11274 inhibited MMP-3 and VEGF, but enhanced PGE2 production by MH7A cells (Figure 4A). We next examined the effect of different doses of SU11274 on MH7A cells. As shown in Figure 4B, SU11274 dose-dependently inhibited MMP-3 and VEGF, but enhanced PGE2 production by MH7A cells (Figure 4B). These results indicate that negative regulation by c-Met signaling independent of the MEK-ERK and PI3K-AKT pathway is also involved in PGE2 production.
Figure 4.
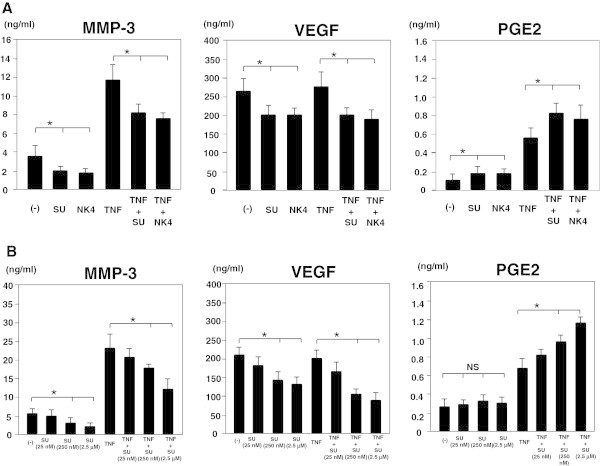
Effects of blocking c-Met signaling on the production of MMP-3, VEGF, and PGE2 by MH7A cells. A. MH7A cells (2 x 104 cells/well) were cultured with or without either NK4 or SU11274 in the presence or absence of 100 ng/ml TNF-α. After 72 hours of incubation, MMP-3, VEGF and PGE2 production was determined by analysis of supernatants using ELISA. B. MH7A cells (2 x 104 cells/well) were cultured with different dose of SU11274 in the presence or absence of 100 ng/ml TNF-α. After 72 hours of incubation, MMP-3, VEGF and PGE2 production was determined by analysis of supernatants using ELISA. Data represent the mean ± SD of three independent experiments. *P < 0.01. NS, not significant.
Discussion
We demonstrated that HGF and its receptor, c-Met, are expressed in synovial tissues and that both HGF and c-Met expression is increased in RA patients, but not OA patients. Synovial expansion is dependent on new blood vessel formation, which is important for the delivery of oxygen, nutrients, and inflammatory cells to the lesions of RA (Szekanecz and Koch 2008, Scot et al. 2010, Szekanecz et al. 2010). It has been reported that HGF and c-Met are expressed in synovial tissues in patients with RA (Nagashima et al. 2001). HGF in the synovial fluid of patients with RA is produced by synovial cells and is related to disease activity (Yukioka et al. 1994). Taken together, HGF may contribute to the development of RA by its potent angiogenic activities, which stimulate synovial cell proliferation.
We demonstrated that inhibition of c-Met signaling resulted in decreased VEGF and MMP-3 production by MH7A cells. VEGF is a potent angiogenic factor that is pivotal in RA pathogenesis. The role for HGF in VEGF production has been implicated in some cell types such as squamous cell carcinoma and synovial cells. It was demonstrated that c-Met signaling activates the PI3-AKT signaling pathway leading to up-regulation of VEGF expression (Dong et al. 2001, Lin et al. 2012). The present study demonstrated that c-Met signaling activates both the MEK-ERK and PI3-AKT signaling pathways leading to up-regulation of VEGF expression in the synovial cell line.
Esposito et al. (2009) have studied the anti-fibrogenic mechanisms of HGF in which increasing MMP expression promoted collagen degradation (Esposito et al. 2009). Addition of HGF to fibroblast cultures of scleroderma patients significantly decreased collagen production and increased MMP-1 expression and activity (Jinnin et al. 2005). Addition of HGF to keloid fibroblastic cells significantly increased the mRNA expression of MMP-1 and MMP-3 in a dose-dependent manner (Lee et al. 2011). This means that HGF may have a therapeutic effect on keloids that is similar to pathologic dermal fibrosis seen in scleroderma (Jinnin et al. 2005, Lee et al. 2011). In a previous study, we demonstrated that HGF gene transfection improved dermal sclerosis in mouse model of scleroderma (Iwasaki et al. 2006). The present results that the blocking c-Met signaling using the c-Met inhibitor (SU11274) inhibited MMP-3 production by MH7A cells provide support for the previous findings.
c-Met is a proto-oncogene that encodes a protein known as the HGF receptor (Liu et al. 1994, Zarnegar and Michalopoulos 1995). Upon HGF stimulation, c-Met induces c-Met kinase catalytic activity, which triggers transphosphorylation of tyrosines Tyr-1234 and Tyr-1235. These two tyrosines engage various signal transducers to initiate a whole spectrum of biological activities driven by c-Met. Two major signaling pathways, MEK/ERK and PI3/AKT, are activated by c-Met (Bottaro et al. 1991, Graziani et al. 1991, O'Brien et al. 2004, Mahtouk et al. 2010). We observed that both ERK1/2 and AKT phosphorylation was enhanced by HGF and was suppressed by the c-Met inhibitor. We also observed that both MEK and PI3K inhibitors suppressed VEGF and MMP-3 production. These results indicate that both c-Met-MEK/ERK and c-Met-PI3/AKT pathways are involved in the production of MMP-3 and VEGF by MH7A cells.
In contrast to the inhibitory effect of the c-Met inhibitor on MMP-3 and VEGF production, blocking c-Met signaling enhanced PGE2 production by MH7A cells. Previous work reported conflicting results that c-Met signaling can promote PGE2 production in colorectal cancer cells via COX-2 up-regulation and 15-PGDH down-regulation at the protein and messenger RNA level (Moore et al. 2009). They demonstrated that both ERK and AKT signals are required for COX-2 protein up-regulation and 15-PGDH down-regulation downstream of c-Met. Furthermore, SU11274 reduced COX-2 expression and increased 15-PGDH expression in high c-Met-expressing cells (Moore et al. 2009). We still do not know the mechanism by which the c-Met inhibitor enhances PGE2 production. Supporting our findings, Zhang et al., performed in vitro experiments on rabbit tendon cells and in vivo experiments in a mouse Achilles tendon injury model. They demonstrated that HGF treatment suppressed IL-1β-induced gene expression of COX-1, COX-2, and the production of PGE2 in tendon cell culture. Injection of HGF into wounded mouse Achilles tendons in vivo decreased PGE2 production in the tissues (Zhang et al. 2013).
HGF is known to function both as an anti-inflammatory agent and as an anti-fibrotic regulator (Gong 2008). Its anti-inflammatory action is primarily mediated by the disruption of signaling by the transcription factor, NF-kB, which is a critical regulator of inflammation. This was demonstrated both in an in vivo renal inflammation mouse model (Giannopoulou et al. 2008) and in human chondrocytes in vitro (Bendinelli et al. 2010). Such a c-Met-mediated negative signal for PGE2 production, independent of the c-Met-MEK-ERK and c-Met-PI3-AKT pathways, may be responsible for the anti-inflammatory effects of HGF on PGE2 production observed in this study. However, HGF has strong angiogenic activities which are crucial for synovial cell proliferation in RA. In this article, we suggested that blocking c-Met signaling inhibited angiogenesis and cartilage and bone destruction by the inhibition of VEGF and MMP-3 production by synovial cell line, respectively. We previously reported that blocking c-Met signaling using HGF antagonisit, NK4, inhibited arthritis in RA model of SKG mice (Tsunemi et al. 2013). Therefore, we think that although blocking c-Met signaling may enhances inflammatory responses, it is effective for the treatment of RA by its strong inhibition of angiogenesis and protease-mediated bone destruction.
In conclusion, this is the first report to demonstrate the differential regulation of c-Met signaling pathways for synovial cell function. Blocking c-Met signaling may be useful to inhibit angiogenesis and cartilage and bone destruction by inhibiting VEGF and MMP-3 production, respectively, while it enhances PGE2 production by synovial cells in RA.
Acknowledgements
This work was partially supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan, a Grant-in-Aid for Scientific Research (C).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS and TI conceived the project and drafted the manuscript. ST and SK performed all biochemical experiment. SK and MS performed immunohistochmical analysis and c-Met expression in RA synovium. TI and HS interpreted the results. All authors read and approved the final manuscript.
Contributor Information
Seiji Shibasaki, Email: seiji@huhs.ac.jp.
Sachi Tsunemi, Email: sachi-t@huhs.ac.jp.
Sachie Kitano, Email: sachie@hyo-med.ac.jp.
Masahiro Sekiguchi, Email: masa-s@hyo-med.ac.jp.
Hajime Sano, Email: hsano@hyo-med.ac.jp.
Tsuyoshi Iwasaki, Email: tsuyo-i@huhs.ac.jp.
References
- Akaogi J, Nozaki T, Satoh M, Yamada H. Role of PGE2 and EP receptors in the pathogenesis of rheumatoid arthritis and as a novel therapeutic strategy. Endocr Metab Immune Disord Drug Targets. 2006;4:383–394. doi: 10.2174/187153006779025711. [DOI] [PubMed] [Google Scholar]
- Ben-Av P, Crofford LJ, Wilder RL, Hla T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleukin-1: a potential mechanism for inflammatory angiogenesis. FEBS Lett. 1995;372:83–87. doi: 10.1016/0014-5793(95)00956-A. [DOI] [PubMed] [Google Scholar]
- Bendinelli P, Matteucci E, Dogliotti G, Corsi MM, Banfi G, Maroni P, Desiderio MA. Molecular basis of anti-inflammatory action of platelet-rich plasma on human chondrocytes: mechanisms of NF-kappaB inhibition via HGF. J Cell Physiol. 2010;225:757–766. doi: 10.1002/jcp.22274. [DOI] [PubMed] [Google Scholar]
- Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met protooncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte Growth Factor/Scatter Factor-induced Activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911–5918. [PubMed] [Google Scholar]
- Esposito C, Parrilla B, Cornacchia F, Grosjean F, Mangione F, Serpieri N, Valentino R, Villa L, Arra M, Esposito V, Dal Canton A. The antifibrogenic effect of hepatocyte growth factor (HGF) on renal tubular (HK-2) cells is dependent on cell growth. Growth Factors. 2009;27:173–180. doi: 10.1080/08977190902834077. [DOI] [PubMed] [Google Scholar]
- Giannopoulou M, Dai C, Tan X, Wen X, Michalopoulos GK, Liu Y. Hepatocyte growth factor exerts its anti-inflammatory action by disrupting nuclear factor-kappaB signaling. Am J Pathol. 2008;173:30–41. doi: 10.2353/ajpath.2008.070583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R. Multi-target anti-inflammatory action of hepatocyte growth factor. Curr Opin Investig Drugs. 2008;9:1163–1170. [PubMed] [Google Scholar]
- Graziani A, Gramaglia D, Cantley LC, Comoglio PM. The tyrosinephosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem. 1991;266:22087–22090. [PubMed] [Google Scholar]
- Green MJ, Gough AK, Devlin J, Smith J, Astin P, Taylor D, Emery P. Serum MMP–3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology (Oxford) 2003;4:83–88. doi: 10.1093/rheumatology/keg037. [DOI] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki T, Imado T, Kitano S, Sano H. Hepatocyte growth factor ameliorates dermal sclerosis in the tight-skin mouse model of scleroderma. Arthritis Res Ther. 2006;8:R161. doi: 10.1186/ar2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Mimura Y, Asano Y, Yamane K, Tamaki K. Effects of hepatocyte growth factor on the expression of type I collagen and matrix metalloproteinase-1 in normal and scleroderma dermal fibroblasts. J Invest Dermatol. 2005;124:324–330. doi: 10.1111/j.0022-202X.2004.23601.x. [DOI] [PubMed] [Google Scholar]
- Kitano M, Hla T, Sekiguchi M, Kawahito Y, Yoshimura R, Miyazawa K, Iwasaki T, Sano H, Saba JD, Tam YY. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium. Regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum. 2006;54:742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Park SE, Rah DK. Effects of hepatocyte growth factor on collagen synthesis and matrix metalloproteinase production in keloids. J Korean Med Sci. 2011;26:1081–1086. doi: 10.3346/jkms.2011.26.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YM, Huang YL, Fong YC, Tsai CH, Chou MC, Tang CH. Hepatocyte Growth Factor Increases Vascular Endothelial Growth Factor-A Production in Human Synovial Fibroblasts through c-Met Receptor Pathway. PLoS One. 2012;7:e50924. doi: 10.1371/journal.pone.0050924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Michalopoulos GK, Zarnegar R. Structural and functional characterization of the mouse hepatocyte growth factor gene promoter. J Biol Chem. 1994;269:4152–4160. [PubMed] [Google Scholar]
- Mahtouk K, Tjin EP, Spaargaren M, Pals ST. The HGF/MET pathway as target for the treatment of multiple myeloma and B cell lymphomas. Biochim Biophys Acta. 2010;1806:208–219. doi: 10.1016/j.bbcan.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Martel-Pelletier J, Pelletier JP, Fahmi H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin Arthritis Rheum. 2003;33:155–167. doi: 10.1016/S0049-0172(03)00134-3. [DOI] [PubMed] [Google Scholar]
- Mino T, Sugiyama E, Taki H, Kuroda A, Yamashita N, Maruyama M, Kobayashi M. Interleukin-1alpha and tumor necrosis factor alpha synergistically stimulate prostaglandin E2–dependent production of interleukin-11 in rheumatoid synovial fibroblasts. Arthritis Rheum. 1998;42:2004–2013. doi: 10.1002/1529-0131(199811)41:11<2004::AID-ART16>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Mori H, Okudaira H. Establishment and characterization of novel human rheumatoid fibroblast-like synoviocyte line, MH7A, immortalized with SV40T antigen. J Biochem. 1988;124:1153–1162. doi: 10.1093/oxfordjournals.jbchem.a022233. [DOI] [PubMed] [Google Scholar]
- Moore AE, Greenhoughy A, Robertsy HR, Hicks DJ, Patsos HA, Williams AC, Paraskeva C. HGF/Met signalling promotes PGE2 biogenesis via regulation of COX-2 and 15-PGDH expression in colorectal cancer cells. Carcinogenesis. 2009;30:1796–1804. doi: 10.1093/carcin/bgp183. [DOI] [PubMed] [Google Scholar]
- Nagashima M, Hasegawa J, Kato K, Yamazaki J, Nishigai K, Ishiwata T, Asano G, Yoshino S. Hepatocyte growth factor (HGF), HGF activator, and c-Met in synovial tissues in rheumtoid arthritis and osteoarthritis. J Rheumatol. 2001;28:1772–1778. [PubMed] [Google Scholar]
- O'Brien LE, Tang K, Kats ES, Kats ES, Schutz-Geschwender A, Lipschutz JH, Mostov KE. ERK and MMPs sequentially regulate distinct stages of epithelial tubule development. Dev Cell. 2004;7:21–32. doi: 10.1016/j.devcel.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Pap T, Muller-Ladner U, Gay RE, Gay S. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–367. doi: 10.1186/ar113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kähäri VM. Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J Biol Chem. 2002;277:32360–32368. doi: 10.1074/jbc.M204296200. [DOI] [PubMed] [Google Scholar]
- Sano H, Hla T, Maier JA, Crofford LJ, Case JP, Maciag T, Wilder RL. In vitro cyclooxygenase expression in synovial tissues of patients with rheumatoid arthritis and osteoarthritis and rats with adjuvant and streptococcal cell wall arthritis. J Clin Invest. 1992;89:97–108. doi: 10.1172/JCI115591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Pride YB, Ma P, Gramlich JL, Chu SC, Quinnan LA, Shirazian S, Liang C, Podar K, Christensen JG, Salgia R. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003;63:5462–5469. [PubMed] [Google Scholar]
- Scot DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- Szekanecz Z, Koch AE. Vascular involvement in rheumatic diseases: ‘vascular rheumatology’. Arthritis Res Ther. 2008;10:224. doi: 10.1186/ar2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekanecz Z, Besenyei T, Paragh G, Koch AE. Angiogenesis in rheumatoid arthritis. Autoimmunity. 2009;42:563–573. doi: 10.1080/08916930903143083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekanecz Z, Besenyei T, Szentpétery A, Koch AE. Angiogenesis and vasculogenesis in rheumatoid arthritis. Curr Opin Rheumatol. 2010;22:299–306. doi: 10.1097/BOR.0b013e328337c95a. [DOI] [PubMed] [Google Scholar]
- Tsunemi S, Iwasaki T, Kitano S, Matsumoto K, Takagi-Kimura M, Kubo S, Tamaoki T, Sano H. Molecular targeting of hepatocyte growth factor by an antagonist, NK4, in the treatment of rheumatoid arthritis. Arthritis Res Ther. 2013;22:R75. doi: 10.1186/ar4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walakovits LA, Moore VL, Bhardwaj N, Gallick GS, Lark MW. Detection of stromelysin and collagenase in synovial fluid from patients with rheumatoid arthritis and posttraumatic knee injury. Arthritis Rheum. 1992;3:35–42. doi: 10.1002/art.1780350106. [DOI] [PubMed] [Google Scholar]
- Yukioka K, Inaba M, Furumitsu Y, Yukioka M, Nishino T, Goto H, Nishizawa Y, Morii H. Levels of hepatocyte growth factor in synovial fluid and serum of patients with rheumatoid arthritis and release of hepatocyte growth factor by rheumatoid synovial fluid cells. J Rheumatol. 1994;21:2184–2189. [PubMed] [Google Scholar]
- Zarnegar R, Michalopoulos GK. The many faces of hepatocyte growth factor: from hepatopoiesis to hematopoiesis. J Cell Biol. 1995;129:1177–1180. doi: 10.1083/jcb.129.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Middleton KK, Fu FH, Im HJ, Wang JH. HGF Mediates the Anti-inflammatory Effects of PRP on Injured Tendons. PLoS One. 2013;8:e67303. doi: 10.1371/journal.pone.0067303. [DOI] [PMC free article] [PubMed] [Google Scholar]