

Abstract
The differential diagnosis of pyrexia, lung nodules and granulomas includes tuberculosis, vasculitis and rarely a malignancy. In countries where tuberculosis or histoplasmosis is endemic, these are the first consideration and often ruled out by microbiological investigations. Vasculitis like granulomatosis with polyangitis (Wegener’s granulomatosis), Churg strauss syndrome and sarcoidosis, which are the second consideration, are ruled out by serological investigations. Confirmation of malignancy merits histopathology. This case highlights how a rare diagnosis of pulmonary lymphomatoid granulomatosis was reached after an open lung biopsy. The following case also describes the natural history of this rare disease as it showed transient spontaneous remission but ultimately required therapy.
Keywords: Pyrexia, Lung nodules, Granulomas, Pulmonary lymphomatoid granulomatosis
Case report
A 25 year old male presented with eight months history of intermittent fever, dyspnea on exertion and fatigability. He had non productive cough, loss of appetite and weight loss. There was no past history or contact with a case of tuberculosis. There was no history suggestive of vasculitis. There was no history of substance abuse or high risk behavior. General physical examination did not reveal any skin lesions or lymphadenopathy. Chest examination revealed bilateral bibasilar crackles with bronchial breathing. Spleen was palpable 6 cm below costal margin. His chest x-ray and computed tomography (CT) scan (Fig. 1a and b) showed multiple bilateral lung nodules of variable sizes with irregular margins and left pneumothorax. There was no hilar or mediastinal lymphadenopathy.
Fig. 1.
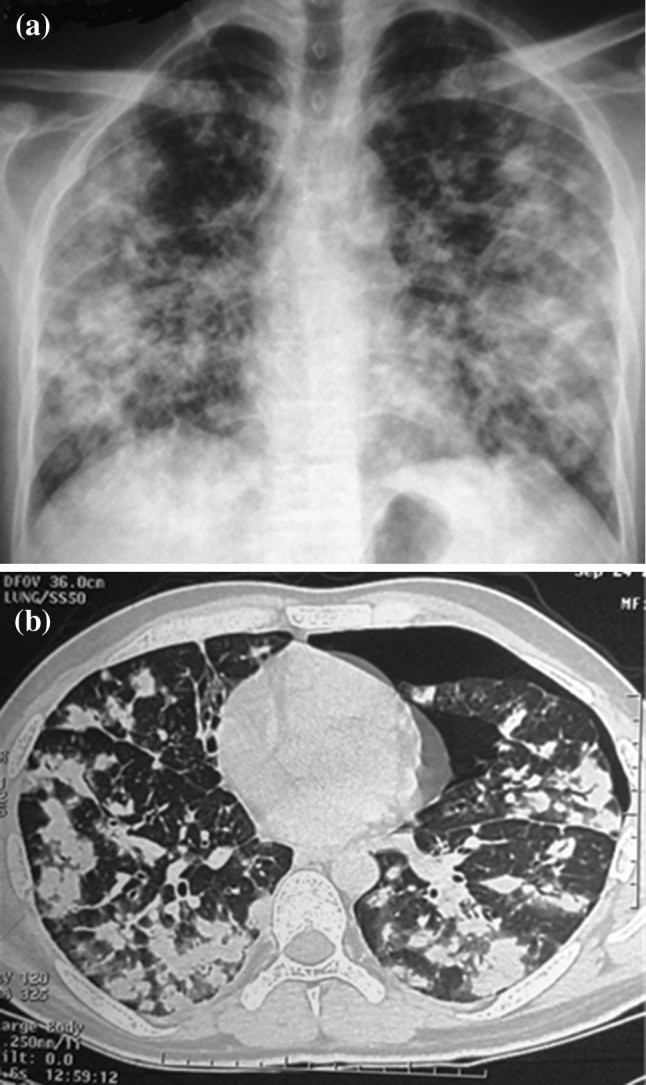
a CXR and b CECT chest at baseline showing bilateral lung nodules of varying sizes
Hemogram showed thrombocytopenia (68 × 109/L) and leucopenia (1.7 × 109/L). Lactate dehydrogenase (LDH) was elevated to three times the upper limit of normal (978 g/L). All bacterial cultures in blood, sputum and urine culture were sterile. Sputum for acid fast bacillus on two occasions and fungus was negative. Viral work-up HBs Ag, anti-HCV, HIV and vasculitis work-up ANA, ANCA was also negative. Investigations for sarcoidosis; serum calcium, 24 h urinary calcium and serum ACE level were within normal range. A transthoracic echocardiography did not show evidence of vegetations. A fibreoptic bronchoscopy with bronchoalveolar lavage and transbronchial biopsy was inconclusive. A bone marrow done at this time point showed hypercellular marrow and didn’t add to the diagnosis.
He was initially managed with antibiotics and left intercostal tube drainage for pneumothorax. As diagnosis was not reached, he was advised to undergo open lung biopsy, which he declined. On follow-up he had spontaneous partial remission in the pulmonary nodules and cytopenias. Six months later he had recurrence of symptoms with progression of the pulmonary nodules. A whole body fluoro-deoxy glucose-positron emission tomography (FDG-PET) scan showed intense uptake in these nodules (maximum SUV 23) with uptake in an ill-defined subcutaneous soft tissue overlying L3 vertebra (Fig. 2). A fine needle aspiration cytology at this lesion was inconclusive. This time he underwent open lung biopsy. The histopathology (Fig. 3) revealed a diagnosis of grade I lymphomatoid granulomatosis. Though he had bicytopenia, bone marrow was not involved. The cause of the cytopenias was thought be due to the disease process per se or possible hypersplenism. Immunodeficiency work-up revealed normal immunoglobulins, T cell lymphopenia, negative Nitro blue tetrazolium test, negative CMV and EBV serology.
Fig. 2.
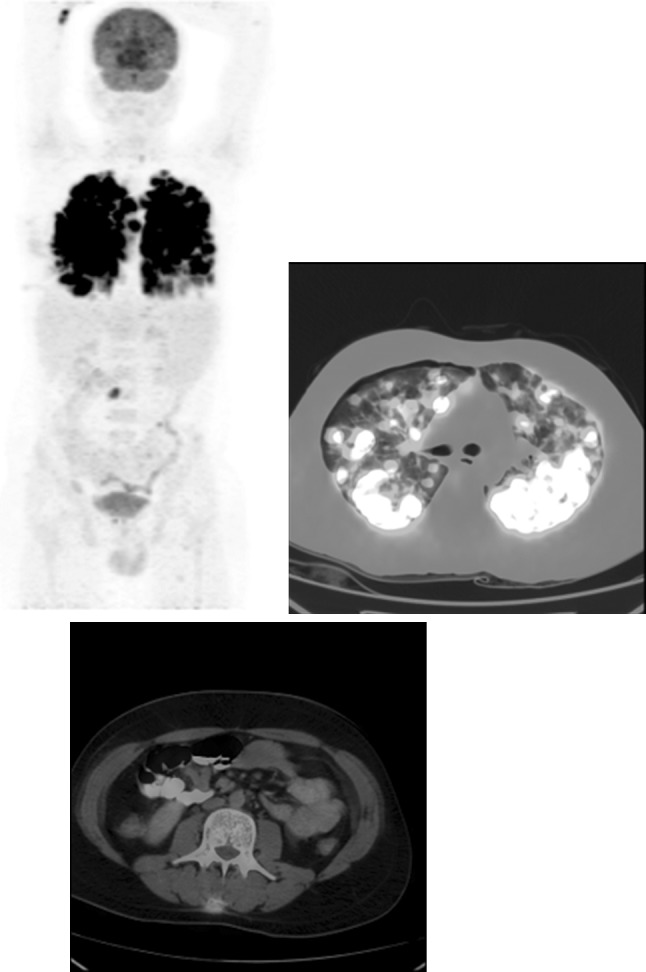
Baseline PET-CT showing intense uptake in the lung nodules (maximum SUV 23) with uptake in an ill-defined subcutaneous soft tissue overlying L3 vertebra
Fig. 3.
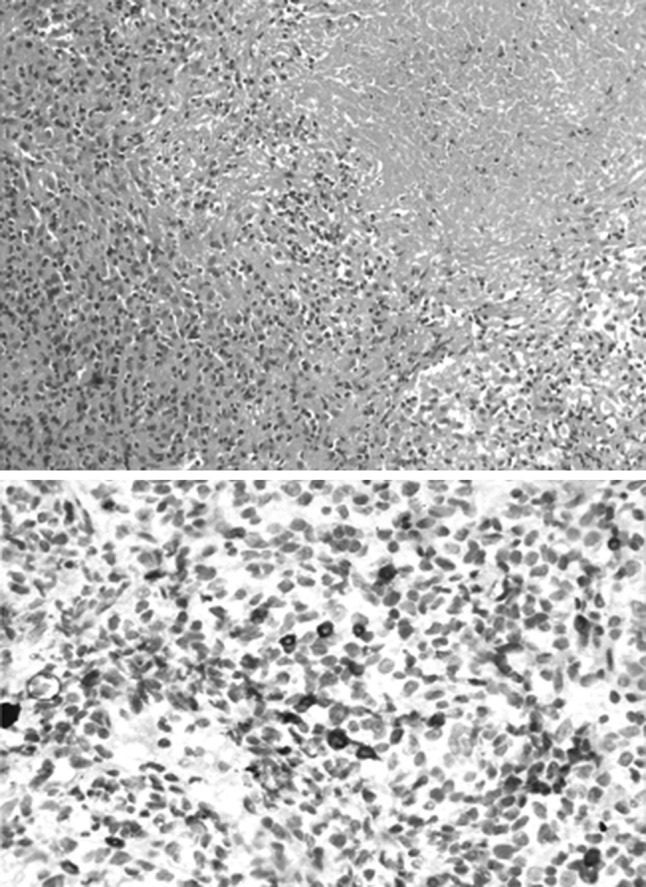
Histopathology shows large nodules composed of collection of lymphocytes and histiocytes admixed with occasional atypical cells. One of the nodules shows infiltration of these lymphoid cells into the walls of thickened blood vessels and large area of necrosis. The immunohistochemistry showed the atypical cells positive for CD20 (shown) and CD79a. These cells were negative for CD30, CD56, S-100 and CD1a immunostains (not shown). Rest of the lymphohistiocytic collection was composed of CD3 positive T cells and CD88 positive macrophages
He was initially treated with single agent prednisolone (60 mg/m2/day), considering the low grade; which was tapered gradually and stopped after 6 months as he achieved a complete remission (CR). However he relapsed 2 months later. This time he was given six cycles of CHOP-21. Rituximab could not be given in view of financial limitations. End of treatment PET-CT (Fig. 4) showed a complete metabolic response though residual nodules were seen. This was taken as CR as per the PERCIST criteria. His counts at the end of treatment were hemoglobin 120 g/L, platelet of 160 × 109/L and WBC of 4.4 × 109/L. His spleen had regressed and was not palpable. He continues to be in remission 10 months after the last cycle.
Fig. 4.
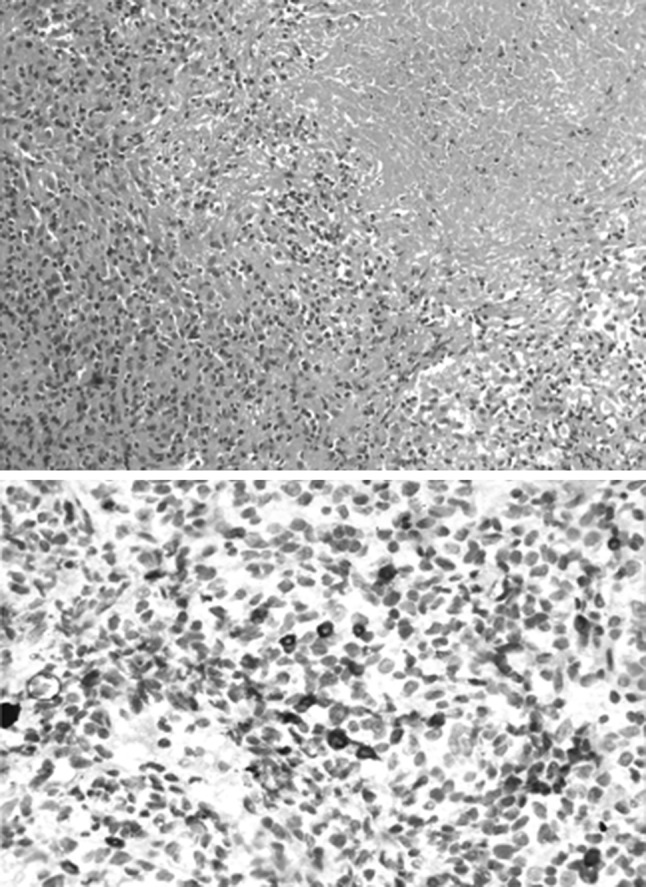
Follow-up PET-CT (End of treatment) showing complete metabolic response
Discussion
Lymphomatoid granulomatosis (LYG) was first described by Liebow et al. as a clinicopathological entity closely mimicking Wegener’s granulomatosis [1]. For the next 20 years this clinical entity remained in controversy for its exact etiology. Immunophenotyping and molecular techniques like PCR and EBV encoded RNA in situ hybridization techniques (EBER ISH) have confirmed it to be an EBV associated mature B-cell neoplasm [2]. Viral proteins normally induce cytotoxic T cell response, which appears to be deficient in LYG. Lymphomatoid granulomatosis commonly occurs in the setting of immunodeficiency.
Lymphomatoid granulomatosis is a rare, multisystem lymphoproliferative disorder. The usual presentation is between 4th and 6th decade of life and twice as common in males. There is a usual delay of 4–8 months in reaching the diagnosis. Lung is most commonly involved organ (80–90 %) followed by skin up to 50 % and nervous system in 30 %. They may occur together at presentation or in sequential or in isolation [3, 4]. Patients may present with constitutional symptoms or related to the organ involvement. Cough, chest pain and dyspnea are common chest symptoms. Skin manifestations include erythematous papules, nodules and ulcerations. Neurological involvement can present as focal deficits or peripheral neuropathy. Liver and renal involvements are usually subclinical. Peripheral lymphadenopathy, splenomegaly and bone marrow involvement are less common. Up to 4 % are diagnosed incidentally. Chest imaging shows lung nodules of variable sizes. Spontaneous regression (20 %) and migration of the lesions is also described in literature [4, 5].
The morphology of LYG consists of a triad that comprises of polymorphic lymphoid infiltrate, transmural infiltration of vasculature by lymphoid cells with appearance of angiitis and focal areas of necrosis within lymphoid infiltrates. WHO classification has graded LYG into three grades on the basis of clonality, cytologic atypia and EBV positivity with clinical implications. Grade I morphology shows polymorphous lymphoid infiltrate with rare or absent large lymphoid cells. EBV + cells are less than 5 per high power field (HPF). Grade III shows more than 50 EBV + large atypical B cells per HPF in confluent sheets, and is considered equivalent of diffuse large B cell lymphoma. Grade II shows 5–50 EBV + cells/HPF and may behave aggressively [6].
In most patients the disease is aggressive with median survival of 2 years. The 5 year mortality is 60–90 % [7]. Poor prognostic predictors are age less than 25 years, high grade histology, CNS involvement and presence of hepatomegaly and leukocytosis [3]. Treatment of LYG is not well established owning to its rarity as well as unpredictable course after therapy. Treatment of grade I LYG is observation or steroids. Grade III LYG is treated like diffuse B cell lymphoma. Rituximab [8] and Interferon [9] have also been tried. Autologous stem cell transplantation has also been tried in refractory disease [10].
The patient in this report was an immunocompetent, young male presenting with constitutional symptoms, cytopenias and predominant lung involvement, concurrently. This can be a diagnostic challenge and invasive investigations may be required to reach the diagnosis, often delaying treatment initiation. This case highlights that though grade I LYG may have spontaneous remission and even respond to steroids, they may eventually require chemotherapy to achieve a sustained CR.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Liebow AA, Carrington CR, Friedman PJ. Lymphomatoid granulomatosis. Hum Pathol. 1972;3:457–558. doi: 10.1016/S0046-8177(72)80005-4. [DOI] [PubMed] [Google Scholar]
- 2.Jaffe ES (2009) The 2008 WHO classification of lymphomas: implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program 523–531 [DOI] [PMC free article] [PubMed]
- 3.Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360–373. doi: 10.1002/1097-0142(197901)43:1<360::AID-CNCR2820430151>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Fauci AS, Haynes BF, Costa J, Katz P, Wolff SM. Lymphomatoid Granulomatosis. Prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68–74. doi: 10.1056/NEJM198201143060203. [DOI] [PubMed] [Google Scholar]
- 5.Sheehy N, Bird B, O’Briain DS, Daly P, Wilson G. Synchronous regression and progression of pulmonary nodules on chest CT in untreated lymphomatoid granulomatosis. Clin Radiol. 2004;59:451–454. doi: 10.1016/j.crad.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:e35–e48. doi: 10.1097/PAS.0b013e3181fd8781. [DOI] [PubMed] [Google Scholar]
- 7.Gitelson E, Al-Saleem T, Smith MR. Review: lymphomatoid granulomatosis: challenges in diagnosis and treatment. Clin Adv Hematol Oncol. 2009;7:68–70. [PubMed] [Google Scholar]
- 8.Rao R, Vugman G, Leslie WT, Loew J, Venugopal P. Lymphomatoid granulomatosis treated with rituximab and chemotherapy. Clin Adv Hematol Oncol. 2003;1:658–660. [PubMed] [Google Scholar]
- 9.Wilson WH, Kingma DW, Raffeld M, Wittes RE, Jaffe ES. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531–4537. [PubMed] [Google Scholar]
- 10.Johnston A, Coyle L, Nevell D. Prolonged remission of refractory lymphomatoid granulomatosis after autologous hemopoietic stem cell transplantation with post-transplantation maintenance interferon. Leuk Lymphoma. 2006;47:323–328. doi: 10.1080/10428190500284262. [DOI] [PubMed] [Google Scholar]