

Abstract
The metal binding preferences of most metalloproteins do not match their metal requirements. Thus, metallation of an estimated 30% of metalloenzymes is aided by metal delivery systems, with ∼25% acquiring preassembled metal cofactors. The remaining ∼70% are presumed to compete for metals from buffered metal pools. Metallation is further aided by maintaining the relative concentrations of these pools as an inverse function of the stabilities of the respective metal complexes. For example, magnesium enzymes always prefer to bind zinc, and these metals dominate the metalloenzymes without metal delivery systems. Therefore, the buffered concentration of zinc is held at least a million-fold below magnesium inside most cells.
Keywords: Copper, Iron, Manganese, Nickel, Zinc, Irving-Williams Series, Metal Sensors, Metallochaperone, Metalloenzymes, Polydisperse Buffer
Introduction
This narrative sets out, with examples, how cells assist metallation. Such assistance is vital because the physical and chemical properties of proteins tend to select essential divalent metal ions with a ranked order of preference that follows the Irving-Williams series (1).
Competitive metals must be kept out of binding sites for the weaker binding ions. Cupric ions are at the top of the series, although their order with respect to zinc can flip (2). In the reducing conditions of the cytoplasm, cuprous (Cu+) rather than cupric (Cu2+) ions are expected to predominate, but these ions can also form tight complexes, especially with sites that contain sulfur ligands (3). In the periplasm of bacterial cells, ferric (Fe3+) rather than ferrous (Fe2+) ions often dominate (4). Ferric ions are retained in solution in organic complexes that can be exceptionally tight and include binding proteins such as the ferric-binding protein (Fbp) in the bacterial periplasm (5).
Because proteins are not rigid, the scope for steric selection of metal cofactors is imperfect. Mismetallation can exploit a subset of ligands and/or distort the native binding geometry. Typically, a protein becomes inactive if one or more residues of an active metal site are recruited to an alternative site, perhaps with alternative geometry, by a more competitive metal. For example, glyoxalase of Clostridium acetobutylicum (GlxI) is activated by nickel or cobalt, both of which assume octahedral geometries, whereas zinc binds tightly in trigonal bipyramidal geometry and inactivates this isoform of the enzyme (6).
Correct metallation in vivo is favored because the cytoplasm is a metal-controlled environment. For example, two periplasmic cupins (manganese MncA and cupric CucA) from a model cyanobacterium bind metal via analogous ligand sets within analogous folds (Fig. 1), yet in vivo they acquire different metals. MncA and CucA both show in vitro metal preferences that match the Irving-Williams series, which is especially problematic for MncA. A 10,000× and 100,000× excess of manganese is required at MncA folding in order for manganese to outcompete cupric or zinc ions, respectively (7). Cuprous ions can also outcompete manganese. Manganese MncA has oxalate decarboxylase activity, whereas neither the zinc nor the copper forms are active (7). CucA is a Sec substrate that folds in the periplasm on secretion, whereas MncA is a Tat substrate. The Tat system translocates prefolded proteins, and hence MncA folds within the cytoplasm before export (7, 8). In this way, MncA entraps manganese before exposure to copper and zinc in the periplasm. In the cytoplasm, at the site of MncA folding, copper and zinc must be at least 10,000 and 100,000× less available than manganese. This must reflect the relative buffered concentrations of these three metals plus, hypothetically, a manganese delivery system for MncA.
FIGURE 1.
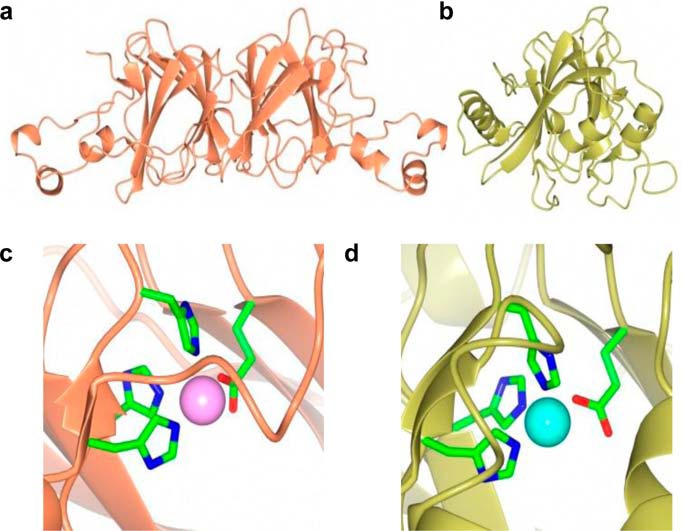
Metallation is governed by metal availability for MncA and CucA. a, Mn(II)-MncA global fold. b, Cu(II)-CucA global fold. Both proteins adopt a cupin architecture, with MncA composed of two cupin domains. c, MncA N-terminal Mn(II)-binding site. d, CucA Cu(II)-binding site. Both proteins coordinate their metals with identical ligand sets, with a water molecule in the open coordination position (this position is occupied by acetate in the C-terminal Mn(II)-binding site of MncA). MncA and CucA both prefer to bind copper rather than manganese in vitro, but MncA folds and traps manganese in the metal-regulated environment of the cytoplasm. Protein Data Bank (PDB) codes: 2VQA and 2XL7.
When Metals Compete with Other Metals for Proteins
Metal availability within cells is restricted such that proteins compete with other molecules, including other proteins, for limited pools of the most competitive metals. Dudev and Lim (9) have assessed the physical and chemical properties of metals and proteins that influence metal preferences. These include valence, ionic radius, coordination geometry, ligand number, second-shell ligands, effects of the protein matrix, and ligand characteristics (net charge, dipole moment and polarizability, charge-donating/-accepting ability, and denticity) (9). Despite these opportunities to tune metal preferences, in vitro metallation is typically aberrant when essential metals simply compete with each other for proteins (7).
Zinc and magnesium are the most commonly utilized metal cofactors (∼16 and ∼9% of all enzymes, respectively) (10), and they dominate the subset of metalloenzymes lacking a defined delivery system, representing ∼78% of this group (Table 1). Empirically, zinc is known to replace magnesium to inactivate enzymes including β-galactosidase (11), tyrosine kinases (12), and magnesium alkaline phosphatase (13, 14). The calculated free energies for replacing magnesium with zinc in rigid or flexible sites implies that zinc will always be favored over magnesium in mono- and binuclear binding pockets, with ΔG for replacement in flexible, neutral sites ranging from −10 to −29 kcal mol−1 (15). The incorporation of magnesium into chlorophyll to metallate chlorophyll-binding proteins is a special case that exploits delivery systems and is therefore considered separately in a later section of this minireview.
TABLE 1.
Types of metal sites and metal delivery pathways in Metal MACiE
| Metal and site type | Example enzyme from Metal MACiEa | Delivery pathway/chaperone | % of Metal MACiE totalb,c |
|---|---|---|---|
| Magnesium | |||
| Mononuclear | Adenylate cyclase (M0058) | None known | 38 |
| Trinuclear (magnesium) | Trichodiene synthase (M0262) | None known | 3 |
| Manganese | |||
| Mononuclear | Xylose isomerase (M0308) | None known | 8 |
| Trinuclear (manganese or zinc) | Deoxyribonuclease IV (M0011) | None known | <1 |
| Iron | |||
| Mononuclear | Catechol 2,3-dioxygenase (M0034) | None known | 3 |
| Dinuclear (FeFe) | Ferredoxin hydrogenase (M0127) | HydE/G provide iron as [FeS], production of which is dependent on CyaY | <1 |
| Dinuclear (NiFe) | Cytochrome c3 hydrogenase (M0126) | Assembly of cyano-, carbonyl-coordinated iron occurs on HypD; source of iron is unknown | <1 |
| Dinuclear (ZnFe) | Purple acid phosphatase (M0043) | None known | <1 |
| Heme | Ubiquinol-cytochrome c reductase (M0208) | Iron chelatase | 7 |
| Iron-sulfur cluster | Aldehyde oxidase (M0105) | CyaY | 14 |
| Cobalt | |||
| Mononuclear | Thiocyanate hydrolase (M0284) | None known | 2 |
| Cobalmin | Methionine synthase (M0268) | CbiX | 2 |
| Nickel | |||
| Dinuclear (NiFe) | Cytochrome c3 hydrogenase (M0126) | HypA/ HypB/ SlyD | <1 |
| Dinuclear (NiNi) | Urease (M0087) | UreE/ UreG | <1 |
| Factor-430 | Coenzyme B sulfoethylthiotransferase (M0156) | None known | <1 |
| Copper | |||
| Mononuclear | Copper-zinc SOD (M0138) | CCS (and others) | 2d; 1e |
| Dinuclear (CuCu) | Tyrosinase (M0125) | Atx1 (and others) | 1 |
| Dinuclear (CuMo) | Carbon-monoxide dehydrogenase (M0107) | None known | <1 |
| Zinc | |||
| Mononuclear | Alcohol dehydrogenase (M0256) | None known | 11 |
| Dinuclear (ZnZn) | Beta lactamase (M0015) | None known | 2 |
| Dinuclear (ZnFe) | Purple acid phosphatase (M0043) | None known | <1 |
| Trinuclear (zinc) | Phospholipase C (M0027) | None known | 1 |
| Molybdenum | |||
| Molybdopterin | Xanthine dehydrogenase (M0139) | MoeA | 2 |
| FeMo cofactor | Nitrogenase (M0212) | CyaY, NifH | <1 |
| Dinuclear (CuMo) | Carbon-monoxide dehydrogenase (M0107) | None known | <1 |
a Metal MACiE identifier shown in parentheses.
b Total excludes calcium enzymes represented in Metal MACiE.
c Hetero-dinuclear sites count as one site for each metal ion, and homo di- and tri-nuclear sites count as one site.
d Known delivery pathways.
e Unknown delivery pathways.
Iron and manganese are the next most common cofactors estimated to be exploited by ∼8 and ∼6% of enzymes (10). These ions account for most (∼18%) of the remaining fraction of metalloenzymes that are devoid of delivery systems, noting that another subset of iron enzymes does have metal delivery systems and iron is commonly found in preassembled cofactors. The divalent ions of manganese and iron have similar ligand affinities, radii, coordination preferences, and solvation free energies, creating a distinct challenge for proteins to discern between these elements when they compete for a site (9).
Uncertain Metallation in Vivo and Cambialistic Proteins
With a few pioneering exceptions (16, 17), the extent of mismetallation in vivo is unknown. Current methods for native metalloproteomics are neither global nor high throughput (7, 18), and so the extent of post-translational regulation through metallation is unclear. The picture is further complicated because multiple metals support catalysis in so-called cambialistic enzymes. Acireductone dioxygenase (ARD)2 from Klebsiella oxytoca is currently a rare example of an enzyme that can catalyze two different reactions dependent upon metal occupancy (19). Iron·ARD is widespread, and the nickel·ARD-dependent pathway has been observed in Bacillus subtilis and Escherichia coli, but both forms have been recovered from K. oxytoca. However, there is currently no evidence that both forms of the enzyme confer a selective advantage to K. oxytoca. Fractional occupancies of ARD with nickel and iron remain to be investigated in vivo, as does the tantalizing possibility that metallation is switched to match metabolic need.
Conformationally Trapped Metals and Opportunities for Proofreading of Metallation
There is scope for mismetallated proteins to be selectively degraded or recycled or to remain in a partially unfolded state. A subset of metal cofactors becomes kinetically trapped in proteins. The correct geometry can stabilize the fold, offering, in effect, the potential for proofreading of metal occupancy based upon second coordination shell interactions. For example, manganese in the copper·cupin CucA is readily replaced upon incubation with copper, but in the structurally related manganese·cupin MncA, manganese becomes trapped at folding and refractory to subsequent replacement by copper (7). Thus, folding and metal trapping are uncoupled from manganese binding to CucA, where this is mismetallation, but coupled to manganese binding in MncA. To date, in vitro biochemical studies of metal binding preferences of proteins have not included protein folding chaperones such as Hsp70 or its co-chaperones and nucleotide exchange factors. Association of chaperones with exposed hydrophobic patches of nascent proteins impacts upon the energetics of protein folding (20), but it remains to be tested whether or not this sometimes imposes a bias in favor of the correct metal.
Metal Delivery Pathways
Fidelity in metallation with two competitive metals, nickel and copper, is typically assisted by metallochaperones (21–23). The term “metallochaperone” describes a collection of proteins, for a diversity of metals, which differ in their biochemical mechanisms. Known nickel chaperones, which include HypB, interact with a battery of other proteins with consumption of nucleotide cofactors aiding metal insertion (21, 22). When Helicobacter pylori HypB aberrantly binds zinc its GTPase, activity is not triggered, and in this way, cofactor delivery becomes selective for nickel (24). Known copper chaperones do not require nucleotide cofactors. Both copper and nickel chaperones introduce a kinetic bias into the partitioning of metals by engaging in specific protein-protein interactions that recognize the correct partners (23). Such interactions also orientate the donor and acceptor ligands to encourage facile ligand exchange (25).
Preassembled complex metal cofactors include cobalamin (cobalt), iron-sulfur clusters, heme and siroheme (iron), molybdopterin (molybdenum), F430 (nickel), and chlorophyll (magnesium). Discrimination between these more elaborate molecular assemblies as opposed to individual metal ions at cofactor selection is less challenging, but nonetheless may be aided by delivery proteins. For example, monothiol glutaredoxins (Grxs) and BolA proteins play roles in [FeS] cluster delivery as well as iron sensing (26), with yeast strains deficient in Grx3 and Grx4 exhibiting defects in multiple iron-dependent enzymes (27, 28); NarJ assists in the insertion of molybdopterin into nitrate reductase in E. coli cells (29), and CcmE functions as a heme chaperone in the periplasm of E. coli, delivering its cargo to CcmF for insertion into cytochrome c (30).
Metallochaperones that contribute toward fidelity in partitioning metals during complex cofactor assembly include chelatases for heme, cobalamin, and chlorophyll (31, 32) and MoeA for molybdopterin (33). Ferrochelatases, for example, can catalyze the insertion of metals other than iron into tetrapyrroles, such that zinc protoporphyrin IX becomes diagnostic for some iron deficiencies (34). The metal preferences and metallation of metallochaperones warrant investigation.
The majority of copper proteins are secreted, and copper efflux from the cytosol is driven by P1-type ATPases that acquire copper from metallochaperones such as Atx1 (35, 36). Exactly how copper is then handed to nascent proteins post-secretion is the topic of current investigations. Oddly, CucA in the cyanobacterial periplasm has impaired metallation in mutants missing copper-transporting P1-type ATPases (CtaA and PacS), and the mutant periplasm is devoid of CucA but enriched with low Mr copper complexes (37). Thus, copper is routed via the cytoplasm and the cyanobacterial copper chaperone Atx1, before export via a P1-type ATPase to load CucA. Moreover, secretion of CucA seems to be coupled to copper efflux (37). A subset of P1-type ATPases that have tight Km and low Vmax does not confer copper resistance but appears to support metal delivery to nascent cupro-proteins (38). There is evidence of interaction between E. coli periplasmic copper chaperone CusF and P1-type ATPase CopA, whereas periplasmic copper chaperone CueP is required for metallation of SodCII in Salmonella enterica sv. Typhimurium (39, 40).
Evaluating the Contribution of Delivery Pathways to Metallation
To estimate the fractions of metalloproteins that bind preassembled cofactors or are otherwise metallated via metallochaperones, the Metal MACiE database has been interrogated. Metal MACiE is a manually curated catalogue of enzymes that require metals for their catalytic mechanisms and for which a protein structure has been determined (41). Metal ions solely performing structural roles in proteins that are not enzymes are not annotated in Metal MACiE. This is liable to lead to an under-representation of zinc, which is widely used in zinc fingers (42). With such limitations in mind, Metal MACiE can be used to make first approximations of the proportions of enzymes with various metal centers. Table 1 lists the types of sites in the database, noting where proteins are known to assist in metal delivery directly to the enzyme (exemplified by nickel and copper), to a subcellular compartment containing the enzyme (exemplified by copper in the secretory system or periplasm), or to preformed metal cofactors. In total, 30% of metalloenzymes within the database are estimated to lie at the end of such delivery pathways, and metalloenzymes are estimated to account for almost half of all enzymes (43).
It is uncertain where most metallochaperones acquire metal and to what extent their relative metal affinities correspond to the metal requirements of the delivery pathways. Cyanobacteria are useful models for exploring partitioning among metallochaperones. In common with other photosynthetic organisms, they have a high demand for metals (44), but they also have delivery proteins for an especially wide range of metals: Atx1 for copper to thylakoids (45), UreE and HypA/B for nickel to urease and hydrogenase (46), ferrochelatase for iron to heme and siroheme (47), magnesium chelatase for magnesium to chlorophyll (48), CbiX for cobalt to cobalamin (plants in contrast do not make cobalamin) (49), MoeA for molybdenum to molybdopterin, CyaY for iron to iron-sulfur clusters, and possibly PratA for manganese to photosystem II (50). A set of metal competition experiments between the purified cyanobacterial metallochaperones could establish whether or not their relative metal affinities simply enable metals to partition to the correct delivery pathway. This in turn would resolve the metallation challenge for ∼30% of metalloenzymes.
Alternatively, metallochaperones might directly acquire metal from importers assisted by specific protein interactions. The idea that inward metal transport is coupled to the loading of delivery pathways, to channel metals to sites of metalloenzyme assembly, is widely envisioned but sparsely evidenced. Notably, analyses of yeast mutants did not identify any single copper donor for either of two copper metallochaperones (51). Nonetheless, there is evidence that the copper chaperone for superoxide dismutase (CCS) can interact with membranes and with the copper importer Ctr1 (52), and metal transfer to Atx1 has also been observed in vitro using a cytosolic domain of Ctr1 (53). Nickel imported by the Nik system is destined for hydrogenase and largely unavailable to nickel-responsive transcriptional regulators (54), which might also suggest direct handover of nickel to HypA/B. However, evidence that the substrate for the Nik importer is a nickel-histidine complex provides an alternative explanation for these observations if HypA/B can preferentially acquire nickel from nickel-histidine (55). There is evidence that a mitochondrial iron importer mitoferrin-1 interacts with a ferrochelatase for heme biogenesis (56). This iron supply pathway cannot be “hardwired” exclusively for iron if zinc protoporphyrin IX accumulates under iron deficiency (34). Iron-sulfur clusters are the targets for surplus cobalt and copper (57–60). Both cobalt and copper directly destabilize the assembled cluster on the scaffold proteins and, at least for cobalt, it is known that the resultant mixed cluster can be delivered to apo-proteins (58, 59). Thus, imperfect metal preferences of delivery systems can sometimes propagate mismetallation.
Metallochaperone-catalyzed delivery of the more competitive metals, such as nickel and copper, enables cells to more efficiently cofactor a subset of proteins with these ions. However, viewed from a different perspective, such metal delivery supports metallation at low buffered concentrations sufficient to exclude these elements from binding sites for metals lower down the Irving-Williams series (1). For example, cyanobacterial mutants missing the copper metallochaperone Atx1 show phenotypes indicative of the mismetallation of binding sites for other metals with copper (61).
The Set Points for Metal Homeostasis
The buffered (rather than total) set points for metals can vary between cell types and intracellular compartments and throughout the lifetime of a cell. Nonetheless, magnesium appears to be universally held at ∼10−3 m inside cells (Fig. 2, gray bars), about 10 times less than the concentration in sea water and 10 times more than typical concentrations in fresh water (62, 63). Proteins that require ferrous ions often exhibit affinities of ∼10−7 m, which is suggested to match the ferrous concentration in the sulfide-rich anaerobic conditions when life first evolved (64). By determining the ferrous affinity of glutathione (glutathione has a concentration of ∼2–10 mm within the cytoplasm), and assuming that this complex is a major component of the cytosolic iron pool, a value in the region of 10−6 to 10−7 m for the buffered concentration of ferrous iron is plausible (65) (Fig. 2, gray bars).
FIGURE 2.
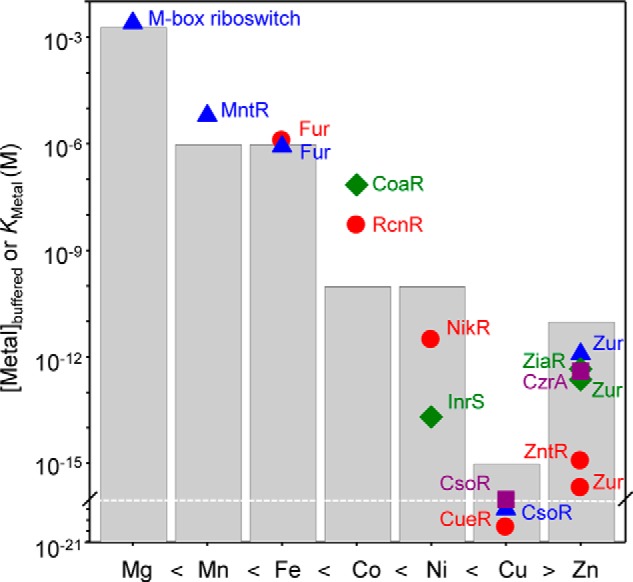
Correlation between buffered set points and metal sensor affinities. Shown are graphical representations of estimated intracellular buffered metal concentrations (gray bars) for magnesium, manganese, iron, cobalt, nickel, copper, and zinc (62, 65, 66, 72, 73, 90, 91) and correlation with KMetal of cytosolic metal sensors for their cognate metal, including Fur (92), RcnR (93), NikR (94), CueR (89), Zur (88), and ZntR (88), from E. coli (red circles); the M-box riboswitch (95), MntR (96), Fur (96), CsoR (97), and Zur (98), from B. subtilis (blue triangles); CoaR (84), InrS (83), Zur (85), and ZiaR (85) from Synechocystis PCC 6803 (green diamonds); and CsoR (99) and CzrA (100) from Staphylococcus aureus (purple squares). It is hypothesized that KMetal of metal sensors maintains the set points for buffered metal concentrations as an inverse function of the Irving-Williams series.
The cytosolic concentration of manganese has been estimated to be comparable with ferrous iron (66, 67) (Fig. 2, gray bars). However, manganese concentrations may be elevated within organelles such as the chloroplast or mitochondria where there is high demand. In a bacterial cytosol, the concentration of manganese can vary. For example, in response to oxidants, manganese is elevated to correctly metallate manganese superoxide dismutase (SOD) (16). Nickel- and cobalt-requiring enzymes are thought to have been more prevalent in early anaerobic life, and Fraústo da Silva and Williams (62) suggest that these two metals are unlikely to have ever exceeded 10−10 m in the cytosol (Fig. 2, gray bars).
Zinc-binding sites in most proteins have affinities that are typically 10−11 m or tighter (68). The use of either synthetic or genetically encoded zinc-responsive fluorophores has placed buffered zinc concentrations within the cytosol of bacteria and eukaryotic cells in the 10−12 to 10−10 m range (69–72). Buffered cytosolic copper concentrations have been estimated to be ∼10−15 m or less using copper-responsive fluorophores (73, 74) (Fig. 2, gray bars). In yeast, copper zinc SOD1 has a copper affinity of 10−15 m but requires the CCS metallochaperone for activation in vivo. Consequently, it was inferred that copper must be buffered below 10−15 m (75). CCS additionally catalyzes the formation of a vital disulfide bond within SOD1 (76), providing an alternative explanation for inactivity of SOD1 in CCS-deficient cells.
What sustains these different buffered metal concentrations? An expectation is that this relates to detection thresholds of sensors that control homeostasis for the respective metals. There are pitfalls in the estimation of KMetal, especially for tighter binding elements (77), generating a jumble of erroneous values. Nonetheless, mindful of this caveat, a remarkable correlation exists between estimates of KMetal for metal sensors and estimates for buffered cytosolic metal concentrations (Fig. 2). This observation is consistent with the intracellular set point for metal homeostasis being a function of these sensor affinities. By setting the metal affinities of metal sensors such that those for the most competitive metals are the tightest, the control of metal efflux, metal influx, and metal sequestration and the switching of metabolism to spare limiting metals are thus primed to maintain the buffered metal concentrations as an inverse function of the Irving-Williams series. Under this regime, subtle differences in the relative metal preferences of metalloenzymes now become sufficient to enable correct in vivo metallation.
How a Cell's Set of Metal Sensors Acts in Concert to Discern Metals One from Another
The actions of metal sensors help maintain buffered metal concentrations, and these concentrations in turn influence which metals are acquired by ∼70% of metalloenzymes. Thus, the metal specificity of metal sensors becomes a dominant factor in the fidelity of metallation. The proportion also becomes even higher than 70% if some metallochaperones are metallated from buffered metal pools. Metal-sensing, DNA-binding transcriptional regulators have been extensively characterized in bacteria (78, 79) and identified for copper, iron, and zinc in yeast (80, 81). However, where metal affinities have been measured for multiple metals, the metal preferences of bacterial metal sensor proteins again tend to simply abide by the Irving-Williams series (78, 79, 82).
Affinity, Access (Kinetics), and Allostery
A series of publications in the first decade of this century revealed that metal specificity of metal sensors can be determined by three factors. First, metal affinity contributes toward metal selectivity. Second, the allosteric mechanism connecting metal binding to altered DNA binding or to gene activation can respond selectively to different metals. Finally, the kinetics of access can differ for different sensors, for example due to delivery proteins (10, 82).
Relative Affinity, Access and Allostery
Since 2010 it has become evident that affinity, allostery, and access operate as relative parameters in a set of sensors (83–85). Such observations are now possible because sufficiently large numbers of bacterial metal sensors have been characterized. Metal selectivity is now seen to result from the concerted actions of a cell's complement of metal sensors. In this manner, specificity is not constrained by absolute metal preferences (10, 82). The best sensor in the set is the sensor that responds. What defines the best in the set for each metal?
Recent studies of the metal sensors of the model organism Synechocystis PCC 6803 exemplify the contributions of relative affinity, relative allostery, and relative access. By examining one sensor from each family of metal sensors present in this organism, the parameter correlating with selective metal detection was found to vary from metal to metal (Fig. 3). Importantly, the absolute metal preferences, as reflected in KMetal values of InrS (nickel-responsive efflux derepressor), CoaR (cobalt-responsive efflux activator), and ZiaR and Zur (zinc-responsive efflux derepressor and influx corepressor, respectively) (61, 83, 86, 87), do not universally match their metal specificities in vivo. Rather, the detection of nickel correlates with relative nickel affinity, and the detection of zinc correlates with relative free energy coupling DNA binding to zinc binding (relative allostery), but a substantial kinetic contribution is invoked in the selective detection of cobalt (relative access) (83–85) (Fig. 3).
FIGURE 3.
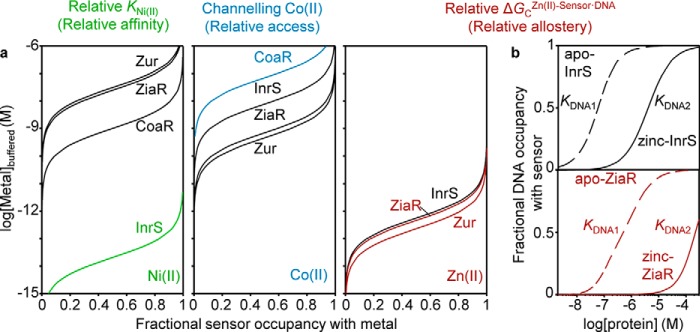
Relative affinity, relative access, and relative allostery in a complement of metal sensors influences the metals detected in vivo. a, calculated fractional occupancy of InrS, Zur, ZiaR, and CoaR with Ni(II), Zn(II), and Co(II) as the concentration of these elements changes: Fractional occupancy: θ= [Metal]buffered/(KMetal + [Metal]buffered) using published KMetal (83–85). b, fractional occupancy of specific DNA (top) with apo- (dashed) and zinc-InrS (solid) and (bottom), apo- (dashed) and zinc-ZiaR (solid), as a function of protein concentration. Coupling free energy: ΔGC = −RTln(KDNA2/KDNA1). The simulated curves were generated using published KDNA values (85), [DNA] = 10 nm. The selective detection of nickel correlates with relative nickel affinity, of zinc with relative ΔGC for zinc, but a major kinetic contribution (channeling) is invoked for cobalt.
To elaborate, InrS possesses the tightest nickel affinity in this set of metal sensors (83). Thus, as the buffered concentration of nickel rises, provided the distribution of nickel among the sensors approximates to the thermodynamic equilibrium state, InrS will trigger nickel efflux before the concentration becomes sufficiently high for nickel to aberrantly bind to any of the other sensors (Fig. 3a) (83). This assumes roughly equivalent numbers of molecules of each sensor per cell (a parameter that in the future needs to be measured). Cognizant of the challenges in determining protein-metal affinities and noting the weak KNi(II) of ZiaR and Zur, a series of interprotein competition experiments also confirmed that nickel partitions from each of the other sensors to InrS (83).
In contrast to nickel, cobalt affinities do not correlate with in vivo specificities; rather, cobalt-sensing CoaR has the weakest KCo(II) of the set of sensors (84), (Fig. 3a). Moreover, in vitro, cobalt promotes DNA association by Zur and DNA dissociation by ZiaR, yet neither ZiaR nor Zur responds to cobalt in vivo under conditions in which CoaR responds (84). This implies that cobalt is channeled to CoaR and away from ZiaR and Zur with their tighter cobalt affinities. There is evidence that CoaR is membrane-associated, and cobalt acquisition may involve channeling via the cobalamin biosynthetic complex, which is also membrane-associated. Additionally, there is evidence that CoaR may not solely sense cobalt directly, but also detect an intermediate in the B12 assembly pathway (84). In summary, CoaR has preferential access to the cobalt effector relative to ZiaR and Zur.
The zinc affinity of InrS is comparable with the sensory sites of ZiaR and Zur (Fig. 3a), yet following prolonged zinc exposure, ZiaR responds but InrS does not. Critically, although the allosteric mechanism of InrS is capable of responding to zinc, the coupling free energy linking zinc binding to DNA binding (ΔGCzinc·sensor·DNA) is greater for ZiaR than for InrS (85), (Fig. 3b). In short, zinc is a more effective derepressor of ZiaR than of InrS. Thus, at some equivalent fractional zinc occupancies, a greater proportion of InrS relative to ZiaR will be bound to DNA. InrS can thereby repress its gene target, whereas the ZiaR target remains derepressed. This exemplifies how relative coupling free energy ΔGC, that is relative allosteric effectiveness, in a complement of metal sensors can also dictate selectivity (Fig. 3b).
Improbable Kinetics and Associative Metallation
Metal affinities of metal sensors for the most competitive metals such as nickel, zinc, and copper are so tight that it is not credible for metal partitioning to and from solution to reach equilibrium in a viable timeframe. The off-rates are too slow. However, this assumes dissociative metal exchange. As an alternative, associative metal exchange can occur to/from labile metal sites of proteins (including metal sensors) and components of a polydisperse buffer. This ill-defined buffer is composed of small molecules such as amino acids, glutathione, organic acids, and inorganic ligands, plus weak adventitious ligands on the surface of macromolecules, specific buffering proteins, and a subset of the delivery proteins. Rates of metal exchange in cells can thus be unexpectedly fast, and can swiftly approach the equilibrium state. Moreover, such a process of associative ligand exchange through a polydisperse buffer can operate at buffered concentrations below 10−9 m, the theoretical threshold for one atom per cell volume in a bacterium such as E. coli (88).
For the most competitive metals, the fully hydrated pool is indeed estimated to be below 10−9 m and thus equates to less than one (free) atom per cell at any instant (88, 89) (Fig. 2). In relation to Fig. 3 and the example in the preceding section, InrS does transiently respond to zinc in vivo, whereas the response of ZiaR is persistent. The buffered concentration of zinc would have to fall below 10−11 m for a protein with the KZn(II) of InrS to have less than full zinc occupancy to restore repression. Under these conditions, persistent ZiaR must therefore detect a pool of exchangeable zinc that is buffered at least 2 orders of magnitude below ∼10−9 m (85). One explanation is that ZiaR is metallated through associative ligand exchange with a polydisperse buffer rather than depending upon a hydrated pool of zinc ions. By way of illustration, the equations in Fig. 4 represent the transfer of zinc from InrS to ZiaR (i) by a dissociative process requiring the slow release of zinc from InrS to the hydrated state, and (ii) by potentially swift associative exchange with ligands of a buffer.
FIGURE 4.
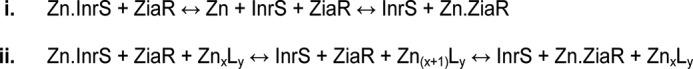
Associative ligand exchange with a polydisperse buffer. i, the transfer of zinc from InrS to ZiaR via a dissociative release of zinc from InrS to a hydrated state. ii, the transfer of zinc from InrS to ZiaR by (potentially swift) associative ligand exchange via a partly (x) zinc-saturated number of ligands (y) of a polydisperse buffer (L).
Prospective: The Elements of Biotechnology and Biomedicine
With such a large proportion of enzymes requiring metals, discord between their metal binding preferences and metal requirements has implications for biological chemistry, as well as applications in biomedicine and biotechnology. For example, knowledge of the in vivo metallation states of components of metabolic and signaling networks is required to improve the accuracy of systems biology computations. Synthetic biology aims to engineer cells for new purposes. Success may often depend upon an ability to coincidentally rewire the circuitry for enzyme metallation.
This work was supported by Biotechnology and Biological Sciences Research Council (BBSRC) research grants BB/K00817X/1 and BB/J017787/1. This is the first article in the Thematic Minireview series “Metals in Biology 2014.”
- ARD
- acireductone dioxygenase
- SOD
- superoxide dismutase
- CCS
- copper chaperone for superoxide dismutase.
REFERENCES
- 1. Irving H., Williams R. J. (1948) Order of stability of metal complexes. Nature 162, 746–747 [Google Scholar]
- 2. Johnson D. A., Nelson P. G. (1995) Factors determining the ligand field stabilization energies of the hexaaqua 2+ complexes of the first transition series and the Irving-Williams order. Inorg. Chem. 34, 5666–5671 [DOI] [PubMed] [Google Scholar]
- 3. Davis A. V., O'Halloran T. V. (2008) A place for thioether chemistry in cellular copper ion recognition and trafficking. Nat. Chem. Biol. 4, 148–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schalk I. J., Yue W. W., Buchanan S. K. (2004) Recognition of iron-free siderophores by TonB-dependent iron transporters. Mol. Microbiol. 54, 14–22 [DOI] [PubMed] [Google Scholar]
- 5. Parker Siburt C. J., Mietzner T. A., Crumbliss A. L. (2012) FbpA — a bacterial transferrin with more to offer. Biochim. Biophys. Acta 1820, 379–392 [DOI] [PubMed] [Google Scholar]
- 6. Suttisansanee U., Lau K., Lagishetty S., Rao K. N., Swaminathan S., Sauder J. M., Burley S. K., Honek J. F. (2011) Structural variation in bacterial glyoxalase I enzymes: investigation of the metalloenzyme glyoxalase I from Clostridium acetobutylicum. J. Biol. Chem. 286, 38367–38374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tottey S., Waldron K. J., Firbank S. J., Reale B., Bessant C., Sato K., Cheek T. R., Gray J., Banfield M. J., Dennison C., Robinson N. J. (2008) Protein-folding location can regulate manganese-binding versus copper- or zinc-binding. Nature 455, 1138–1142 [DOI] [PubMed] [Google Scholar]
- 8. Kudva R., Denks K., Kuhn P., Vogt A., Müller M., Koch H. G. (2013) Protein translocation across the inner membrane of Gram-negative bacteria: the Sec and Tat dependent protein transport pathways. Res. Microbiol. 164, 505–534 [DOI] [PubMed] [Google Scholar]
- 9. Dudev T., Lim C. (2014) Competition among metal ions for protein binding sites: determinants of metal ion selectivity in proteins. Chem. Rev. 114, 538–556 [DOI] [PubMed] [Google Scholar]
- 10. Waldron K. J., Rutherford J. C., Ford D., Robinson N. J. (2009) Metalloproteins and metal sensing. Nature 460, 823–830 [DOI] [PubMed] [Google Scholar]
- 11. Fernandes S., Geueke B., Delgado O., Coleman J., Hatti-Kaul R. (2002) β-Galactosidase from a cold-adapted bacterium: purification, characterization and application for lactose hydrolysis. Appl. Microbiol. Biotechnol. 58, 313–321 [DOI] [PubMed] [Google Scholar]
- 12. Sun G., Budde R. J. (1999) Substitution studies of the second divalent metal cation requirement of protein tyrosine kinase CSK. Biochemistry 38, 5659–5665 [DOI] [PubMed] [Google Scholar]
- 13. Ciancaglini P., Pizauro J. M., Curti C., Tedesco A. C., Leone F. A. (1990) Effect of membrane moiety and magnesium ions on the inhibition of matrix-induced alkaline phosphatase by zinc ions. Int. J. Biochem. 22, 747–751 [DOI] [PubMed] [Google Scholar]
- 14. Hung H. C., Chang G. G. (2001) Differentiation of the slow-binding mechanism for magnesium ion activation and zinc ion inhibition of human placental alkaline phosphatase. Protein Sci. 10, 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang T. Y., Dudev T., Lim C. (2008) Mononuclear versus binuclear metal-binding sites: metal-binding affinity and selectivity from PDB survey and DFT/CDM calculations. J. Am. Chem. Soc. 130, 3844–3852 [DOI] [PubMed] [Google Scholar]
- 16. Imlay J. A. (2014) The mismetallation of enzymes during oxidative stress. J. Biol. Chem. 289, 28121–28128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang M., Parker M. J., Stubbe J. (2014) Choosing the right metal: case studies of class I ribonucleotide reductases. J. Biol. Chem. 289, 28104–28111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cvetkovic A., Menon A. L., Thorgersen M. P., Scott J. W., Poole F. L., 2nd, Jenney F. E., Jr., Lancaster W. A., Praissman J. L., Shanmukh S., Vaccaro B. J., Trauger S. A., Kalisiak E., Apon J. V., Siuzdak G., Yannone S. M., Tainer J. A., Adams M. W. (2010) Microbial metalloproteomes are largely uncharacterized. Nature 466, 779–782 [DOI] [PubMed] [Google Scholar]
- 19. Dai Y., Wensink P. C., Abeles R. H. (1999) One protein, two enzymes. J. Biol. Chem. 274, 1193–1195 [DOI] [PubMed] [Google Scholar]
- 20. Hartl F. U., Hayer-Hartl M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 [DOI] [PubMed] [Google Scholar]
- 21. Higgins K. A., Carr C. E., Maroney M. J. (2012) Specific metal recognition in nickel trafficking. Biochemistry 51, 7816–7832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaluarachchi H., Chan Chung K. C., Zamble D. B. (2010) Microbial nickel proteins. Nat. Prod. Rep. 27, 681–694 [DOI] [PubMed] [Google Scholar]
- 23. Robinson N. J., Winge D. R. (2010) Copper metallochaperones. Annu. Rev. Biochem. 79, 537–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sydor A. M., Lebrette H., Ariyakumaran R., Cavazza C., Zamble D. B. (2014) Relationship between Ni(II) and Zn(II) coordination and nucleotide binding by the Helicobacter pylori [NiFe]-hydrogenase and urease maturation factor HypB. J. Biol. Chem. 289, 3828–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banci L., Bertini I., Cantini F., Felli I. C., Gonnelli L., Hadjiliadis N., Pierattelli R., Rosato A., Voulgaris P. (2006) The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat. Chem. Biol. 2, 367–368 [DOI] [PubMed] [Google Scholar]
- 26. Mapolelo D. T., Zhang B., Randeniya S., Albetel A. N., Li H., Couturier J., Outten C. E., Rouhier N., Johnson M. K. (2013) Monothiol glutaredoxins and A-type proteins: partners in Fe-S cluster trafficking. Dalton Trans. 42, 3107–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ojeda L., Keller G., Muhlenhoff U., Rutherford J. C., Lill R., Winge D. R. (2006) Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces cerevisiae. J. Biol. Chem. 281, 17661–17669 [DOI] [PubMed] [Google Scholar]
- 28. Mühlenhoff U., Molik S., Godoy J. R., Uzarska M. A., Richter N., Seubert A., Zhang Y., Stubbe J., Pierrel F., Herrero E., Lillig C. H., Lill R. (2010) Cytosolic monothiol glutaredoxins function in intracellular iron sensing and trafficking via their bound iron-sulfur cluster. Cell Metab. 12, 373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vergnes A., Pommier J., Toci R., Blasco F., Giordano G., Magalon A. (2006) NarJ chaperone binds on two distinct sites of the aponitrate reductase of Escherichia coli to coordinate molybdenum cofactor insertion and assembly. J. Biol. Chem. 281, 2170–2176 [DOI] [PubMed] [Google Scholar]
- 30. San Francisco B., Kranz R. G. (2014) Interaction of holoCcmE with CcmF in heme trafficking and cytochrome c biosynthesis. J. Mol. Biol. 426, 570–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schubert H. L., Raux E., Wilson K. S., Warren M. J. (1999) Common chelatase design in the branched tetrapyrrole pathways of heme and anaerobic cobalamin synthesis. Biochemistry 38, 10660–10669 [DOI] [PubMed] [Google Scholar]
- 32. Tanaka R., Tanaka A. (2007) Tetrapyrrole biosynthesis in higher plants. Annu. Rev. Plant Biol. 58, 321–346 [DOI] [PubMed] [Google Scholar]
- 33. Sandu C., Brandsch R. (2002) Evidence for MoeA-dependent formation of the molybdenum cofactor from molybdate and molybdopterin in Escherichia coli. Arch. Microbiol. 178, 465–470 [DOI] [PubMed] [Google Scholar]
- 34. Labbé R. F., Vreman H. J., Stevenson D. K. (1999) Zinc protoporphyrin: a metabolite with a mission. Clin. Chem. 45, 2060–2072 [PubMed] [Google Scholar]
- 35. Larin D., Mekios C., Das K., Ross B., Yang A. S., Gilliam T. C. (1999) Characterization of the interaction between the Wilson and Menkes disease proteins and the cytoplasmic copper chaperone, HAH1p. J. Biol. Chem. 274, 28497–28504 [DOI] [PubMed] [Google Scholar]
- 36. Lin S. J., Pufahl R. A., Dancis A., O'Halloran T. V., Culotta V. C. (1997) A role for the Saccharomyces cerevisiae ATX1 gene in copper trafficking and iron transport. J. Biol. Chem. 272, 9215–9220 [PubMed] [Google Scholar]
- 37. Waldron K. J., Firbank S. J., Dainty S. J., Pérez-Rama M., Tottey S., Robinson N. J. (2010) Structure and metal loading of a soluble periplasm cuproprotein. J. Biol. Chem. 285, 32504–32511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raimunda D., González-Guerrero M., Leeber B. W., 3rd, Argüello J. M. (2011) The transport mechanism of bacterial Cu+-ATPases: distinct efflux rates adapted to different function. BioMetals 24, 467–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Padilla-Benavides T., George Thompson A. M., McEvoy M. M., Argüello J. M. (2014) Mechanism of ATPase-mediated Cu+ export and delivery to periplasmic chaperones: the interaction of Escherichia coli CopA and CusF. J. Biol. Chem. 289, 20492–20501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Osman D., Patterson C. J., Bailey K., Fisher K., Robinson N. J., Rigby S. E., Cavet J. S. (2013) The copper supply pathway to a Salmonella Cu,Zn-superoxide dismutase (SodCII) involves P1B-type ATPase copper efflux and periplasmic CueP. Mol. Microbiol. 87, 466–477 [DOI] [PubMed] [Google Scholar]
- 41. Andreini C., Bertini I., Cavallaro G., Holliday G. L., Thornton J. M. (2009) Metal-MACiE: a database of metals involved in biological catalysis. Bioinformatics 25, 2088–2089 [DOI] [PubMed] [Google Scholar]
- 42. Klug A. (2010) The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 79, 213–231 [DOI] [PubMed] [Google Scholar]
- 43. Andreini C., Bertini I., Cavallaro G., Holliday G. L., Thornton J. M. (2008) Metal ions in biological catalysis: from enzyme databases to general principles. J. Biol. Inorg. Chem. 13, 1205–1218 [DOI] [PubMed] [Google Scholar]
- 44. Blaby-Haas C. E., Merchant S. S. (2014) Lysosome-related organelles as mediators of metal homeostasis. J. Biol. Chem. 289, 28129–28136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tottey S., Rondet S. A., Borrelly G. P., Robinson P. J., Rich P. R., Robinson N. J. (2002) A copper metallochaperone for photosynthesis and respiration reveals metal-specific targets, interaction with an importer, and alternative sites for copper acquisition. J. Biol. Chem. 277, 5490–5497 [DOI] [PubMed] [Google Scholar]
- 46. Hoffmann D., Gutekunst K., Klissenbauer M., Schulz-Friedrich R., Appel J. (2006) Mutagenesis of hydrogenase accessory genes of Synechocystis sp. PCC 6803: additional homologues of hypA and hypB are not active in hydrogenase maturation. FEBS J. 273, 4516–4527 [DOI] [PubMed] [Google Scholar]
- 47. Funk C., Vermaas W. (1999) A cyanobacterial gene family coding for single-helix proteins resembling part of the light-harvesting proteins from higher plants. Biochemistry 38, 9397–9404 [DOI] [PubMed] [Google Scholar]
- 48. Jensen P. E., Gibson L. C., Henningsen K. W., Hunter C. N. (1996) Expression of the chlI, chlD, and chlH genes from the Cyanobacterium Synechocystis PCC6803 in Escherichia coli and demonstration that the three cognate proteins are required for magnesium-protoporphyrin chelatase activity. J. Biol. Chem. 271, 16662–16667 [DOI] [PubMed] [Google Scholar]
- 49. Leech H. K., Raux E., McLean K. J., Munro A. W., Robinson N. J., Borrelly G. P., Malten M., Jahn D., Rigby S. E., Heathcote P., Warren M. J. (2003) Characterization of the cobaltochelatase CbiXL: evidence for a 4Fe-4S center housed within an MXCXXC motif. J. Biol. Chem. 278, 41900–41907 [DOI] [PubMed] [Google Scholar]
- 50. Stengel A., Gügel I. L., Hilger D., Rengstl B., Jung H., Nickelsen J. (2012) Initial steps of photosystem II de novo assembly and preloading with manganese take place in biogenesis centers in Synechocystis. Plant Cell 24, 660–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Portnoy M. E., Schmidt P. J., Rogers R. S., Culotta V. C. (2001) Metal transporters that contribute copper to metallochaperones in Saccharomyces cerevisiae. Mol. Genet. Genomics 265, 873–882 [DOI] [PubMed] [Google Scholar]
- 52. Pope C. R., De Feo C. J., Unger V. M. (2013) Cellular distribution of copper to superoxide dismutase involves scaffolding by membranes. Proc. Natl. Acad. Sci. U.S.A. 110, 20491–20496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao Z., Wedd A. G. (2002) A C-terminal domain of the membrane copper pump Ctr1 exchanges copper(I) with the copper chaperone Atx1. Chem. Commun. (Camb.) 21, 588–589 [DOI] [PubMed] [Google Scholar]
- 54. Rowe J. L., Starnes G. L., Chivers P. T. (2005) Complex transcriptional control links NikABCDE-dependent nickel transport with hydrogenase expression in Escherichia coli. J. Bacteriol. 187, 6317–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chivers P. T., Benanti E. L., Heil-Chapdelaine V., Iwig J. S., Rowe J. L. (2012) Identification of Ni-(l-His)2 as a substrate for NikABCDE-dependent nickel uptake in Escherichia coli. Metallomics 4, 1043–1050 [DOI] [PubMed] [Google Scholar]
- 56. Chen W., Dailey H. A., Paw B. H. (2010) Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 116, 628–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Macomber L., Imlay J. A. (2009) The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 8344–8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chillappagari S., Seubert A., Trip H., Kuipers O. P., Marahiel M. A., Miethke M. (2010) Copper stress affects iron homeostasis by destabilizing iron-sulfur cluster formation in Bacillus subtilis. J. Bacteriol. 192, 2512–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ranquet C., Ollagnier-de-Choudens S., Loiseau L., Barras F., Fontecave M. (2007) Cobalt stress in Escherichia coli: the effect on the iron-sulfur proteins. J. Biol. Chem. 282, 30442–30451 [DOI] [PubMed] [Google Scholar]
- 60. Thorgersen M. P., Downs D. M. (2007) Cobalt targets multiple metabolic processes in Salmonella enterica. J. Bacteriol. 189, 7774–7781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tottey S., Patterson C. J., Banci L., Bertini I., Felli I. C., Pavelkova A., Dainty S. J., Pernil R., Waldron K. J., Foster A. W., Robinson N. J. (2012) Cyanobacterial metallochaperone inhibits deleterious side reactions of copper. Proc. Natl. Acad. Sci. U.S.A. 109, 95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fraústo da Silva J. J. R., Williams R. J. P. (1991) The Biological Chemistry of the Elements: The Inorganic Chemistry of Life, Oxford University Press, Oxford [Google Scholar]
- 63. Grubbs R. D. (2002) Intracellular magnesium and magnesium buffering. BioMetals 15, 251–259 [DOI] [PubMed] [Google Scholar]
- 64. Anbar A. D. (2008) Oceans. Elements and evolution. Science 322, 1481–1483 [DOI] [PubMed] [Google Scholar]
- 65. Hider R. C., Kong X. L. (2011) Glutathione: a key component of the cytoplasmic labile iron pool. BioMetals 24, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 66. Williams R. J. (1982) Free manganese (II) and iron (II) cations can act as intracellular cell controls. FEBS Lett. 140, 3–10 [DOI] [PubMed] [Google Scholar]
- 67. McNaughton R. L., Reddi A. R., Clement M. H., Sharma A., Barnese K., Rosenfeld L., Gralla E. B., Valentine J. S., Culotta V. C., Hoffman B. M. (2010) Probing in vivo Mn2+ speciation and oxidative stress resistance in yeast cells with electron-nuclear double resonance spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 107, 15335–15339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maret W. (2004) Zinc and sulfur: a critical biological partnership. Biochemistry 43, 3301–3309 [DOI] [PubMed] [Google Scholar]
- 69. Krezel A., Maret W. (2006) Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 11, 1049–1062 [DOI] [PubMed] [Google Scholar]
- 70. Bozym R. A., Thompson R. B., Stoddard A. K., Fierke C. A. (2006) Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem. Biol. 1, 103–111 [DOI] [PubMed] [Google Scholar]
- 71. Vinkenborg J. L., Nicolson T. J., Bellomo E. A., Koay M. S., Rutter G. A., Merkx M. (2009) Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat. Methods 6, 737–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang D., Hurst T. K., Thompson R. B., Fierke C. A. (2011) Genetically encoded ratiometric biosensors to measure intracellular exchangeable zinc in Escherichia coli. J. Biomed. Opt. 16, 087011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huang C. P., Fofana M., Chan J., Chang C. J., Howell S. B. (2014) Copper transporter 2 regulates intracellular copper and sensitivity to cisplatin. Metallomics 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dodani S. C., Domaille D. W., Nam C. I., Miller E. W., Finney L. A., Vogt S., Chang C. J. (2011) Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and x-ray fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 5980–5985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rae T. D., Schmidt P. J., Pufahl R. A., Culotta V. C., O'Halloran T. V. (1999) Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808 [DOI] [PubMed] [Google Scholar]
- 76. Furukawa Y., Torres A. S., O'Halloran T. V. (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xiao Z., Wedd A. G. (2010) The challenges of determining metal-protein affinities. Nat. Prod. Rep. 27, 768–789 [DOI] [PubMed] [Google Scholar]
- 78. Reyes-Caballero H., Campanello G. C., Giedroc D. P. (2011) Metalloregulatory proteins: metal selectivity and allosteric switching. Biophys. Chem. 156, 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Helmann J. D. (2014) Specificity of metal sensing: iron and manganese homeostasis in Bacillus subtilis. J. Biol. Chem. 289, 28112–28120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rutherford J. C., Bird A. J. (2004) Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot. Cell 3, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li L., Bagley D., Ward D. M., Kaplan J. (2008) Yap5 is an iron-responsive transcriptional activator that regulates vacuolar iron storage in yeast. Mol. Cell. Biol. 28, 1326–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Waldron K. J., Robinson N. J. (2009) How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 7, 25–35 [DOI] [PubMed] [Google Scholar]
- 83. Foster A. W., Patterson C. J., Pernil R., Hess C. R., Robinson N. J. (2012) Cytosolic Ni(II) sensor in cyanobacterium: nickel detection follows nickel affinity across four families of metal sensors. J. Biol. Chem. 287, 12142–12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Patterson C. J., Pernil R., Dainty S. J., Chakrabarti B., Henry C. E., Money V. A., Foster A. W., Robinson N. J. (2013) Co(ll)-detection does not follow Kco(ll) gradient: channelling in Co(ll)-sensing. Metallomics 5, 352–362 [DOI] [PubMed] [Google Scholar]
- 85. Foster A. W., Pernil R., Patterson C. J., Robinson N. J. (2014) Metal specificity of cyanobacterial nickel-responsive repressor InrS: cells maintain zinc and copper below the detection threshold for InrS. Mol. Microbiol. 92, 797–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rutherford J. C., Cavet J. S., Robinson N. J. (1999) Cobalt-dependent transcriptional switching by a dual-effector MerR-like protein regulates a cobalt-exporting variant CPx-type ATPase. J. Biol. Chem. 274, 25827–25832 [DOI] [PubMed] [Google Scholar]
- 87. Thelwell C., Robinson N. J., Turner-Cavet J. S. (1998) An SmtB-like repressor from Synechocystis PCC 6803 regulates a zinc exporter. Proc. Natl. Acad. Sci. U.S.A. 95, 10728–10733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Outten C. E., O'Halloran T. V. (2001) Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 292, 2488–2492 [DOI] [PubMed] [Google Scholar]
- 89. Changela A., Chen K., Xue Y., Holschen J., Outten C. E., O'Halloran T. V., Mondragón A. (2003) Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science 301, 1383–1387 [DOI] [PubMed] [Google Scholar]
- 90. Wang D., Hosteen O., Fierke C. A. (2012) ZntR-mediated transcription of zntA responds to nanomolar intracellular free zinc. J. Inorg. Biochem. 111, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Alatossava T., Jütte H., Kuhn A., Kellenberger E. (1985) Manipulation of intracellular magnesium content in polymyxin B nonapeptide-sensitized Escherichia coli by ionophore A23187. J. Bacteriol. 162, 413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mills S. A., Marletta M. A. (2005) Metal binding characteristics and role of iron oxidation in the ferric uptake regulator from Escherichia coli. Biochemistry 44, 13553–13559 [DOI] [PubMed] [Google Scholar]
- 93. Iwig J. S., Leitch S., Herbst R. W., Maroney M. J., Chivers P. T. (2008) Ni(II) and Co(II) sensing by Escherichia coli RcnR. J. Am. Chem. Soc. 130, 7592–7606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chivers P. T., Sauer R. T. (2002) NikR repressor: high-affinity nickel binding to the C-terminal domain regulates binding to operator DNA. Chem. Biol. 9, 1141–1148 [DOI] [PubMed] [Google Scholar]
- 95. Dann C. E., 3rd, Wakeman C. A., Sieling C. L., Baker S. C., Irnov I., Winkler W. C. (2007) Structure and mechanism of a metal-sensing regulatory RNA. Cell 130, 878–892 [DOI] [PubMed] [Google Scholar]
- 96. Ma Z., Faulkner M. J., Helmann J. D. (2012) Origins of specificity and cross-talk in metal ion sensing by Bacillus subtilis Fur. Mol. Microbiol. 86, 1144–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ma Z., Cowart D. M., Scott R. A., Giedroc D. P. (2009) Molecular insights into the metal selectivity of the copper(I)-sensing repressor CsoR from Bacillus subtilis. Biochemistry 48, 3325–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ma Z., Gabriel S. E., Helmann J. D. (2011) Sequential binding and sensing of Zn(II) by Bacillus subtilis Zur. Nucleic Acids Res. 39, 9130–9138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Grossoehme N., Kehl-Fie T. E., Ma Z., Adams K. W., Cowart D. M., Scott R. A., Skaar E. P., Giedroc D. P. (2011) Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J. Biol. Chem. 286, 13522–13531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pennella M. A., Arunkumar A. I., Giedroc D. P. (2006) Individual metal ligands play distinct functional roles in the zinc sensor Staphylococcus aureus CzrA. J. Mol. Biol. 356, 1124–1136 [DOI] [PubMed] [Google Scholar]