

Background: Glycosyltransferase activities of LARGE are required for α-dystroglycan function.
Results: This study reveals that LARGE/LARGE2 glucuronyltransferase activity correlates with α-dystroglycan laminin binding activity in Largemyd and Large2−/− mice.
Conclusion: Both LARGE and LARGE2 contribute to the functional modification of α-dystroglycan in vivo.
Significance: Our results provide the first strong evidence that LARGE2 is involved in α-dystroglycan modification in vivo.
Keywords: Extracellular Matrix, Glycosyltransferase, Laminin, Muscular Dystrophy, Post-translational Modification (PTM), LARGE, Dystroglycan, Glucuronyltransferase
Abstract
Mutations in the LARGE gene have been identified in congenital muscular dystrophy (CMD) patients with brain abnormalities. Both LARGE and its paralog, LARGE2 (also referred to as GYLTL1B) are bifunctional glycosyltransferases with xylosyltransferase (Xyl-T) and glucuronyltransferase (GlcA-T) activities, and are capable of forming polymers consisting of [-3Xyl-α1,3GlcAβ1-] repeats. LARGE-dependent modification of α-dystroglycan (α-DG) with these polysaccharides is essential for the ability of α-DG to act as a receptor for ligands in the extracellular matrix. Here we report on the endogenous enzymatic activities of LARGE and LARGE2 in mice and humans, using a newly developed assay for GlcA-T activity. We show that normal mouse and human cultured cells have endogenous LARGE GlcA-T, and that this activity is absent in cells from the Largemyd (Large-deficient) mouse model of muscular dystrophy, as well as in cells from CMD patients with mutations in the LARGE gene. We also demonstrate that GlcA-T activity is significant in the brain, heart, and skeletal muscle of wild-type and Large2−/− mice, but negligible in the corresponding tissues of the Largemyd mice. Notably, GlcA-T activity is substantial, though reduced, in the kidneys of both the Largemyd and Large2−/− mice, consistent with the observation of α-DG/laminin binding in these contexts. This study is the first to test LARGE activity in samples as small as cryosections and, moreover, provides the first direct evidence that not only LARGE, but also LARGE2, is vital to effective functional modification of α-DG in vivo.
Introduction
Modification of α-dystroglycan (α-DG)4 by the like-acetylglucosaminyltransferase (LARGE) is required for (a) α-DG binding to laminin-G domain-containing extracellular matrix ligands such as laminin, agrin, and neurexin, (b) the maintenance of sarcolemmal integrity in skeletal muscle, (c) proper structure and function of the central nervous system, and (d) the interaction between the basement membrane and epithelial cells (1), and is thus referred to as “functional modification.” Mutations in the genes encoding LARGE (2) and other proteins that are involved directly or indirectly in O-mannosyl glycan synthesis on α-DG [protein O-mannosyltransferase 1 (POMT1) (3), POMT2 (4), protein O-mannose β-1,2-N-acetylglucosaminyltransferase 1 (POMGnT1) (5), fukutin (6), fukutin-related protein (FKRP) (7), ISPD (isoprenoid synthase domain containing) (8, 9), GTDC2 (10), β1,3-N-acetylglucosaminyltransferase (B3GNT1) (11), B3GALNT2 (12), TMEM5 (transmembrane protein 5) (13), and SGK196 (14)] have been identified in the dystroglycanopathies. These are congenital muscular dystrophies (CMDs) that are characterized by perturbed glycosylation and reduced ligand binding of α-DG, and are often associated with brain abnormalities (15). The Largemyd mouse, a spontaneous model of muscular dystrophy, has a mutation in the Large gene and results in defective α-DG glycosylation (16).
Functional modification of, and ligand binding to, α-DG require the phosphorylation of O-linked mannose. LARGE, fukutin, and FKRP have all been implicated in the modification pathway that assembles the ligand-binding moiety on this phosphorylated O-mannosyl glycan (17). β3GnT1, which is involved in the synthesis of poly-N-acetyllactosamine (18) and whose sequence is closely related to the second domain of LARGE (19), has been shown to contribute to the synthesis of the laminin-binding glycan of α-DG through formation of a complex with LARGE (20). Moreover, a study using a forward genetic screen demonstrated that β3GnT1 is required for functional glycosylation of α-DG in vivo; mice expressing a mutant form of this protein exhibited axonal-guidance defects like those observed in ISPD or an epiblast-specific DG mutant mouse (21). Recently, SGK196 was found to be a protein O-mannose kinase (designated as POMK) that generates [GalNAc-β3-GlcNAc-β4-(phosphate-6-)Man], a modification required for the functional glycosylation of α-DG (22). POMK phosphorylates the O-linked mannose only when the latter is extended with GalNAc-β3-GlcNAc-β, a unique disaccharide structure produced by sequential actions of GTDC2 and B3GALNT2. However, the precise molecular functions of the glycosyltransferase β3GnT1, and the other proteins fukutin, FKRP, and ISPD, as well as that of TMEM5, in α-DG glycosylation are not known.
Both LARGE and its paralog LARGE2 are bifunctional glycosyltransferases that have xylosyltransferase (Xyl-T) and glucuronyltransferase (GlcA-T) activities and the ability to polymerize Xyl-GlcA repeats (23–25). The overexpression of either of the LARGE paralogs in cultured cells enhances the functional modification of α-DG (26–30). Moreover, the overexpression of LARGE circumvents defects in the modification of α-DG in patient cells from several distinct types of CMD (30). A recent study revealed that the ability of LARGE to catalyze the α-DG modification is dependent on the availability of O-mannosyl phosphate acceptor sites (9). The LARGE paralogs have different tissue expression profiles; LARGE is widely expressed, with levels highest in brain, heart, and skeletal muscle, whereas LARGE2 is highly expressed in kidney and placenta, but undetectable in brain (28, 29). The ability of LARGE2 to generate polymeric Xyl-GlcA repeats and to enhance the functional modification of α-DG in cultured cells suggested that, like LARGE, this protein is involved in the physiological function of α-DG (24, 25). However, there is no direct in vivo evidence for this, and LARGE2 has not been reported to be involved in dystroglycanopathies or any other diseases.
We have developed an enzyme assay that enables us to measure endogenous LARGE enzymatic activity in cultured human lymphoblasts and myoblasts, mouse fibroblasts and tissues, and even from cryosections of both human and mouse skeletal muscle. Our results demonstrate that LARGE enzymatic activity is directly involved in the functional modification of α-DG, and provide strong evidence that LARGE2 is indeed also involved in this modification in vivo.
EXPERIMENTAL PROCEDURES
Materials
LARGEdTM and LARGE2dTM were described previously (23, 24). Mouse skin fibroblasts from wild-type (WT) C57/BL6 and Largemyd mice, and human Epstein-Barr virus-immortalized lymphoblasts from the CMD patient (Patient #1) were described previously (17, 31). Human control myoblasts were purchased from Cook MyoSite (Pittsburgh, Lot P201014-50M). Primary myoblasts from Patient #2 were cultured using standard method. Patient #2: 7-year-old girl, pregnancy, delivery, and neonatal period were normal. Developmental delays were suspected at 1 year. At 7 years she had minimal weakness, and was verbal with cognitive delays. MRI revealed thickened and pachygyric appearing frontal cortex and elevated creatine kinase at 2290 units/liter. Muscle biopsy revealed mild dystrophic changes and reduced staining for the glycan epitope of α-DG (IIH6). Control human quadriceps muscle biopsies were taken during diagnostic investigations of adult patients and were studied with patient consent. The biopsies had no histopathologic abnormalities.
Generation of Large2 (Gyltl1b) Knock-out Mice
An embryonic stem (ES) cell line (JM8A3.N1) with a targeted mutation in Large2 (Gyltl1btm1(KOMP)Wtsi) was obtained from the KOMP Repository at the University of California Davis. In the targeted ES cells (CloneID EPD0387_6_B09), Large2 exons 4–11 are replaced with a LacZ/neo-containing targeting cassette. Large2 knock-out mice were generated by blastocyst injection of targeted ES cells and chimeric mice were bred with C57BL/6 to select for germline transmission. Large2 knock-out mice were viable and obtained at the expected 1:3 Mendelian ratio when heterozygous mice were bred. No breeding or fertility problems were observed in the homozygous null mice. The genotype was confirmed by PCR. A 390 bp wild-type fragment was generated using primer LG2.wtF1 (5′-AGCTTCTACTGCCACCCAAGT-3′) in combination with LG2.wtR2 (5′-GGGTAATAATCTCTCGGCTGG-3′). Primer pair LG2.wtF1 and LAR3 (5′-CACAACGGGTTCTTCTGTTAGTCC-3′) amplified a 497bp mutant PCR fragment. The Large2-null mice appeared healthy and were indistinguishable from their heterozygous or control littermates with respect to body weight and life span when aged up to one year (data not shown). Also no increased spontaneous tumor load was observed in the null mice.
Preparation of Microsomal Membranes
Cells or mouse tissues were homogenized with ten volumes of 20 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, pH 7.4 and 0.25 m sucrose containing protease inhibitors (0.6 μg/ml of pepstatin A, 0.5 μg/ml of aprotinin, 0.5 μg/ml of leupeptin, 0.75 mm of benzamidine, 0.1 mm of phenylmethanesulfonyl fluoride, 0.4 μg/ml of calpain inhibitor, and 0.4 μg/ml of calpeptin). After centrifugation at 6,000 × g, the supernatant was ultracentrifuged at 100,000 × g and the resultant pellet (microsomal membranes) was solubilized in 20 mm MOPS buffer, pH 6.5 and 1% Triton X-100 for the enzymatic assay.
Enzymatic Assay
The HPLC-based enzymatic assay for LARGEdTM and LARGE2dTM was performed using GlcA-β-MU as the acceptor for Xyl-T activity, and Xyl-α1,3-GlcA-β-MU for GlcA-T activity as described previously (23, 24). For the assessment of metal dependence, a buffer at an optimal pH for each activity was used: LARGEdTM Xyl-T with 0.1 m sodium acetate, pH 5.0, LARGEdTM GlcA-T with 0.1 m MOPS, pH 6.5, LARGE2dTM Xyl-T with 0.1 m MOPS, pH 7.5, and LARGE2dTM GlcA-T with 0.1 m sodium acetate, pH 5.5. For the assessment of endogenous LARGE GlcA-T activity in cells or tissues, solubilized microsomes were incubated in a volume of 25 μl for 3 h at 37 °C, with 0.4 mm Xyl-α1,3-GlcA-β-MU and 10 mm UDP-GlcA in 0.1 m MES buffer, pH 6.0, at 5 mm MnCl2, 5 mm MgCl2, and 0.5% Triton X-100. The reaction was terminated by adding 25 μl of 0.1 m EDTA and boiling for 5 min, and the supernatant was analyzed using an LC-18 column. The GlcA-T activity was assessed by subtracting the background observed in the negative control sample without donor sugar, and normalized against the amount of protein measured using the DC Protein Assay (Bio-Rad). For the GlcA-T activity in cryosections of mouse or human quadriceps muscles, wheat-germ agglutinin (WGA)-agarose beads incubated with the Triton X-100 extract of proteins from the sections were directly used in the assay. The activity was assessed by subtracting the nonspecific background observed in the WGA-agarose incubated with the Largemyd muscle sections, and normalized to tissue volume (cross sectional area x number of sections x thickness of sections).
Mass Spectrometry (MS) Analysis
MS analysis of the enzymatic product was conducted on an Agilent 6520A Quadrupole Time of Flight mass spectrometer as described previously (24).
Pull-down Assay, Glycoprotein Enrichment, Immunoblotting, and Laminin Overlay
A pull-down assay using α-DG-Fc fusion constructs with or without the T192M substitution was performed as described previously (32), on lysates from TSA201 cells expressing myc+His6-tagged mouse LARGE2 (24). Glycoprotein enrichment using WGA-agarose beads, immunoblotting, and laminin overlay were performed as described previously (17, 23). Antibodies to the α-DG glycan (IIH6 and VIA4–1) and core (sheep5) epitopes were described previously (33). IIH6 and VIA4–1 are available from the Developmental Studies Hybridoma Bank at the University of Iowa.
RESULTS
Metal Dependence of Xyl-T and GlcA-T Activities of LARGE and LARGE2
In a previous study of LARGE and LARGE2 (24), using recombinant enzymes that lack their transmembrane domains (LARGEdTM and LARGE2dTM), we found that these proteins have distinct pH optima for both their Xyl-T and GlcA-T activities. To further optimize the conditions for assaying the endogenous enzymes, we tested the metal dependence of each activity, using Xyl-α1,3-GlcA-β-MU and GlcA-β-MU as acceptors for the GlcA-T and Xyl-T assays, respectively (24). The DXD motifs (Asp-X-Asp; X, any amino acid) required for metal-ion binding and catalysis are conserved in the LARGE paralogs, and many glycosyltransferases with this motif require Mn2+ as a cofactor (34, 35). We assayed LARGE and LARGE2 using a buffer containing either a metal ion or EDTA at 5 mm, at the pH that is optimal for each activity. For the Xyl-T activities (Fig. 1 left), both LARGE paralogs require a metal ion, with LARGE activity dependent on specifically Mn2+; notably, LARGE2 activity was lower yet substantial in the presence of Mg2+ or Ca2+. In contrast, the GlcA-T activities (Fig. 1, right) of both the LARGE paralogs were substantial in the absence of any metal ion (enzyme preparations were subjected to extensive buffer exchange to remove residual metal ions before the assay was carried out), yet both activities were completely inhibited by addition of EDTA. Mn2+ enhanced the activities of both LARGE and LARGE2 substantially. However, whereas Mg2+ and Ca2+ were as effective as Mn2+ for LARGE activity, they had no effect on LARGE2 activity.
FIGURE 1.
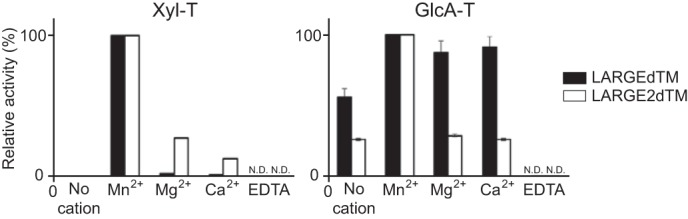
Metal dependence of Xyl-T and GlcA-T activities of LARGEdTM and LARGE2dTM. Data from two independent experiments carried out in the presence or absence of 5 mm of each metal ion or EDTA are shown as relative activity (%). The Xyl-T and GlcA-T activities in the presence of Mn2+ were arbitrarily set at 100%. ND, not detected.
Endogenous LARGE GlcA-T Activity in Cultured Cells
Using the disaccharide acceptor Xyl-α1,3-GlcA-β-MU, we tested for endogenous LARGE GlcA-T activity (Fig. 2A) in cultured cells. First, we used fibroblasts isolated from skeletal muscle of WT or Largemyd mice (in the latter, a mutation in the Large gene causes defects in α-DG glycosylation) (16). The microsomal membranes were extracted from these fibroblasts and solubilized in 1% Triton X-100, conditions compatible with the GlcA-T assay for LARGEdTM (data not shown). When these solubilized samples were incubated with Xyl-α1,3-GlcA-β-MU and UDP-GlcA in a buffer at pH 6.5 (the optimum for LARGE GlcA-T activity (24)), a specific product peak (P) was observed; its retention time was the same as that of its counterpart in the control reaction with LARGEdTM, on both an amide column (Fig. 3A, gradient elution) and an LC-18 column (Fig. 3B, isocratic elution). The elution profiles are consistent with the reaction of LARGEdTM, whose product peak was confirmed as GlcA-added Xyl-α1,3-GlcA-β-MU by MS (Fig. 2, B–D). The reaction was specific because the product peak required the presence of both UDP-GlcA (+donor, Fig. 3, A and B) and LARGE (no such peak was detected using Largemyd fibroblasts). Product detection was more sensitive when the sample was purified on the LC-18 column (Fig. 3B) versus amide column (Fig. 3A). We were able to detect specific GlcA-T activity in the control fibroblasts with as few as 250,000 cells.
FIGURE 2.

LARGE GlcA-T reaction on the disaccharide acceptor. A, scheme depicting the reaction that LARGE GlcA-T activity catalyzes on the acceptor Xyl-α1,3-GlcA-β-MU. B and C, HPLC profile of the reaction product LARGEdTM generates from Xyl-α1,3-GlcA-β-MU and UDP-GlcA, as assessed by amide column (B) and LC-18 column (C). P, product. S, unreacted substrate. Dotted line, %B buffer. D, quadrupole time of flight (Q/TOF)-MS analysis of the product peak isolated in C. Star and diamond indicate Xyl and GlcA, respectively.
FIGURE 3.

The WT, but not the Largemyd, fibroblasts have LARGE GlcA-T activity. A and B, representative HPLC profiles from the amide column (A) and the LC-18 column (B) of the product obtained from the reaction. LARGEdTM served as the control enzyme, solubilized microsomes extracted from the WT or Largemyd mouse fibroblasts as the source of endogenous enzyme, Xyl-α1,3-GlcA-β-MU as substrate, and UDP-GlcA as the donor sugar. P, product. S, unreacted substrate. Dotted line, % buffer B. Asterisks indicate nonspecific peak observed regardless of presence or absence of the donor substrate.
We also applied our assay to microsomes prepared from human Epstein-Barr virus-transformed lymphoblasts from a control individual and a CMD patient (Patient #1) in whom abnormal splicing of the LARGE transcript is accompanied by markedly reduced α-DG glycosylation (31). We detected specific GlcA-T activity in the control lymphoblasts but no or very little activity in the patient cells (Fig. 4, A and B). In addition, we examined human myoblasts from a control individual and the other CMD patient (Patient #2) who has compound heterozygous mutations in the LARGE gene. The patient has a novel 74 kb deletion including exons 4–6 in one allele, and a known missense G1525A (E509K) mutation in exon 13 in the other allele (2). We were able to detect specific GlcA-T activity in the control myoblasts and not in the patient cells (Fig. 4C). Thus, the assay detects specifically the endogenous GlcA-T activity of LARGE in cultured cells.
FIGURE 4.

CMD patient lymphoblasts are deficient for LARGE GlcA-T activity. A, representative HPLC profiles from the LC-18 column of the product obtained from reactions in which solubilized microsomes extracted from control or CMD Patient #1 lymphoblasts served as the source of enzyme and Xyl-α1,3-GlcA-β-MU as substrate, in the presence (top) or absence (bottom) of the donor sugar UDP-GlcA. P, product. S, unreacted substrate. Dotted line, % buffer B. Asterisks indicate nonspecific peak observed regardless of presence or absence of the donor substrate. B, GlcA-T activity on the solubilized microsomes extracted from the control or the CMD Patient #1 lymphoblasts. Relative activity (%) with respect to the control, and the standard deviation in triplicate experiments, are shown. C, representative HPLC profiles of the GlcA-T assay on solubilized microsomes extracted from control or CMD Patient #2 myoblasts. Note that the elution profiles are different from those in A, because a slightly lower concentration of buffer B was used for the separation.
LARGE GlcA-T Activity in Mouse Tissues
We next tested for measurable endogenous LARGE GlcA-T activity in mouse tissues. First, we examined microsomes prepared from the brains of WT mice. The GlcA-T activity was linearly dependent on time, the amount of protein, and the presence of acceptor (Xyl-α1,3-GlcA-β-MU) (Fig. 5, A–C) and showed a typical saturation curve for the donor (UDP-GlcA) (Fig. 5D). The activity was specific to LARGE, as it was reduced to 43.5% in the Largemyd heterozygous mouse (+/−) versus WT (+/+) brain sample, and was undetectable in the homozygous Largemyd mouse (−/−) brain sample (Fig. 5E). Next we assessed the total LARGE- and LARGE2-derived GlcA-T activity in the tissues of WT, Largemyd, and Large2−/− mice, using a buffer at pH 6.0 (at which both LARGE paralogs show substantial GlcA-T activity (24)). We observed specific GlcA-T activity in both the WT and Large−/− brains but none in the Largemyd brain (Fig. 6, A and B). Results for heart and skeletal muscle were similar, showing that LARGE2 is not substantially active in these tissues. In the case of kidney, in contrast, both the Largemyd and the Large2−/− mice had reduced but some levels of enzyme activity (Fig. 6B). These results are consistent with the expression profiles of the LARGE paralogs in these tissues (28, 29).
FIGURE 5.

GlcA-T activity of endogenous LARGE in mouse brain. A–D, LARGE GlcA-T activity on solubilized mouse brain membranes as a function of time (A), amount of protein (B), concentration of acceptor Xyl-α1,3-GlcA-β-MU (C), and donor UDP-GlcA (D). E, specific GlcA-T activity on brain membranes extracted from the WT (+/+, n = 3), heterozygous (+/−, n = 3), and homozygous Largemyd (−/−, n = 1) mice. Data are shown as relative activity (%).
FIGURE 6.

The GlcA-T activity in mouse tissues from WT, Largemyd, and Large2−/− mice. A, representative HPLC (LC-18 column) of the product obtained from the LARGE GlcA-T reaction using solubilized microsomes extracted from the brain of each mouse. P, product. S, unreacted substrate. Dotted line, %B buffer. B, specific GlcA-T activity on solubilized microsomes extracted from tissues of the WT, Largemyd, or Large2−/− mice. Data obtained from three individual animals are shown, with error bars indicating the standard deviations.
Physical Interaction between LARGE2 and α-DG
The N-terminal domain of α-DG must be present for LARGE to functionally modify the α-DG mucin-like domain (26). Recently, a limb-girdle muscular dystrophy patient was found to have a missense mutation that results in substitution T192M (Thr-192→Met) within the α-DG N terminus, and it was suggested that the accompanying failure in functional modification of α-DG is a consequence of disruption of the molecular interaction between α-DG and LARGE (32). Using α-DG-Fc fusion proteins with or without the T192M substitution, we tested the interaction with LARGE2 by a pull-down assay, as examined with LARGE in the previous study. While both Fc5 (α-DG-Fc) and Fc2 (α-DG N terminus-Fc) bound to LARGE2, Fc proteins harboring the T192M substitution bound much less efficiently (Fig. 7). These data support the notion that, like LARGE, LARGE2 is involved in the functional modification of α-DG, and that the T192M substitution also prevents this modification.
FIGURE 7.
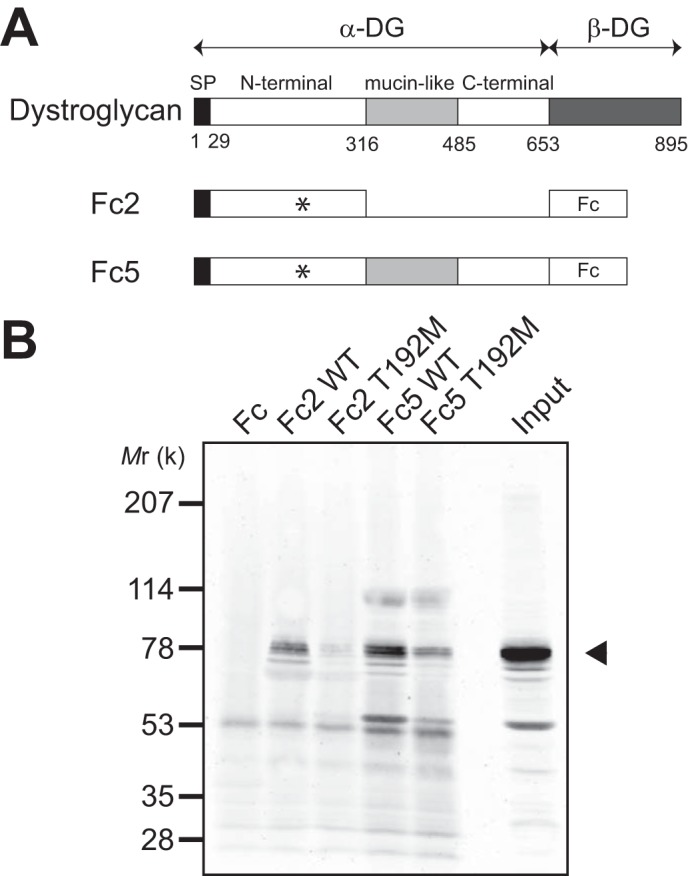
Molecular interaction between α-DG and LARGE2. A, schematic representation of the α-DG-Fc fusion constructs used in the pull-down assay. The position of Thr-192 is indicated by an asterisk. B, representative data from pull-down of myc+His6-tagged LARGE2 with the α-DG-Fc fusion constructs, with or without the mucin and C-terminal domains, and with or without the T192M substitution (in the N-terminal domain). Immunoblotting with anti-myc antibody demonstrates that LARGE2 binds the N-terminal domain of α-DG, and that this binding is perturbed by the T192M mutation. Arrowhead indicates the position of myc+His6-tagged LARGE2.
Functional Modification of α-DG in Mouse Tissues
We examined the functional modification of α-DG in mouse tissues by immunoblotting WGA-enriched glycoproteins from the WT, Largemyd, and Large2−/− mice using antibodies to the α-DG glycan epitopes (IIH6 and VIA4–1), and by carrying out a laminin overlay assay. In the cases of skeletal muscle, heart, and brain, the functional modification of α-DG in the Large2−/− mouse was comparable to that in the WT mouse (Fig. 8). The samples from the corresponding Largemyd tissues showed no reactivity to IIH6 or VIA4–1, or laminin binding capacity, even though the α-DG core protein was expressed at high levels (Fig. 8). In the kidneys, in contrast, the functional modification was robust only in the WT samples; it was diminished in both the Largemyd and the Large2−/− samples, with respect to both intensity of staining and molecular weight (Fig. 8). Thus, like LARGE, LARGE2 contributes to the functional modification of α-DG in the kidney.
FIGURE 8.
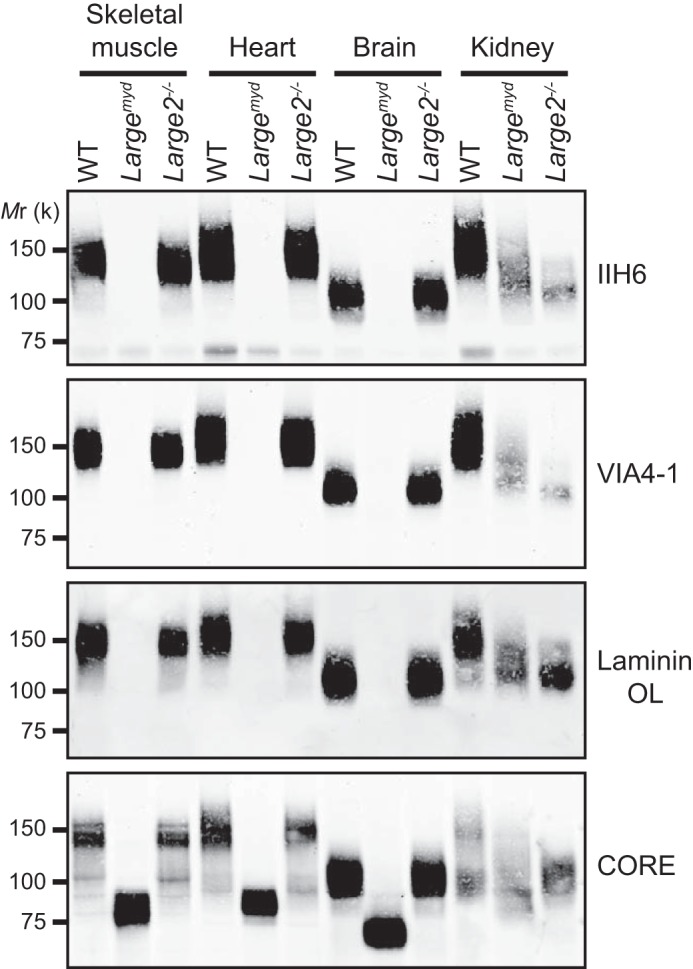
Functional glycosylation of α-DG in tissues of the WT, Largemyd, and Large2−/− mice. Immunoblotting or laminin overlay (OL) of WGA-enriched glycoproteins extracted from tissues of the WT, Largemyd, or Large2−/− mouse. The α-DG antibodies used are: IIH6, VIA4–1 (recognize glycan epitopes) and CORE (recognizes a core protein epitope). Mr, relative molecular mass.
LARGE GlcA-T Activity in Mouse and Human Skeletal Muscle Sections
In the course of carrying out this study, we found that we could enrich specifically the GlcA-T activity of endogenous LARGE by purification with WGA-agarose. When solubilized membranes from rabbit brain were incubated with WGA-agarose, almost all of the LARGE GlcA-T activity bound to the beads, and the activity was specifically eluted from the beads by N-acetylglucosamine (Fig. 9A). Given the potential usefulness of assessing LARGE GlcA-T activity from biopsy sections, we took advantage of this enrichment to adapt our assay to measuring LARGE GlcA-T activity from relatively small amounts of mouse and human skeletal muscle. We first subjected extracts of 30 quadriceps muscle cryosections of 10-μm thickness (section area; 18∼42 mm2) to WGA enrichment. Subsequent measurement of the specific GlcA-T activity as for the experiments above revealed that the activity in the mouse and human skeletal muscle sections was comparable, and that no activity was present in the sections from Largemyd mice (Fig. 9B). Similar results were obtained when the assay was repeated with the other sets of extracts from human and mouse quadriceps muscle cryosections.
FIGURE 9.

LARGE GlcA-T activity in WGA-enriched extract from skeletal-muscle cryosections. A, HPLC profiles of unbound (Void) and eluate (Elu) fractions of WGA-agarose-treated rabbit brain-membrane extracts. P, product. S, unreacted substrate. Dotted line, % buffer B. B, quantification of GlcA-T activity on the WGA-enriched extracts of skeletal-muscle cryosections. Relative activity (%) with respect to mouse samples, and standard deviation (n = 3 for human controls, n = 5 control animals for mouse, n = 3 for Largemyd) are shown.
DISCUSSION
Given that the expression levels of endogenous glycosyltransferases in the Golgi apparatus are generally low, it has been a challenge to detect the endogenous LARGE proteins in cells and tissues. Our modified assay, which enables us to measure the endogenous enzymatic activities of LARGE and LARGE2 in cultured cells and tissues, revealed that LARGE2 has the capacity to functionally modify α-DG in a physiologically relevant setting.
Our initial characterization of the metal dependence of the enzymatic activities of LARGE and LARGE2 indicates that, as is the case for the pH optima of the Xyl-T and GlcA-T activities (24), their metal binding requirements also differ. This is consistent with what we know about the other glycosyltransferases that contribute to the functional modification of α-DG. For example, POMT activity does not require metal cations whereas POMGnT1 does and, moreover, has a strong preference for Mn2+ (36–38). In the cases of LARGE and LARGE2, both the Xyl-T and GlcA-T activities have a preference for Mn2+. Notably, although the Xyl-T activities showed an almost complete dependence on the cation, as did the GlcA-T activity of LARGE2, the GlcA-T activity of LARGE was substantial in the presence of either Mg2+ or Ca2+. The physiological underpinnings of this difference remain to be determined.
For measuring the endogenous activity, we used Xyl-α1,3-GlcA-β-MU as the acceptor in the GlcA-T assay for the LARGE proteins and found it to be a good acceptor substrate. The disaccharide acceptor probably has an advantage over the simpler monosaccharide acceptors (such as the Xyl-α-pNP and GlcA-β-MU used in the initial characterization of enzymatic activities of LARGE) with respect to the specificity and sensitivity of the detection in contexts where enzyme is limiting. This is in contrast to our initial study in which abundant recombinant enzyme was used for the assay (23), and was crucial for developing an assay that could be carried out on small samples such as those obtained from patient biopsies.
We used the GlcA-T assay to compare activities among control and Largemyd mouse fibroblasts, as well as human lymphoblasts from a control and a CMD patient (Patient #1) who has an abnormal transcript of LARGE. The Large gene in the Largemyd mouse has an intragenic deletion of exons 4–7 which causes a frameshift and premature stop codon before the first catalytic domain in the resultant mRNA (16). The CMD patient is homozygous for a LARGE allele in which an intra-chromosomal duplication is inserted into intron 10, and thus most of the transcripts are truncated immediately after exon 10 (31) and produce a protein that does not contain the second catalytic domain, i.e. the GlcA-T domain (23). The finding of no or only very low GlcA-T activity in these cells accounts for the defective glycosylation of α-DG in the mutant mouse and patient. In the case of the other CMD patient (Patient #2), one of the alleles has a novel 74 kb deletion including exons 4–6, and the other allele has a known missense G1525A (Glu509Lys) mutation (2). When LARGE was coexpressed with α-DG in cultured cells, the Glu509Lys mutant failed to enhance the functional modification (28). Our assay data is consistent with those previous reports; the patient cells had no or very low GlcA-T activity with the mutations.
Dystroglycanopathies can be caused by mutations in multiple genes related to O-mannosyl glycan synthesis, and these lead to an extremely broad clinical spectrum and relatively poor correlations between genotype and phenotype (15). An enzymatic assay of POMGnT1 activity has been carried out on extracts of muscle biopsies from Muscle-Eye-Brain disease patients and various controls, as well as on fibroblasts and lymphoblasts from the patients and carriers (39, 40). Similarly, assays have been developed for both the POMGnT1 and POMT activities, and have been used on lymphoblasts and fibroblasts from dystroglycanopathy patients, including some that have not been genetically characterized (41, 42). These reports indicate that enzymatic assays of known glycosyltransferases are viable tools for rapid and sensitive screening for dystroglycanopathies. Our enzymatic assay for LARGE activity, which can be applied to cryosections of tissue biopsies as well as fibroblasts, immortalized lymphoblasts and myoblasts, is expected to likewise be a powerful and complementary tool that will identify patients with pathogenic mutations of LARGE.
Our analysis of tissues from WT, Largemyd, and Large2−/− mice using the LARGE GlcA-T assay demonstrated a good correlation between enzymatic activity and the tissue expression profiles of the Large transcripts (i.e. robust expression of Large but not Large2 transcript in mouse brain, heart, and skeletal muscle, and substantial expression of both transcripts in kidney (28, 29)). Since Large2 expression is highest in kidney we analyzed the null mice for kidney disease. Our tests for proteinuria as an indicator of kidney disease revealed no overt abnormalities in the null mice (data not shown). Considering the overlap in function and tissue-specific expression of Large and Large2, it does not come as a surprise that a lack of Large2 expression can be tolerated and functionally compensated by the wide expression of the paralog Large. Such genetic redundancy has been widely accepted to account for the lack of phenotypes in many knockouts (43). Consistent with redundancy as an explanation for the lack of an apparent phenotype in the Large2−/− mice, a study of a mouse model of Fukuyama-type congenital muscular dystrophy, which is caused by a mutation in the fukutin gene, has indicated that even a small amount of IIH6-reactive α-DG is sufficient to maintain integrity of the skeletal muscle (44). On the other hand, it must also be mentioned that in contrast to previous reports suggesting functions for DG in the kidney (45, 46), a study using a conditional DG allele with a variety of Cre promoters suggests that DG does not contribute significantly to kidney development or function (47). Given that DG is the only known substrate for modification by LARGE or LARGE2, these findings raise questions about the physiological significance of the LARGE paralogs in the kidney. An alternative possibility is that LARGE or LARGE2 adds the laminin-binding glycan to an as yet unknown substrate(s), as suggested by a study based on adenovirus-mediated LARGE overexpression in DG-deficient cells (48), and that this aspect of LARGE activity may account for their physiological relevance in the kidney.
In conclusion, our results from immunoblotting kidney extracts from WT and mutant mice, as well as our pull-down assay for the interaction between the α-DG N terminus and LARGE2, provide strong evidence that LARGE2 contributes to the functional modification of α-DG in vivo.
Acknowledgments
We thank all members of the Campbell laboratory for fruitful discussions and technical support, Dr. Harry Schachter for valuable comments on the manuscript, the University of Iowa Proteomics Facility and Developmental Studies Hybridoma Bank for their services, and the DNA and cell bank of Genethon (Evry, France) where the blood lymphocytes of Patient #1 have been immortalized. The Large2 (Gyltl1b) knock-out mice used for this research project were generated from a targeted ES cell line obtained from the NCRR-NIH-supported Knock-Out Mouse Project (KOMP) Repository.
This work was supported in part by the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant (1 U54 NS053672, to K. P. C., S. A. M., and T. W.), the Muscular Dystrophy Association, and an American Recovery and Reinvestment Act (ARRA) Go Grant (1 RC2 NS069521-01, to K. P. C. and T. W.).
- α-DG
- α-dystroglycan
- B3GNT1
- β1,3-N-acetylglucosaminyltransferase
- CMD
- congenital muscular dystrophy
- FKRP
- fukutin-related protein
- GlcA
- glucuronic acid
- GlcA-β-MU
- 4-methylumbelliferyl-β-d-glucuronide
- GlcA-T
- glucuronyltransferase
- HPLC
- high-performance liquid chromatography
- ISPD
- isoprenoid synthase domain containing
- MOPS
- 3-(N-morpholino)propanesulfonic acid
- MS
- mass spectrometry
- POMGnT1
- protein O-mannose β-1,2-N-acetylglucosaminyltransferase 1
- POMT
- protein O-mannosyltransferase
- Xyl
- xylose
- Xyl-T
- xylosyltransferase
- LARGE
- like-acetylglucosaminyltransferase.
REFERENCES
- 1. Barresi R., Campbell K. P. (2006) Dystroglycan: from biosynthesis to pathogenesis of human disease. J. Cell Sci. 119, 199–207 [DOI] [PubMed] [Google Scholar]
- 2. Longman C., Brockington M., Torelli S., Jimenez-Mallebrera C., Kennedy C., Khalil N., Feng L., Saran R. K., Voit T., Merlini L., Sewry C. A., Brown S. C., Muntoni F. (2003) Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of α-dystroglycan. Hum. Mol. Genet. 12, 2853–2861 [DOI] [PubMed] [Google Scholar]
- 3. Beltrán-Valero de Bernabe D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W. B., Cormand B., Lehesjoki A. E., Cruces J., Voit T., Walsh C. A., van Bokhoven H., Brunner H. G. (2002) Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 71, 1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Reeuwijk J., Janssen M., van den Elzen C., Beltran-Valero de Bernabé D., Sabatelli P., Merlini L., Boon M., Scheffer H., Brockington M., Muntoni F., Huynen M. A., Verrips A., Walsh C. A., Barth P. G., Brunner H. G., van Bokhoven H. (2005) POMT2 mutations cause α-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 42, 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida A., Kobayashi K., Manya H., Taniguchi K., Kano H., Mizuno M., Inazu T., Mitsuhashi H., Takahashi S., Takeuchi M., Herrmann R., Straub V., Talim B., Voit T., Topaloglu H., Toda T., Endo T. (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, Dev. Cell 1, 717–724 [DOI] [PubMed] [Google Scholar]
- 6. Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M., Hamano K., Sakakihara Y., Nonaka I., Nakagome Y., Kanazawa I., Nakamura Y., Tokunaga K., Toda T. (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394, 388–392 [DOI] [PubMed] [Google Scholar]
- 7. Brockington M., Blake D. J., Prandini P., Brown S. C., Torelli S., Benson M. A., Ponting C. P., Estournet B., Romero N. B., Mercuri E., Voit T., Sewry C. A., Guicheney P., Muntoni F. (2001) Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am. J. Hum. Genet. 69, 1198–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roscioli T., Kamsteeg E. J., Buysse K., Maystadt I., van Reeuwijk J., van den Elzen C., van Beusekom E., Riemersma M., Pfundt R., Vissers L. E., Schraders M., Altunoglu U., Buckley M. F., Brunner H. G., Grisart B., Zhou H., Veltman J. A., Gilissen C., Mancini G. M., Delrée P., Willemsen M. A., Ramadža D. P., Chitayat D., Bennett C., Sheridan E., Peeters E. A., Tan-Sindhunata G. M., de Die-Smulders C. E., Devriendt K., Kayserili H., El-Hashash O. A., Stemple D. L., Lefeber D. J., Lin Y. Y., van Bokhoven H. (2012) Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 44, 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Willer T., Lee H., Lommel M., Yoshida-Moriguchi T., de Bernabe D. B., Venzke D., Cirak S., Schachter H., Vajsar J., Voit T., Muntoni F., Loder A. S., Dobyns W. B., Winder T. L., Strahl S., Mathews K. D., Nelson S. F., Moore S. A., Campbell K. P. (2012) ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 44, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Manzini M. C., Tambunan D. E., Hill R. S., Yu T. W., Maynard T. M., Heinzen E. L., Shianna K. V., Stevens C. R., Partlow J. N., Barry B. J., Rodriguez J., Gupta V. A., Al-Qudah A. K., Eyaid W. M., Friedman J. M., Salih M. A., Clark R., Moroni I., Mora M., Beggs A. H., Gabriel S. B., Walsh C. A. (2012) Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet. 91, 541–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buysse K., Riemersma M., Powell G., van Reeuwijk J., Chitayat D., Roscioli T., Kamsteeg E. J., van den Elzen C., van Beusekom E., Blaser S., Babul-Hirji R., Halliday W., Wright G. J., Stemple D. L., Lin Y. Y., Lefeber D. J., van Bokhoven H. (2013) Missense mutations in β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum. Mol. Genet. 22, 1746–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stevens E., Carss K. J., Cirak S., Foley A. R., Torelli S., Willer T., Tambunan D. E., Yau S., Brodd L., Sewry C. A., Feng L., Haliloglu G., Orhan D., Dobyns W. B., Enns G. M., Manning M., Krause A., Salih M. A., Walsh C. A., Hurles M., Campbell K. P., Manzini M. C., Stemple D., Lin Y. Y., Muntoni F. (2013) Mutations in B3GALNT2 Cause Congenital Muscular Dystrophy and Hypoglycosylation of α-Dystroglycan. Am. J. Hum. Genet. 92, 354–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vuillaumier-Barrot S., Bouchet-Séraphin C., Chelbi M., Devisme L., Quentin S., Gazal S., Laquerrière A., Fallet-Bianco C., Loget P., Odent S., Carles D., Bazin A., Aziza J., Clemenson A., Guimiot F., Bonnière M., Monnot S., Bole-Feysot C., Bernard J. P., Loeuillet L., Gonzales M., Socha K., Grandchamp B., Attié-Bitach T., Encha-Razavi F., Seta N. (2012) Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am. J. Hum. Genet. 91, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jae L. T., Raaben M., Riemersma M., van Beusekom E., Blomen V. A., Velds A., Kerkhoven R. M., Carette J. E., Topaloglu H., Meinecke P., Wessels M. W., Lefeber D. J., Whelan S. P., van Bokhoven H., Brummelkamp T. R. (2013) Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340, 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Godfrey C., Foley A. R., Clement E., Muntoni F. (2011) Dystroglycanopathies: coming into focus. Curr. Opin. Genet. Dev. 21, 278–285 [DOI] [PubMed] [Google Scholar]
- 16. Grewal P. K., Holzfeind P. J., Bittner R. E., Hewitt J. E. (2001) Mutant glycosyltransferase and altered glycosylation of α-dystroglycan in the myodystrophy mouse. Nat. Genet. 28, 151–154 [DOI] [PubMed] [Google Scholar]
- 17. Yoshida-Moriguchi T., Yu L., Stalnaker S. H., Davis S., Kunz S., Madson M., Oldstone M. B., Schachter H., Wells L., Campbell K. P. (2010) O-mannosyl phosphorylation of α-dystroglycan is required for laminin binding. Science 327, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasaki K., Kurata-Miura K., Ujita M., Angata K., Nakagawa S., Sekine S., Nishi T., Fukuda M. (1997) Expression cloning of cDNA encoding a human β-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc. Natl. Acad. Sci. U.S.A. 94, 14294–14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peyrard M., Seroussi E., Sandberg-Nordqvist A. C., Xie Y. G., Han F. Y., Fransson I., Collins J., Dunham I., Kost-Alimova M., Imreh S., Dumanski J. P. (1999) The human LARGE gene from 22q12.3-q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc. Natl. Acad. Sci. U.S.A. 96, 598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bao X., Kobayashi M., Hatakeyama S., Angata K., Gullberg D., Nakayama J., Fukuda M. N., Fukuda M. (2009) Tumor suppressor function of laminin-binding α-dystroglycan requires a distinct β3-N-acetylglucosaminyltransferase. Proc. Natl. Acad. Sci. U.S.A. 106, 12109–12114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wright K. M., Lyon K. A., Leung H., Leahy D. J., Ma L., Ginty D. D. (2012) Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron 76, 931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoshida-Moriguchi T., Willer T., Anderson M. E., Venzke D., Whyte T., Muntoni F., Lee H., Nelson S. F., Yu L., Campbell K. P. (2013) SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 341, 896–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M. E., Yu L., Campbell K. P. (2012) Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inamori K., Hara Y., Willer T., Anderson M. E., Zhu Z., Yoshida-Moriguchi T., Campbell K. P. (2013) Xylosyl- and glucuronyltransferase functions of LARGE in α-dystroglycan modification are conserved in LARGE2. Glycobiology 23, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashikov A., Buettner F. F., Tiemann B., Gerardy-Schahn R., Bakker H. (2013) LARGE2 generates the same xylose- and glucuronic acid-containing glycan structures as LARGE. Glycobiology 23, 303–309 [DOI] [PubMed] [Google Scholar]
- 26. Kanagawa M., Saito F., Kunz S., Yoshida-Moriguchi T., Barresi R., Kobayashi Y. M., Muschler J., Dumanski J. P., Michele D. E., Oldstone M. B., Campbell K. P. (2004) Molecular recognition by LARGE is essential for expression of functional dystroglycan. Cell 117, 953–964 [DOI] [PubMed] [Google Scholar]
- 27. Brockington M., Torelli S., Prandini P., Boito C., Dolatshad N. F., Longman C., Brown S. C., Muntoni F. (2005) Localization and functional analysis of the LARGE family of glycosyltransferases: significance for muscular dystrophy. Hum. Mol. Genet. 14, 657–665 [DOI] [PubMed] [Google Scholar]
- 28. Fujimura K., Sawaki H., Sakai T., Hiruma T., Nakanishi N., Sato T., Ohkura T., Narimatsu H. (2005) LARGE2 facilitates the maturation of α-dystroglycan more effectively than LARGE. Biochem. Biophys. Res. Commun. 329, 1162–1171 [DOI] [PubMed] [Google Scholar]
- 29. Grewal P. K., McLaughlan J. M., Moore C. J., Browning C. A., Hewitt J. E. (2005) Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 15, 912–923 [DOI] [PubMed] [Google Scholar]
- 30. Barresi R., Michele D. E., Kanagawa M., Harper H. A., Dovico S. A., Satz J. S., Moore S. A., Zhang W., Schachter H., Dumanski J. P., Cohn R. D., Nishino I., Campbell K. P. (2004) LARGE can functionally bypass α-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat. Med. 10, 696–703 [DOI] [PubMed] [Google Scholar]
- 31. Clarke N. F., Maugenre S., Vandebrouck A., Urtizberea J. A., Willer T., Peat R. A., Gray F., Bouchet C., Manya H., Vuillaumier-Barrot S., Endo T., Chouery E., Campbell K. P., Mégarbané A., Guicheney P. (2011) Congenital muscular dystrophy type 1D (MDC1D) due to a large intragenic insertion/deletion, involving intron 10 of the LARGE gene. Eur. J. Hum. Genet. 19, 452–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hara Y., Balci-Hayta B., Yoshida-Moriguchi T., Kanagawa M., Beltrán-Valero de Bernabé D., Gündesli H., Willer T., Satz J. S., Crawford R. W., Burden S. J., Kunz S., Oldstone M. B., Accardi A., Talim B., Muntoni F., Topalolu H., Diner P., Campbell K. P. (2011) A dystroglycan mutation associated with limb-girdle muscular dystrophy. N. Engl. J. Med. 364, 939–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michele D. E., Barresi R., Kanagawa M., Saito F., Cohn R. D., Satz J. S., Dollar J., Nishino I., Kelley R. I., Somer H., Straub V., Mathews K. D., Moore S. A., Campbell K. P. (2002) Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418, 417–422 [DOI] [PubMed] [Google Scholar]
- 34. Wiggins C. A., Munro S. (1998) Activity of the yeast MNN1 α-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc. Natl. Acad. Sci. U.S.A. 95, 7945–7950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Unligil U. M., Zhou S., Yuwaraj S., Sarkar M., Schachter H., Rini J. M. (2000) X-ray crystal structure of rabbit N-acetylglucosaminyltransferase I: catalytic mechanism and a new protein superfamily. EMBO J. 19, 5269–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang W., Betel D., Schachter H. (2002) Cloning and expression of a novel UDP-GlcNAc:α-D-mannoside β1,2-N-acetylglucosaminyltransferase homologous to UDP-GlcNAc:α-3-D-mannoside β1,2-N-acetylglucosaminyltransferase I. Biochem. J. 361, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Manya H., Chiba A., Yoshida A., Wang X., Chiba Y., Jigami Y., Margolis R. U., Endo T. (2004) Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. U.S.A. 101, 500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takahashi S., Sasaki T., Manya H., Chiba Y., Yoshida A., Mizuno M., Ishida H., Ito F., Inazu T., Kotani N., Takasaki S., Takeuchi M., Endo T. (2001) A new β-1,2-N-acetylglucosaminyltransferase that may play a role in the biosynthesis of mammalian O-mannosyl glycans. Glycobiology 11, 37–45 [DOI] [PubMed] [Google Scholar]
- 39. Vajsar J., Zhang W., Dobyns W. B., Biggar D., Holden K. R., Hawkins C., Ray P., Olney A. H., Burson C. M., Srivastava A. K., Schachter H. (2006) Carriers and patients with muscle-eye-brain disease can be rapidly diagnosed by enzymatic analysis of fibroblasts and lymphoblasts. Neuromuscul. Disord. 16, 132–136 [DOI] [PubMed] [Google Scholar]
- 40. Zhang W., Vajsar J., Cao P., Breningstall G., Diesen C., Dobyns W., Herrmann R., Lehesjoki A. E., Steinbrecher A., Talim B., Toda T., Topaloglu H., Voit T., Schachter H. (2003) Enzymatic diagnostic test for Muscle-Eye-Brain type congenital muscular dystrophy using commercially available reagents. Clin. Biochem. 36, 339–344 [DOI] [PubMed] [Google Scholar]
- 41. Manya H., Bouchet C., Yanagisawa A., Vuillaumier-Barrot S., Quijano-Roy S., Suzuki Y., Maugenre S., Richard P., Inazu T., Merlini L., Romero N. B., Leturcq F., Bezier I., Topaloglu H., Estournet B., Seta N., Endo T., Guicheney P. (2008) Protein O-mannosyltransferase activities in lymphoblasts from patients with α-dystroglycanopathies. Neuromuscul. Disord. 18, 45–51 [DOI] [PubMed] [Google Scholar]
- 42. Lommel M., Cirak S., Willer T., Hermann R., Uyanik G., van Bokhoven H., Körner C., Voit T., Barić I., Hehr U., Strahl S. (2010) Correlation of enzyme activity and clinical phenotype in POMT1-associated dystroglycanopathies. Neurology 74, 157–164 [DOI] [PubMed] [Google Scholar]
- 43. Barbaric I., Miller G., Dear T. N. (2007) Appearances can be deceiving: phenotypes of knockout mice. Brief Funct. Genomic Proteomic 6, 91–103 [DOI] [PubMed] [Google Scholar]
- 44. Kanagawa M., Nishimoto A., Chiyonobu T., Takeda S., Miyagoe-Suzuki Y., Wang F., Fujikake N., Taniguchi M., Lu Z., Tachikawa M., Nagai Y., Tashiro F., Miyazaki J., Tajima Y., Takeda S., Endo T., Kobayashi K., Campbell K. P., Toda T. (2009) Residual laminin-binding activity and enhanced dystroglycan glycosylation by LARGE in novel model mice to dystroglycanopathy. Hum. Mol. Genet. 18, 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Durbeej M., Larsson E., Ibraghimov-Beskrovnaya O., Roberds S. L., Campbell K. P., Ekblom P. (1995) Non-muscle α-dystroglycan is involved in epithelial development. J. Cell Biol. 130, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vogtländer N. P., Visch H. J., Bakker M. A., Berden J. H., van der Vlag J. (2009) Ligation of α-dystroglycan on podocytes induces intracellular signaling: a new mechanism for podocyte effacement? PLoS One 4, e5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jarad G., Pippin J. W., Shankland S. J., Kreidberg J. A., Miner J. H. (2011) Dystroglycan does not contribute significantly to kidney development or function, in health or after injury. Am. J. Physiol. Renal Physiol. 300, F811–F820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Z., Zhang P., Hu H. (2011) LARGE expression augments the glycosylation of glycoproteins in addition to α-dystroglycan conferring laminin binding. PLoS One 6, e19080. [DOI] [PMC free article] [PubMed] [Google Scholar]