

Background: Opening and closing of the CFTR channel involve conformational changes in the channel pore.
Results: Different metal bridges stabilize the channel in the open or closed configuration.
Conclusion: Opening and closing result from relative lateral movement of different transmembrane segments.
Significance: This work defines the three-dimensional conformational changes underlying channel opening and closing.
Keywords: ABC Transporter, Chloride Channel, Cysteine-mediated Cross-linking, Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), Ion Channel
Abstract
Opening and closing of the cystic fibrosis transmembrane conductance regulator are controlled by ATP binding and hydrolysis by the cytoplasmic nucleotide-binding domains. Different conformational changes in the channel pore have been described during channel opening and closing; however, the relative importance of these changes to the process of gating the pore is not known. We have used patch clamp recording to identify high affinity Cd2+ bridges formed between pairs of pore-lining cysteine residues introduced into different transmembrane α-helices (TMs). Seven Cd2+ bridges were identified forming between cysteines in TMs 6 and 12. Interestingly, each of these Cd2+ bridges apparently formed only in closed channels, and their formation stabilized the closed state. In contrast, a single Cd2+ bridge identified between cysteines in TMs 1 and 12 stabilized the channel open state. Analysis of the pattern of Cd2+ bridge formation in different channel states suggests that lateral separation and convergence of different TMs, rather than relative rotation or translation of different TMs, is the key conformational change that causes the channel pore to open and close.
Introduction
Cystic fibrosis is caused by loss-of-function mutations in the cystic fibrosis transmembrane conductance regulator (CFTR),2 a phosphorylation-regulated, nucleotide-gated Cl− channel found in the apical membrane of many epithelial cell types (1). CFTR is a member of the ATP-binding cassette (ABC) family of membrane transport proteins, with an archetypal architecture of two membrane-spanning domains (MSDs), each made up of six transmembrane α-helices (TMs) and two cytoplasmic nucleotide-binding domains (NBDs) (see Fig. 1). CFTR also contains a unique cytoplasmic regulatory domain. Opening and closing of phosphorylated CFTR channels are controlled by ATP binding and hydrolysis by a dimer of the two NBDs; this is presumed to result in the opening and closing of a gate within the MSDs that allows or prevents the transmembrane movement of Cl− and other small anions (2, 3). However, the overall structure of the channel pore formed by the MSDs, the location of the gate, and the structural rearrangements that occur within the pore region that control gate opening and closing are not known.
FIGURE 1.
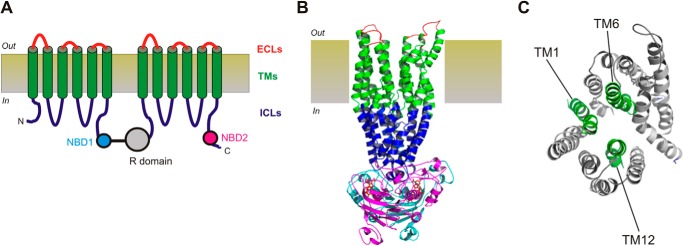
Putative structure of the CFTR channel pore region. A, graphic model of CFTR structure, consisting of 12 TMs (green), two NBDs (NBD1, cyan; NBD2, magenta), the cytoplasmic R domain (gray), six extracellular loops (ECLs, red), and four intracellular loops (ICLs, blue). B, so-called “channel-like” atomic homology model of CFTR structure (6), based on the bacterial ABC protein Sav1866 and showing the approximate extent of the extracellular loops, TMs, intracellular loops, and NBDs using the same color scheme as in A. The R domain is absent from this model. C, location of putative pore-lining TMs 1, 6, and 12 within the MSDs of this same homology model, viewed from the extracellular side of the membrane. CFTR model structures were visualized with PyMOL (33) using coordinates provided by Ref. 6.
The channel pore is lined by a subset of the TMs, with TMs 1, 6, and 12 playing particularly important functional roles (2, 3) (see Fig. 1). At the functional level, the pore is divided into a central narrow region flanked by wider inner and outer vestibules (3). The contributions of different TMs, and specific amino acid side chains coming from different TMs, to each of these functionally distinct, two-dimensional regions of the pore has been reviewed recently (3); however, the three-dimensional structure of the pore is less well defined experimentally. Three-dimensional atomic homology models of CFTR have been generated based on related, non-ion channel ABC proteins (4–6); however, the structures of the pore suggested by these models show some important discrepancies from functional models based on direct experimental evidence (3).
The three-dimensional structure of the pore is not static but presumably changes as the channel opens and closes. Indeed, different conformational changes of the pore during channel gating have been proposed (2, 3). These include local constrictions/dilations of the pore (7–10), as well as rotational (11, 12) and translational (7) movements of individual TMs. However, although experimental evidence for these different kinds of movement has been put forward, the relative mechanistic importance of these movements for the process of channel opening and closing is not known. Evidence for conformational changes in individual TMs within the inner vestibule of the pore during channel gating is mainly two-dimensional in nature, coming from state-dependent differences in the accessibility of cytoplasmically applied cysteine reactive methanethiosulfonate reagents to cysteine side chains introduced into TMs 1 (8, 9, 13, 14), 6 (8, 9, 11, 15), 11 (10), and 12 (12, 16). However, how the relative positions of these TM changes in three-dimensional space during channel gating have not previously been investigated.
Direct information concerning the three-dimensional structure of the pore has come from disulfide cross-linking experiments (7, 13, 17), as well as from experiments in which anionic channel blocker binding was altered by introduction of positively charged amino acid side chains (17, 18). Previously, we have used the oxidizing agent copper(II)-o-phenanthroline to show that disulfide bonds can be formed between cysteine side chains introduced into different TMs, and we used this information to infer the relative proximity of residues in these different TMs lining the inner vestibule of the pore (13, 17). However, because these disulfide bonds are essentially irreversible under non-reducing conditions, this approach has several disadvantages, such as the potential for disulfide bonds to form during rare or unnatural channel conformations when normally distant cysteine side chains transiently approach close together. Furthermore, the irreversible nature of disulfide bonds makes it difficult to investigate state-dependent changes in the proximity of pairs of cysteine side chains. An alternative approach is to use weaker Cd2+ bridges that are more likely to catch the channel in native conformations (19–22). An additional advantage is that these weaker, reversible interactions can break and reform, allowing channel gating to proceed relatively normally (22) and so potentially providing information on the relative propensity of Cd2+ bridges to form in different gating states. In the present study, we have investigated the potential of Cd2+ to form metal bridges between nearby pore-lining cysteine side chains introduced into TMs 1, 6, and 12 lining a localized region of the inner vestibule of the channel pore close to an anionic blocker-binding site (17, 18). In particular, we have taken advantage of the dynamic nature of Cd2+ bridges to associate the formation of these bridges with different channel conformational states. Our results show that Cd2+ bridges form selectively between a limited number of cysteine pairs in TMs 6 and 12, suggesting a relative alignment of these two TMs. Interestingly, each of these bridges appears to form only in closed channels, and they stabilize the channel in the closed state. In contrast, a single identified Cd2+ bridge between TMs 1 and 12 stabilized the channel in the open state. These results support a model in which relative lateral movement of different TMs toward and away from each other in the inner vestibule, rather than helical rotation or translation of individual TMs, is required for the channel to open and close.
EXPERIMENTAL PROCEDURES
Experiments were carried out in baby hamster kidney cells transiently transfected with CFTR. In this study, we have used a human CFTR variant in which all cysteines had been removed by mutagenesis (as described in Ref. 23) and that includes a mutation in NBD1 (V510A) to increase protein expression in the cell membrane (24). This Cys-less variant, which we have used in previous studies of pore conformational change (7–9), has functional properties that are very similar to those of wild-type CFTR (25). Additional mutations were introduced into the Cys-less background using the QuikChange site-directed mutagenesis system (Agilent Technologies, Santa Clara, CA) and verified by DNA sequencing. In some cases, cysteine mutants were combined with the NBD2 mutation E1371Q, which we (7–9) and others (12, 26) have used to abolish ATP-dependent channel gating and lock CFTR channels in the open state. In our expression system, the open probability of channels bearing the E1371Q mutation is >95% (7).
To investigate potential Cd2+ bridges formed between pore-lining cysteine side chains exposed in the inner vestibule of the CFTR pore, we combined individual cysteines that we previously found to be accessible to cytoplasmically applied methanethiosulfonate reagents in three important pore-lining TMs: TM1 (K95C, Q98C) (13), TM6 (I344C, V345C, M348C, A349C) (15), and TM12 (M1140C, S1141C, T1142C, Q1144C, W1145C, V1147C, N1148C) (16), to generate a total of 50 double cysteine mutants (8 TM1:TM6; 14 TM1:TM12; 28 TM6:TM12).
Macroscopic CFTR currents were recorded using patch clamp recordings from inside-out membrane patches, as described in detail recently (17). Following patch excision and recording of background currents, CFTR channels were activated by exposure to protein kinase A catalytic subunit (PKA; 20 nm) plus MgATP (1 mm) in the intracellular solution. In some cases (see Fig. 4), patches were subsequently treated with 2 mm sodium pyrophosphate (PPi) to stabilize the channel open state (27, 28), as described previously in studies of state-dependent modification of Cys-less CFTR (8, 15). Both intracellular (bath) and extracellular (pipette) solutions contained (in mm): 150 NaCl, 2 MgCl2, 10 N-tris[hydroxymethyl]methyl-2-aminoethanesulfonate, pH 7.4. Current traces were filtered at 100 Hz using an eight-pole Bessel filter, digitized at 250 Hz, and analyzed using pCLAMP software (Molecular Devices, Sunnyvale, CA). CFTR current amplitudes were monitored during brief voltage deflections (to ±50 mV) from a holding potential of 0 mV applied every 6 s (10). In some cases, the identity of CFTR currents in inside-out patches was confirmed by their sensitivity to the specific inhibitor CFTRinh-172 (5 μm). Different concentrations of CdCl2 were applied directly to the cytoplasmic face of inside-out patches from stock solutions made up in normal intracellular solution. In those cases where exposure to Cd2+ caused inhibition of current amplitude, concentration-inhibition relationships were fitted by the equation.
 |
FIGURE 4.
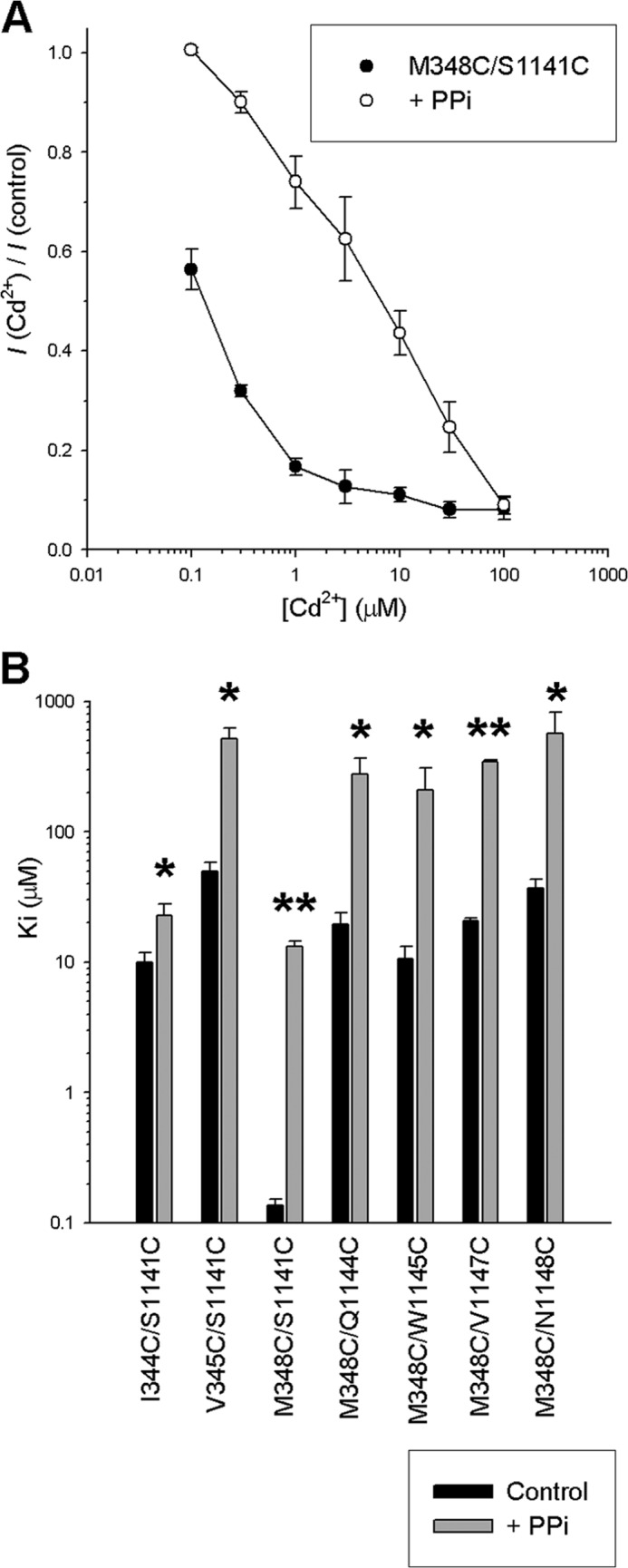
Effect of increasing channel open probability with PPi on Cd2+ sensitivity of TM6:TM12 double cysteine mutant channels. A, mean fractional current remaining following the addition of different concentrations of Cd2+ in M348C/S1141C channels in the presence of PKA and ATP (●) or following activation by PKA and ATP followed by treatment with 2 mm PPi to maximize channel open probability (○). B, mean Ki values estimated for the double cysteine mutant channels listed, either in the presence of PKA and ATP (black bars) or following treatment with PPi (gray bars). Asterisks indicate a significant difference in the presence of PPi (*, p < 0.05; **, p < 0.00005). Error bars indicate the means ± S.E. from 3–7 patches.
Experiments were carried out at room temperature (21–24 °C). The values are presented as the means ± S.E. Tests of significance were carried out using an unpaired t test, with p < 0.05 being considered statistically significant. All chemicals were from Sigma-Aldrich, except for PKA (Promega, Madison, WI).
RESULTS
Metal Bridge Formation between TMs 6 and 12
Pore-lining TMs 6 and 12 make important functional contributions to the inner vestibule of the pore (2, 3) and occupy symmetrical positions within the MSDs (Fig. 1). To investigate the potential for Cd2+ bridge formation between pore-forming side chains coming from TMs 6 and 12, we introduced cysteines at each of four sites in TM6 (Ile-344, Val-345, Met-348, Ala-349) and seven sites in TM12 (Met-1140, Ser-1141, Thr-1142, Gln-1144, Trp-1145, Val-1147, Asn-1148) that we have previously found to be accessible to cysteine-reactive methanethiosulfonate reagents applied to the cytoplasmic side of the membrane (15, 16). Combination of each single cysteine mutant from each TM then gave a total of 28 double-cysteine mutants containing one cysteine in TM6 and one cysteine in TM12 within the putative inner vestibule of the pore.
Cys-less CFTR itself was weakly inhibited by the addition of millimolar concentrations of Cd2+ to the cytoplasmic side of the membrane (Fig. 2, A and B), presumably acting in a cysteine-independent manner. All 11 single cysteine mutants tested were more strongly inhibited by Cd2+ (Fig. 2, A and B), with estimated Ki values in the range of 70–250 μm (Fig. 3). This suggests that Cd2+ is able to bind to each of these introduced cysteine side chains (29), which is consistent with the proposed pore-lining location of the cysteine residues introduced. The effects of Cd2+ appeared independent of voltage (Fig. 2, A and B).
FIGURE 2.

Effects of Cd2+ on single and double cysteine mutant channels in TMs 6 and 12. A, sample time courses of current activation (by PKA application in the presence of ATP, at time 0) and inhibition by Cd2+ (at the concentrations indicated at the top of each panel) for the named CFTR variants. Currents were monitored using depolarizing voltage ramps (B) and are plotted at −50 mV (●) and +50 mV (○). The identity of CFTR currents was confirmed by sensitivity to CFTRinh-172 (5 μm; indicated by hatched bars marked inh-172). B, sample macroscopic I-V curves for these same four membrane patches, recorded before the addition of Cd2+ (control) and after the addition of the noted concentration of Cd2+. C, sample time courses (upper panels) and I-V curves (lower panels) recorded from similar experiments for the double cysteine mutants I344C/S1141C (left) and M348C/S1141C (right). D and E, concentration-dependent inhibition of the single (filled symbols) and double (open symbols) cysteine mutants channels indicated, indicating the increase in Cd2+ apparent affinity when individual cysteine mutants in TMs 6 and 12 are combined. Data have been fitted as described under “Experimental Procedures.” Mean of data from 3–9 patches. Error bars indicate the means ± S.E.
FIGURE 3.
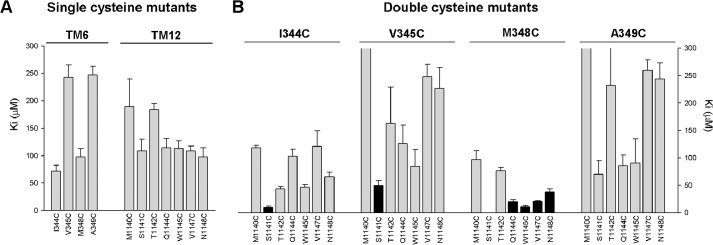
Mean inhibitory effect of Cd2+ on single and double cysteine mutant channels in TMs 6 and 12. Mean Ki values were calculated from fits to data from individual patches as those shown in Fig. 2, D and E. A, single cysteine mutants in TM6 (left) and TM12 (right) as indicated. B, double cysteine mutants that combine one cysteine substituted in TM6 (listed above the panel) with one in TM12 (listed below the panel). Those double mutants that showed a significantly different Ki from that of either of the corresponding single cysteine mutants (p < 0.05) are filled black, whereas those that with no significant difference are filled gray. Note that the Ki for M348C/S1141C (0.14 ± 0.02 μm, n = 5) is too small to be visible on this scale, but was significantly different from either M348C or S1141C alone (p < 0.0005; see also Fig. 2E). Also note that the estimated Ki values for some double mutants were ≥300 μm (V345C/M1140C, 316 ± 38 μm, n = 3; A349C/M1140C, 345 ± 58 μm, n = 3; A349C/T1142C, 231 ± 68 μm, n = 3). Error bars indicate the means ± S.E. from 3–7 patches.
Of the 28 double-cysteine mutants tested, most (twenty-one) did not result in an increase in sensitivity to Cd2+ as compared with the corresponding single-cysteine mutants (Fig. 4). In contrast, the remaining seven double cysteine mutants, namely I344C/S1141C (Fig. 2, C and D), V345C/S1141C, M348C/S1141C (Fig. 2, C and E), M348C/V1144C, M348C/W1145C, M348C/V1147C, and M348C/N1148C, all showed increased sensitivity to Cd2+, leading to a significant decrease in Ki as compared with either of the single cysteine mutants from which they were derived (estimated Ki values < 50 μm; Fig. 3). In theory, this apparent increase in Cd2+ potency could reflect additive effects of two Cd2+ ions bound simultaneously to different cysteine side chains. However, if the inner vestibule of the pore were able to accommodate two Cd2+ ions simultaneously, we would expect most double cysteine mutants to show strong Cd2+ binding, especially where the two cysteine side chains are expected to be relatively far apart (see “Discussion”). Instead, we propose that the relatively small number of double cysteine mutant channels that show strengthened Cd2+ binding results from specific coordination of a single Cd2+ ion interacting simultaneously with both cysteine side chains. This proposal is consistent with recent studies showing that cysteine side chains introduced into ion channel pores bind a single Cd2+ ion and that nearby cysteine side chains then increase the strength of binding of this single Cd2+ ion (29). In effect, those combinations of cysteine pairs that led specifically to a strengthened binding of Cd2+ within the pore (but no other combination of cysteines tested) are proposed to support the formation of Cd2+ bridges between TMs 6 and 12 that impair channel function. In particular, M348C/S1141C was associated with a dramatic increase in sensitivity to Cd2+ (Fig. 2, C and E, and Fig. 3) with an estimated Ki of 0.14 ± 0.02 μm (n = 5).
The finding that Cd2+ bridges between TMs 6 and 12 inhibited channel function suggested to us that these bridges may stabilize the channel closed state. To test this idea, we applied Cd2+ following channel treatment with 2 mm PPi to stabilize the channel open state (see “Experimental Procedures”). In each case, PPi treatment resulted in a weakening of Cd2+ inhibition (Fig. 4A) and a significant increase in Ki (Fig. 4B) of between 2.3-fold (in I344C/S1141C) and 97-fold (in M348C/S1141C). This weakened interaction is consistent with Cd2+ normally interacting with the closed state of these mutant channels to impair channel opening. To confirm this apparent state dependence of Cd2+ bridge formation, we combined selected single and double cysteine mutants with the E1371Q mutation in NBD2 that results in constitutively open channels in our expression system (see “Experimental Procedures”). As shown in Fig. 5, all E1371Q-containing channels tested were only weakly sensitive to inhibition by Cd2+, resulting in a significant increase in Ki both in single cysteine (I344C, M348C, S1141C) and in double cysteine (I344C/S1141C, Fig. 5, A–C; M348C/S1141C, Fig. 5, A, D, and E) mutant channels. However, the effect of the E1371Q mutation was greater in the double cysteine mutants; this gating mutation increased Ki 30-fold in I344C/S1141C (Fig. 5C) and 2500-fold in M348C/S1141C (Fig. 5E). In fact, in striking contrast to normally gating CFTR channels in which these double cysteine mutants showed greatly increased sensitivity to Cd2+ as compared with the corresponding single cysteine mutants (Figs. 3 and Fig. 5, C and E), in E1371Q mutant channels, the Ki for the double cysteine mutants was not significantly different from the single cysteine mutants (Fig. 5, B–E). The finding that these cysteine pairs fail to increase Cd2+ sensitivity in constitutively open channels (relative to single cysteines) suggests that Cd2+ bridge formation effectively does not occur in the open state and supports the idea that the inhibitory effect of Cd2+ bridge formation between TMs 6 and 12 results from a specific interaction with, and resulting stabilization of, the channel closed state.
FIGURE 5.

Effect of the E1371Q mutation on the effects of Cd2+ on single and double cysteine mutant channels in TMs 6 and 12. A, sample time courses and I-V curves illustrating the low Cd2+ sensitivity of constitutively active I344C/S1141C/E1371Q (left panels) and M348C/S1141C/E1371Q (right panels) channels in inside-out patches. Experiments were performed as described in the legend for Fig. 2. Note that although currents were relatively insensitive to Cd2+, they were inhibited by CFTRinh-172 (5 μm; indicated by hatched bars marked inh-172). B–E, mean fractional current remaining following the addition of different concentrations of Cd2+ (B and D) and mean Ki values for the single and double cysteine mutants listed (C and E). In C and E, black bars indicate channels gating normally, and gray bars indicate the corresponding cysteine mutants with the E1371Q mutation; asterisks indicate a significant difference between the two for the same cysteine constructs (p < 0.02). Error bars indicate the means ± S.E. from 3–7 patches.
Metal Bridge Formation between TMs 1 and 6 and 12
In addition to TMs 6 and 12, TM1 also plays an important functional role lining the inner vestibule (3, 13, 14). However, in our hands, fewer side chains from TM1 line the inner vestibule (13), and when investigating potential Cd2+ bridge formation involving this TM, we were limited to cysteines substituted at two positions (Lys-95 and Gln-98). Furthermore, in contrast to TM6:TM12 double cysteine mutants, in several cases, double mutants involving these sites in TM1 failed to express significant currents in inside-out membrane patches. As a result, we were only able to study 9 mutants combining cysteines in TMs 1 and 6, and 10 mutants combining cysteines in TMs 1 and 12.
In striking contrast to cysteine substitution in TMs 6 and 12, we found that K95C channel currents were weakly, but significantly, increased in amplitude by Cd2+ at concentrations of 30–100 μm (Fig. 6). Because K95C is the only single mutant we have studied that involves replacement of a positively charged amino acid side chain by cysteine, and because removal of this positive charge by mutagenesis is known to reduce single channel conductance by ∼85% (17, 18, 30), we considered whether this apparent stimulatory effect of Cd2+ could result from a bound Cd2+ ion acting as a surrogate fixed positive charge in the pore and thereby increasing Cl− conductance. If this were the case, Cd2+ should have a consistent stimulatory effect irrespective of channel gating. However, we found that constitutively open K95C/E1371Q channels were insensitive to Cd2+ at concentrations up to 100 μm (Fig. 6B), as were K95C channels following treatment with 2 mm PPi (data not shown), inconsistent with an effect of Cd2+ on Cl− conductance. Instead, these results suggest that Cd2+ binding to K95C results in a small but significant stabilization of the channel open state, an effect that is not observed in E1371Q channels that are already open almost 100% of the time (7). In contrast to these results with K95C, but in common with TMs 6 and 12 single cysteine mutants, Q98C channels were inhibited by Cd2+ (Fig. 7).
FIGURE 6.

Cadmium increases macroscopic current amplitude in K95C and K95C/S1141C channels in a gating-dependent manner. A, sample time courses and I-V curves showing Cd2+ concentration-dependent increases in macroscopic current amplitude in K95C (left panels) and K95C/S1141C (right panels) channels in inside-out patches. Experiments were performed as described in the legend for Fig. 2. Note that although currents were increased by Cd2+, they were inhibited by CFTRinh-172 (5 μm). B, concentration-dependent increases in current amplitude in K95C were not observed in K95C/E1371Q. Asterisks indicate a significant difference from K95C (p < 0.05). C, Cd2+ causes a significantly greater increase in K95C/S1141C current amplitude as compared with K95C (p < 0.0001); this increase is not observed in K95C/S1141C/E1371Q channels. Error bars indicate the means ± S.E. from 3–6 patches.
FIGURE 7.
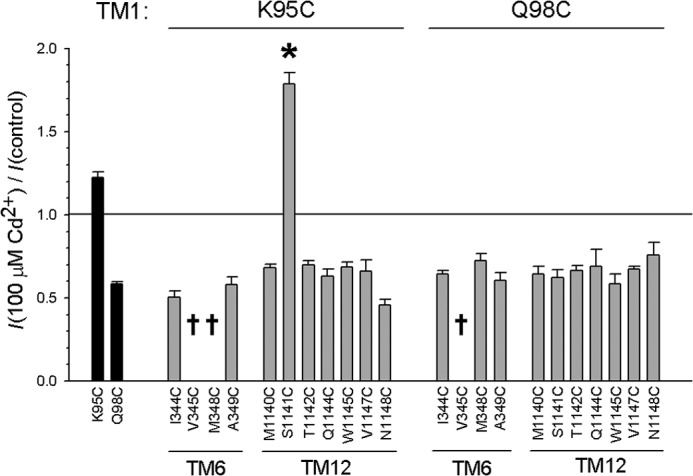
Mean inhibitory effect of Cd2+ on channels bearing cysteine side chains in TM1. Shown is the mean effect of 100 μm Cd2+ on macroscopic current amplitude for two single cysteine mutants (K95C, Q98C; black bars) and 19 double cysteine mutants that combine the TM1 cysteine listed above the panel with the TMs 6 or 12 cysteine listed below. Daggers indicate three mutants that did not express measureable macroscopic current in inside-out patches. Asterisk indicates a significant difference from the corresponding single cysteine mutant in TMs 6 or 12 (p < 0.000005). Error bars indicate the means ± S.E. from 3–5 patches.
Most (18 out of 19) double cysteine mutants including K95C or Q98C were weakly inhibited by Cd2+, consistent with Cd2+ effects on single cysteine mutants in TMs 6 or 12 (or indeed in Q98C) (Fig. 7). Thus, evidence for Cd2+ bridge formation involving TM1 is scant. However, one mutant, K95C/S1141C, was apparently stimulated by Cd2+ (Fig. 6A), and this stimulation was of a greater magnitude than that seen in the K95C single cysteine mutant (Figs. 6C and 7). This suggests that a Cd2+ bridge may form between K95C in TM1 and S1141C in TM12. As with K95C itself, the stimulatory effects of Cd2+ on K95C/S1141C were abolished by the E1371Q mutation (Fig. 6C) or by treatment with PPi (data not shown), suggesting that the observed effects of Cd2+ are due to an increase in channel open probability and a stabilization of the channel open state. Thus, although Cd2+ bridges between TMs 6 and 12 consistently stabilize the closed state, this single Cd2+ bridge between TMs 1 and 12 appears to stabilize the channel in the open state.
DISCUSSION
The inner vestibule of the CFTR channel pore is lined by TMs 1, 6, and 12 (3); however, their relative arrangement in three-dimensional space is not known. Consistent with their known pore-forming roles, cysteine residues introduced into each of 13 putative pore-lining positions in these three TMs resulted in macroscopic current sensitivity to submillimolar concentrations of cytoplasmically applied Cd2+ ions (Figs. 3A and 7) that was not observed in Cys-less background channels (Fig. 2). Similar Cd2+ sensitivity has previously been described for cysteine mutants in other parts of the pore (7, 26) and presumably reflects Cd2+ ion binding to these 13 different pore-exposed cysteine side chains (29). Channels bearing two cysteine residues, one in each of two different TMs, were also sensitive to cytoplasmic Cd2+ (Figs. 3B and 7). In most cases, combining two cysteines in different TMs did not lead to a significant change in apparent Cd2+ binding (Figs. 3B and 7), suggesting that the two cysteine side chains in question were unable to act together to coordinate Cd2+ binding, and also that simultaneous binding of two Cd2+ ions to both cysteines in this localized region of the pore is unlikely. However, in a limited number of cases, double cysteine mutants exhibited tighter Cd2+ binding than either of the single cysteine mutants from which they were derived (Fig. 3). In highly localized regions of ion channel pores, this is generally considered consistent with the formation of a metal bridge between the two cysteine side chains (29). Indeed, the ability of a restricted subset of cysteine pairs specifically to coordinate binding of a single Cd2+ ion in the inner vestibule is consistent with the proposed alignment of TMs 1, 6, and 12 around this region of the pore (Fig. 8A). In contrast, binding of a second Cd2+ ion would be expected to be favored when two cysteine side chains were further apart, when simultaneously bound Cd2+ ions would be expected to experience less steric and electrostatic interference.
FIGURE 8.

Cadmium bridges between different TMs stabilize different channel conformations. A, identification of different Cd2+ bridges formed between different TMs. Primary sequences of TMs 1, 6, and 12 are aligned as described previously (3). (Note: On the figure, the TMs are shown in the order (left to right) 6 - 12 - 1 for visual clarity.) Residues in red are those with pore-lining side chains mutated in the present study; those in black are non-pore-lining. Red lines connect residues that can form Cd2+ bridges following mutation to cysteine. B, relative location and orientation of Met-348 (TM6) with those residues in TM12, which can form Cd2+ bridges with M348C following mutation to cysteine. C, relative location and orientation of Ser-1141 (TM12) with those residues in TMs 1 and 6, which can form Cd2+ bridges with S1141C following mutation to cysteine. In both B and C, individual TMs are illustrated as in the atomic model of Fig. 1, viewed both from the top (upper panels) and from the side (lower panels). D, relative location and orientation of key Cd2+ bridge-forming residues Lys-95 (TM1), Met-348 (TM6), and Ser-1141 (TM12), viewed from above. E, proposed graphic model for Cd2+ bridge formation between the key sites illustrated in D in open and closed states, and proposed implications for conformational rearrangement of these three TMs associated with channel gating. The closed state is stabilized by a Cd2+ bridge between M348C (TM6) and S1141C (TM12), and the open state is stabilized by a Cd2+ bridge between K95C (TM1) and S1141C (TM12). In this model, ATP-dependent channel opening and closing are associated with a relative lateral movement of different TM helices (depicted by red arrows) with no requirement for helical rotation.
Of eight putative Cd2+ bridges identified (out of a total of 50 double cysteine mutants tested), all involved a cysteine substituted either for Met-348 (TM6) or for Ser-1141 (TM12) (Fig. 8A). Furthermore, when cysteine was present at both of these positions, an unusually high affinity Cd2+ bridge was formed (Figs. 2–5). These results suggest that cysteine side chains substituted for Met-348 or Ser-1141 are particularly well positioned to form inter-TM Cd2+ bridges.
Consideration of the different Cd2+ bridges that can form between different TMs may provide some information on the relative alignment and orientation of these TMs. Thus, M348C is able to form Cd2+ bridges with cysteines at multiple positions in TM12 (S1141C, Q1144C, W1145C, V1147C, N1148C) (Fig. 8B), and S1141C is able to form Cd2+ bridges with cysteines both in TM1 (K95C) and in TM6 (I344C, V345C, M348C) (Fig. 8C). The variety of bridges that can be formed from these two positions suggests that some degree of conformational flexibility must exist within these TMs. For example, M348C is able to form Cd2+ bridges with TM12 sites on a broad face of the TM12 helical face (Fig. 8B, top panel), suggesting some degree of relative rotational flexibility or movement, and also across a distance spanning two helical turns (from Ser-1141 to Asn-1148) (Fig. 8B, bottom panel), suggesting also some translational flexibility or movement. Similarly, S1141C is able to form Cd2+ bridges with different sites in TM6 (Fig. 8C). Thus, our results suggest that different TMs surrounding the inner vestibule do not occupy rigid positions relative to one another. Cadmium bridges between cysteine side chains are thought to require sulfur distances <6.5 Å (31).
Seven different Cd2+ bridges were identified as forming between cysteine side chains in TMs 6 and 12 (Figs. 3B and 8A). Interestingly, all of these Cd2+ bridges appear to form preferentially in the closed state and to stabilize the channel in this state. Thus, if channels are stabilized in the open state, either using PPi to manipulate NBD-driven gating (Fig. 4) or using E1371Q channels that cannot hydrolyze ATP (Fig. 5), the inhibitory effects of Cd2+ are greatly reduced, suggesting that Cd2+ binds more strongly to the closed state than to the open state. Most striking in this respect was the M348C/S1141C mutant that appears to bind Cd2+ ∼2500 times more strongly in channels that are gating normally as compared with constitutively open E1371Q channels (Fig. 5). The unusually high apparent Cd2+ binding affinity of this double cysteine mutant (Figs. 3B and 4B), more than 700-fold lower Ki than the corresponding single cysteine mutants (Figs. 2E and 3), suggests that M348C and S1141C are uniquely well positioned to coordinate tight Cd2+ binding in closed channels. However, when the channel is open, these two cysteines do not appear to coordinate Cd2+ at all because the apparent Cd2+ affinity in M348C/S1141C/E1371Q channels appears the same as in M348C/E1371Q or S1141C/E1371Q (Fig. 5, D and E).
In contrast, a single identified Cd2+ bridge between TMs 1 and 12 (K95C/S1141C) appears to stabilize the channel open state, leading to a significant increase in channel current (Fig. 6). Because this effect was abolished in E1371Q channels (Fig. 6), it appears to reflect a Cd2+ bridge-induced change in channel gating rather than channel conductance. No evidence for Cd2+ bridge formation was observed in the limited number of TM1:TM6 double cysteine mutants that could be studied.
If we assume that the effects of Cd2+ bridges on the stability of the channel open and closed states reflects changes in the relative positions of cysteine side chains introduced into different TMs in these states, then our results would suggest that a number of side chains in TMs 6 and 12 are in close proximity in closed channels, and K95C (TM1) and S1141C (TM12) are in close proximity in open channels. Loss of Cd2+ coordination in these TM6:TM12 mutants in the open state would then reflect reduced physical proximity of these specific pairs of cysteine side chains concomitant with channel opening. What kinds of physical rearrangements of the TMs during channel gating might result in these changes in side chain relative positions?
Changes in accessibility of different side chains in TMs 6 and 12 from the cytoplasm during channel opening and closing have been interpreted as reflecting rotational movements of these TMs within the inner vestibule (11, 12). Such rotational movements could also lead to state-dependent changes in the relative positions of different side chains such as those we infer from our results. However, helical rotation alone does not appear to be able to explain the effects of Cd2+ bridges on channel gating. If the only movement of TMs 6 and 12 during channel opening and closing was a relative rotation, then we would expect some Cd2+ bridges to stabilize the closed state, but others (following rotation of the helices) to stabilize the open state. In fact, we found that Cd2+ bridges only stabilize the closed state and that these bridges appear incapable of forming in the open state. A similar argument suggests that relative translational movement of TMs 6 and 12 does not underlie channel opening and closing; if this were the case, some Cd2+ bridges should again be expected to stabilize the open state. Instead, we propose that the most likely physical explanation for this state-dependent Cd2+ bridge formation is that one face of each of TMs 6 and 12 is physically close together when the channel is closed and that these two TMs then move apart when the channel is open, such that the distance between the two TMs in the open state is too great for Cd2+ bridges to form between any pairs of cysteine residues exposed at this level of the pore (Fig. 8, D and E). This does not imply that these helices do not rotate and/or translate during channel opening and closing, only that the rotation and/or translation (at this level) appears coincidental to opening and closing, with physical separation and convergence of these two TMs being the mechanistically necessary conformational change that results in opening and closing. Thus, forcing TMs 6 and 12 to remain in close physical proximity (by formation of Cd2+ bridges between the two) stabilizes the channel closed state (Fig. 8E). Furthermore, this stabilization of the closed state appears relatively independent of the exact location of the Cd2+ bridge (Fig. 8, A–C). As described above, several different Cd2+ bridges can form between M348C (TM6) and TM12 (Fig. 8, A and B), as well as between S1141C (TM12) and TM6; all appear to stabilize the closed state (Figs. 3 and 4). Thus, for the reasons described earlier, there may be some degree of rotational and/or translational flexibility between TMs 6 and 12 in closed channels. On the other hand, maintaining the channel in the open state prevents Cd2+ bridge formation between cysteines in TMs 6 and 12 (Figs. 4 and 5), suggesting that these two TMs have separated during channel opening (Fig. 8E).
In contrast to this consistent pattern of Cd2+ bridge formation between TMs 6 and 12 stabilizing the closed state, a single Cd2+ bridge between K95C (TM1) and S1141C (TM12) appears to stabilize the open state (Fig. 6). Because only a single Cd2+ bridge between these TMs was identified functionally, it is difficult to draw any conclusion regarding conformational rearrangements between these TMs during channel gating. Thus, relative rotation or translational movement of these two TMs could bring these two cysteines into close proximity in the open state and separate them when the channel closes. Alternatively, it has been suggested that Lys-95 is not even exposed to the pore lumen when the channel is closed (14), although this suggestion is at odds with functional evidence that the native positive charge at this position can interact with cytoplasmically applied pore-blocking ions not only in the open state but also in the closed state (32). Finally, this state-dependent Cd2+ bridge formation could result from the same kinds of change in relative proximity of TMs 1 and 12 as we propose for TMs 6 and 12 (Fig. 8E). This kind of model would suggest that TMs 1 and 12 are closer together in open channels and move further apart when the channel closes (Fig. 8E). No evidence was obtained for Cd2+ bridge formation between TMs 1 and 6, and so we cannot draw any inference on proximity of these TMs in either the open or the closed state. Functional evidence suggests that Lys-95, Ile-344, Val-345, and Ser-1141 are all close together in the inner vestibule of the pore (17, 18, 32).
Our results suggest that different TMs converge and separate during channel gating, implying that ATP-dependent channel opening and closing may involve lateral movements of different TMs surrounding the inner vestibule (Fig. 8E). Furthermore, the physical requirement for these movements in order for channel opening and closing to proceed is underscored by the fact that constraining these movements (by the formation of Cd2+ bridges between different TMs) is capable of either stabilizing the closed state (preventing channel opening) or stabilizing the open state (preventing channel closing). Thus, although the conformational changes of the MSDs that underlie channel opening and closing are ultimately controlled by ATP interactions with the NBDs, these results offer encouragement that altering interactions within the MSDs themselves can, in theory, directly influence the overall activity of the CFTR channel. A substance that could influence TM:TM interactions in this way to stabilize the channel open state could theoretically represent a novel mechanism to increase CFTR function in cystic fibrosis patients.
Acknowledgment
We thank Christina Irving for technical assistance.
This work was supported by the Canadian Institutes of Health Research.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ABC
- ATP-binding cassette
- MSD
- membrane-spanning domain
- NBD
- nucleotide-binding domain
- TM
- transmembrane α-helix.
REFERENCES
- 1. Lubamba B., Dhooghe B., Noel S., Leal T. (2012) Cystic fibrosis: insight into CFTR pathophysiology and pharmacotherapy. Clin. Biochem. 45, 1132–1144 [DOI] [PubMed] [Google Scholar]
- 2. Hwang T.-C., Kirk K. L. (2013) The CFTR ion channel: gating, regulation, and anion permeation. Cold Spring Harb. Perspect. Med. 3, a009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Linsdell P. (2014) Functional architecture of the CFTR chloride channel. Mol. Membr. Biol. 31, 1–16 [DOI] [PubMed] [Google Scholar]
- 4. Mornon J.-P., Lehn P., Callebaut I. (2008) Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell. Mol. Life Sci. 65, 2594–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mornon J.-P., Lehn P., Callebaut I. (2009) Molecular models of the open and closed states of the whole human CFTR protein. Cell. Mol. Life Sci. 66, 3469–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dalton J., Kalid O., Schushan M., Ben-Tal N., Villà-Freixa J. (2012) New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J. Chem. Inf. Model. 52, 1842–1853 [DOI] [PubMed] [Google Scholar]
- 7. Wang W., Linsdell P. (2012) Relative movements of transmembrane regions at the outer mouth of the cystic fibrosis transmembrane conductance regulator channel pore during channel gating. J. Biol. Chem. 287, 32136–32146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang W., Linsdell P. (2012) Conformational change opening the CFTR chloride channel pore coupled to ATP-dependent channel gating. Biochim. Biophys. Acta 1818, 851–860 [DOI] [PubMed] [Google Scholar]
- 9. Wang W., Linsdell P. (2012) Alternating access to the transmembrane domain of the ATP-binding cassette protein cystic fibrosis transmembrane conductance regulator (ABCC7). J. Biol. Chem. 287, 10156–10165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang W., El Hiani Y., Rubaiy H. N., Linsdell P. (2014) Relative contribution of different transmembrane segments to the CFTR chloride channel pore. Pflügers Arch. 466, 477–490 [DOI] [PubMed] [Google Scholar]
- 11. Bai Y., Li M., Hwang T.-C. (2010) Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J. Gen. Physiol. 136, 293–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bai Y., Li M., Hwang T.-C. (2011) Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7). J. Gen. Physiol. 138, 495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang W., El Hiani Y., Linsdell P. (2011) Alignment of transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Gen. Physiol. 138, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao X., Bai Y., Hwang T.-C. (2013) Cysteine scanning of CFTR's first transmembrane segment reveals its plausible roles in gating and permeation. Biophys. J. 104, 786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. El Hiani Y., Linsdell P. (2010) Changes in accessibility of cytoplasmic substances to the pore associated with activation of the cystic fibrosis transmembrane conductance regulator chloride channel. J. Biol. Chem. 285, 32126–32140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qian F., El Hiani Y., Linsdell P. (2011) Functional arrangement of the 12th transmembrane region in the CFTR chloride channel pore based on functional investigation of a cysteine-less CFTR variant. Pflügers Arch. 462, 559–571 [DOI] [PubMed] [Google Scholar]
- 17. Zhou J.-J., Li M.-S., Qi. J., Linsdell P. (2010) Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J. Gen. Physiol. 135, 229–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El Hiani Y., Linsdell P. (2012) Tuning of CFTR chloride channel function by location of positive charges within the pore. Biophys. J. 103, 1719–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Webster S. M., Del Camino D., Dekker J. P., Yellen G. (2004) Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature 428, 864–868 [DOI] [PubMed] [Google Scholar]
- 20. Campos F. V., Chanda B., Roux B., Bezanilla F. (2007) Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc. Natl. Acad. Sci. U.S.A. 104, 7904–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li M., Kawate T., Silberberg S. D., Swartz K. J. (2010) Pore-opening mechanism in trimeric P2X receptor channels. Nat. Commun. 1, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henrion U., Renhorn J., Börjesson S. I., Nelson E. M., Schwaiger C. S., Bjelkmar P., Wallner B., Lindahl E., Elinder F. (2012) Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc. Natl. Acad. Sci. U.S.A. 109, 8552–8557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mense M., Vergani P., White D. M., Altberg G., Nairn A. C., Gadsby D. C. (2006) In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 25, 4728–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M.-S., Demsey A. F. A., Qi J., Linsdell P. (2009) Cysteine-independent inhibition of the CFTR chloride channel by the cysteine-reactive reagent sodium (2-sulphonatoethyl) methanethiosulphonate (MTSES). Br. J. Pharmacol. 157, 1065–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holstead R. G., Li M.-S., Linsdell P. (2011) Functional differences in pore properties between wild-type and cysteine-less forms of the CFTR chloride channel. J. Membr. Biol. 243, 15–23 [DOI] [PubMed] [Google Scholar]
- 26. Beck E. J., Yang Y., Yaemsiri S., Raghuram V. (2008) Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J. Biol. Chem. 283, 4957–4966 [DOI] [PubMed] [Google Scholar]
- 27. Gunderson K. L., Kopito R. R. (1994) Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane conductance regulator channel gating. J. Biol. Chem. 269, 19349–19353 [PubMed] [Google Scholar]
- 28. Carson M. R., Winter M. C., Travis S. M., Welsh M. J. (1995) Pyrophosphate stimulates wild-type and mutant cystic fibrosis transmembrane conductance regulator Cl− channels. J. Biol. Chem. 270, 20466–20472 [DOI] [PubMed] [Google Scholar]
- 29. Choi L.-S., Mach T., Bayley H. (2013) Rates and stoichiometries of metal ion probes of cysteine residues within ion channels. Biophys. J. 105, 356–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ge N., Muise C. N., Gong X., Linsdell P. (2004) Direct comparison of the functional roles played by different membrane spanning regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 279, 55283–55289 [DOI] [PubMed] [Google Scholar]
- 31. Rulísek L., Vondrásek J. (1998) Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J. Inorg. Biochem. 71, 115–127 [DOI] [PubMed] [Google Scholar]
- 32. Linsdell P. (2014) State-dependent blocker interactions with the CFTR chloride channel: implications for gating the pore. Pflügers Arch. 10.1007/s00424-014-1501-7 [DOI] [PubMed] [Google Scholar]
- 33. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]