

Background: Flavodiiron proteins (FDPs) are O2 and/or NO reductases.
Results: Point mutations, near the active site of an Entamoeba histolytica O2-selective FDP, result in increased NO reductase activity and faster inactivation in the reaction with O2.
Conclusion: Residues close to the FDPs diiron site modulate the preference toward O2 or NO.
Significance: We unravel molecular determinants of substrate specificity in enzymes affording resistance to oxygen and/or nitric oxide.
Keywords: Flavoprotein, Iron, Nitric Oxide, Nitrosative Stress, Oxidation-Reduction (Redox), Oxidative Stress, Oxygen
Abstract
Flavodiiron proteins (FDPs) are a family of enzymes endowed with bona fide oxygen- and/or nitric-oxide reductase activity, although their substrate specificity determinants remain elusive. After a comprehensive comparison of available three-dimensional structures, particularly of FDPs with a clear preference toward either O2 or NO, two main differences were identified near the diiron active site, which led to the construction of site-directed mutants of Tyr271 and Lys53 in the oxygen reducing Entamoeba histolytica EhFdp1. The biochemical and biophysical properties of these mutants were studied by UV-visible and electron paramagnetic resonance (EPR) spectroscopies coupled to potentiometry. Their reactivity with O2 and NO was analyzed by stopped-flow absorption spectroscopy and amperometric methods. These mutations, whereas keeping the overall properties of the redox cofactors, resulted in increased NO reductase activity and faster inactivation of the enzyme in the reaction with O2, pointing to a role of the mutated residues in substrate selectivity.
Introduction
Flavodiiron proteins (FDPs)4 form a protein family with a relevant role in the response to oxidative and nitrosative stress in a wide range of organisms (1–9), affording protection against oxygen and/or nitric oxide (NO) by reducing these species to water or nitrous oxide (N2O), respectively. In pathogens, FDPs were suggested to enable survival in the hostile environment encountered during host infection, contributing to the detoxification of O2, particularly in the gut (10, 11), and of NO produced by the host immune response (12–16). Although most known FDPs are encoded in the genomes of prokaryotes, several examples are found in multicellular and unicellular eukaryotes (6, 10, 17–21). Among the latter, the enzymes from the anaerobic protozoan pathogens Giardia intestinalis (10), Trichomonas vaginalis (20), and Entamoeba histolytica (Fdp1) (11) have been investigated.
All FDPs share a common structural unit composed by two domains (the flavodiiron core, herein designated FDP-D), which constitutes the signature of these enzymes and defines the simplest class of FDPs (class A according to Ref. 1): an N-terminal metallo-β-lactamase-like domain harboring the catalytic diiron site, followed by a short chain flavodoxin-like domain containing a flavin mononucleotide (FMN) (2, 4, 10, 22–24). The three-dimensional structures revealed also that a “head to tail” dimeric arrangement is a pre-requisite for enzymatic function, allowing a close proximity between the FMN and the diiron center from opposing monomers, whereas within each monomer the minimal distance between the two cofactors is about 30 Å, precluding efficient electron transfer.
The iron ligands are highly conserved (2, 4), except in the enzymes from oxygenic photosynthetic organisms, which have highly variable putative metal ligands (6, 17, 21), and will not be further considered here. In “canonical” FDPs the ligands are arranged in a characteristic motif (H-X-E-X-D-H-X(61)-H-X(18)-D-X(60)-H) with the exception of the Desulfovibrio gigas enzyme (23), where the His adjacent to the Asp ligand is unbound, being replaced by a H2O molecule; the mutation of this His residue to Asn or Ala resulted in no changes in the catalytic properties of the Thermotoga maritima FDP (25). Rubredoxins have been identified as the physiological redox partners for FDPs in a few organisms (reviewed in Ref. 4). However, no rubredoxin-encoding genes have been identified in the genomes of E. histolytica or other protozoa encoding FDPs (26–28) and therefore their physiological partners remain unknown.
FDPs may display distinct substrate specificity: whereas some enzymes are selective for either NO or O2, other FDPs are able to reduce both substrates with similar catalytic efficiencies (e.g. the FDPs from Moorella thermoacetica (8) and Desulfovibrio species (9, 29)). The FDP from Escherichia coli, named flavorubredoxin (FlRd or NorV) (7, 30), is selective toward NO (30, 31). FlRd shares a high amino acid sequence identity with the NO-selective FDP from another enterobacterium, Salmonella typhimurium (32), and indeed this type of FDPs (classified as class B (1)) is highly conserved in enterobacteria. On the other end of the substrate specificity spectrum lie the FDPs from G. intestinalis (10) and E. histolytica (11) that have a marked substrate preference toward O2, together with the FDPs from T. maritima (25) and the methanogenic Archaeon Methanothermobacter marburgensis (22).
The molecular determinants for substrate specificity remain elusive. By comparing the structures of two well characterized enzymes (the O2-selective FDP from G. intestinalis and the NO-reducing enzyme from E. coli), we identified two residues in close spatial proximity to the diiron site that differ between the two enzymes: a tyrosine and a lysine in the protozoan FDP replaced by serine and aspartate, respectively, in the bacterial enzyme. This analysis was extended also to other FDPs with clear substrate selectivity, by comparing their crystallographic structures, or through the construction of three-dimensional models and amino acid structural alignments. We subsequently constructed site-directed mutants of the O2-selective E. histolytica Fdp1, by replacing those residues (E. histolytica Tyr271 and Lys53) by residues found in the NO-reducing enzymes from enterobacteria. Herein, these EhFdp1 variants (two single mutants and a double mutant) were thoroughly characterized in terms of their spectroscopic and kinetic properties. The mutated residues proved to modulate the reactivity with O2 and enhance the reactivity toward NO, supporting the working hypothesis that the residues in these positions contribute to substrate selectivity in FDPs.
EXPERIMENTAL PROCEDURES
Structure Prediction of the E. histolytica EhFdp1 and Amino Acid Sequence Comparisons
The three-dimensional structure of E. histolytica HM-1:IMSS EhFdp1 (XP_656946) was predicted using I-TASSER (33, 34) with the G. intestinalis FDP (PDB code 2Q9U, chain A) as template. The EhFdp1 structural model was subsequently compared with the structures of the FDPs from G. intestinalis (PDB code 2Q9U), E. coli (PDB code 4D02), M. thermoacetica (PDB code 1YCF), D. gigas (PDB code 1E5D), T. maritima (PDB code 1VME), and M. marburgensis (PDB code 2OHI). Additionally, CLUSTALX (35) was used to align the EhFdp1 amino acid sequence against the FDPs sequence alignment profile, obtained from the superimposition of the above mentioned set of FDPs crystallographic structures using Modeler (35, 36).
Protein Expression and Purification
Site-directed mutants of the EhFdp1 were generated at GenScript. Direct sequencing confirmed that only the desired mutations were inserted. Expression in E. coli BL21(DE3) Gold cells and purification of wild-type (WT) and mutated EhFdp1 were carried out as previously described (11). Because the physiological electron donor to the E. histolytica EhFdp1 is presently unknown, we used an artificial reducing system from E. coli: the flavorubredoxin reductase (FlRd-Red) and the rubredoxin domain (Rd-D) of FlRd, that were expressed and purified as described in Ref. 37.
Cofactor Content Determination
The protein concentration was determined by the bicinchoninic acid (38). The flavin content was measured by visible absorption spectroscopy after extraction with trichloroacetic acid and neutralization with ammonium acetate as in (39). Iron was quantitated by the 2,4,6-tripydridyl 1,3,5-triazine (40) and the ferrozine (41) spectrophotometric assays, and by inductively coupled plasma-atomic emission spectroscopy at the Requimte Laboratório de Análises, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa. Results from the three iron quantitation methods were consistent and averaged. The enzyme activities were normalized taking into consideration the iron content of each protein variant.
Oligomeric State Determination
The oligomeric state of the proteins was determined by size exclusion chromatography. The proteins (∼2 mg/ml) were loaded onto a Superdex 200 10/300 GL column (GE Healthcare), previously equilibrated with and eluted with 20 mm Tris-HCl, 150 mm NaCl, 18% (v/v) glycerol, pH 7.5.
EPR and Redox Titrations of the Diiron Center and FMN
The diiron center of the FDPs and the corresponding reduction potentials were investigated by EPR spectroscopy, using a Bruker EMX spectrometer equipped with an Oxford Instruments ESR-900 continuous flow helium cryostat. The FMN reduction potentials were determined by redox titrations monitored by visible absorption spectroscopy, in a Shimadzu UV-1603 spectrophotometer. The proteins (about 50 μm, for the diiron center titrations and 30 μm for the FMN titrations) were anaerobically titrated, under a continuous flush of argon, in 50 mm Tris-HCl, 18% (v/v) glycerol, pH 7.5. Glucose oxidase (280 nm, from Aspergillus niger), catalase (640 nm, from bovine liver), and glucose (1 mm) were added to the reaction mixture to scavenge contaminating oxygen. Redox mediators were used at concentrations of 80 or 0.5 μm for the diiron center or the FMN redox titrations, respectively, as in Ref. 37, and control redox titrations in the absence of the protein were performed to check for possible spectral interferences. The electrodes were calibrated with a quinhydrone-saturated solution at pH 7.0. Redox potentials were normalized against the potential of the standard hydrogen electrode. EPR data were fitted to a Nernst equation for two consecutive one-electron processes. Flavin reduction potentials were determined by singular value decomposition (SVD) analysis, as described below.
Stopped-flow Absorption Spectroscopy
Stopped-flow experiments were carried out in thermostated instrument (DX.17MV, Applied Photophysics, Leatherhead, UK), equipped with a diode-array detector (light path, 1 cm). Reaction buffer was 50 mm Tris-HCl, 18% (v/v) glycerol, pH 7.5. All concentrations reported below are before mixing in the stopped-flow apparatus. The reaction of reduced FDPs with O2 or NO was investigated as follows. Proteins were diluted to a final concentration of about 10 μm, flushed with nitrogen, pre-reduced by addition of NADH (300 μm), E. coli FlRd-Red (0.3 μm), and Rd-D (0.4 μm) in the presence of catalase (640 nm, to scavenge H2O2 possibly produced by the reaction of NADH/FlRd-Red/Rd-D with residual O2 (42, 43), and placed on ice, protected from light to prevent flavin damaging photoreactions. Reduced proteins were then mixed at 5 °C in the stopped-flow apparatus against either O2 (air-equilibrated buffer at 25 °C, ∼240 μm) or NO (200 μm). NO solutions were prepared as in Ref. 44. Time-resolved absorption spectra were recorded with an acquisition time of 1.0 ms/spectrum according to a logarithmic time scale. When necessary, to ensure anaerobic conditions, the reaction buffer was degassed with nitrogen and vacuum cycles, and residual O2 scavenged with glucose oxidase (280 nm), catalase (640 nm), and glucose (1 mm).
To estimate the maximum velocity of O2 consumption (Vmax,O2) by EhFdp1, Rd-D at varying concentrations (from 22 to 342 μm) was pre-reduced with 400 μm NADH, 20 nm FlRd-Red (in the presence of 1.3 μm catalase), and mixed at 25 °C with each EhFdp1 variant (100 nm in FMN) in air-equilibrated buffer. Following rapid mixing, Rd-D reoxidation was monitored at 484 nm. The initial reaction rate calculated from the absorption changes detected soon after mixing (< 100 ms) was normalized to both the extinction coefficient of Rd-D and the concentration of EhFdp1, and divided by 4 (number of electrons required for reduction of O2 to 2H2O) to attain VO2. Appropriate controls of Rd-D direct oxidation by oxygen were performed in the absence of EhFdp1.
Amperometric Measurements of O2 and NO Reductase Activities
The O2 and NO reductase activities of wild-type and mutated EhFdp1 were measured amperometrically with Clark-type electrodes selective for O2 (Oxygraph-2K, Oroboros Instruments, Innsbruck, Austria) or NO (ISO-NOP, World Precision Instruments, Sarasota, FL). The assays were performed in 50 mm Tris-HCl, 18% (v/v) glycerol, pH 7.5. The O2 reductase activity was evaluated at 25 °C in O2-equilibrated buffer (∼1.25 mm), in the presence of NADH (5 mm), E. coli FlRd-Red (0.5 μm), and Rd-D (2.5 μm) acting as electron carriers for EhFdp1. The reaction was initiated by addition of EhFdp1 (50 to 120 nm). Assays were performed in the presence of catalase (640 nm) and superoxide dismutase (240 nm, from bovine erythrocytes). The NO reductase activity was determined under anaerobic conditions in the presence of Rd-D (2 μm), FlRd-Red (1 μm), EDTA (20 μm), and the O2 scavenging system (glucose, glucose oxidase, and catalase). Sequential additions of NO (2 μm to 4 μm) were followed by addition of 1 mm NADH, and the reaction was initiated by addition of EhFdp1 (50 to 120 nm). Activities were normalized to the amount of the active enzyme, i.e. taking into account the iron content.
Data Analysis
Optical data (redox titrations and kinetic) were analyzed with MATLAB software (Mathworks, South Natick, MA). Spectral deconvolution was achieved by SVD analysis combined with curve fitting, as in Ref. 45, or by using the “left matrix division” operator implemented in MATLAB.
RESULTS AND DISCUSSION
Structural Comparison of the Diiron Active Site
Fig. 1A shows an overlay of the structures of the O2-reducing G. intestinalis and NO-reducing E. coli enzymes. At the level of the catalytic center, the two enzymes are very similar: the ligands are strictly conserved, each iron being ligated by the side chains of histidines and aspartates/glutamates. Besides the bridging μ-(hydr)oxo species and the GiD171 carboxylate, Fep (the Fe atom proximal to the FMN of the other monomer) is bound to GiH85, GiE87, and GiH152, whereas Fed (distal to FMN) is bound to GiD89, GiH90, and GiH230 (G. intestinalis FDP numbering). Two striking differences between the two enzymes can be identified: a lysine (G. intestinalis Lys58) replaced by an aspartate (E. coli Asp52) in the E. coli enzyme and a tyrosine (G. intestinalis Tyr267) replaced by a serine (E.coli Ser262) in the E. coli FlRd (Fig. 1A). These tyrosine or serine belong to the other monomer of the head-to-tail dimer.
FIGURE 1.
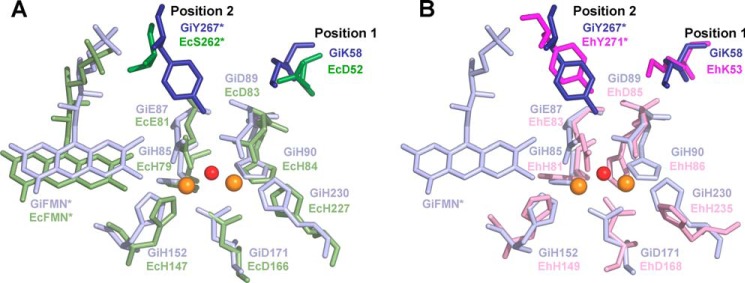
Structural comparison of the diiron sites from FDPs with different substrate selectivity. Structural superimposition of the diiron center regions of the O2-reducing G. intestinalis FDP (PDB code 2Q9U, blue) and panel A, the NO-selective E. coli FDP-D (PDB code 4D02, green), or panel B, the model of E. histolytica EhFdp1 (pink). Only the diiron center and the FMN (from the other monomer) of G. intestinalis FDP are shown. The iron ions and the μ(hydr)oxo-bridging oxygen atom are shown as orange and red spheres, respectively. Positions 1 and 2 correspond to residues mutated in E. histolytica EhFdp1. *, denotes the residues from the other monomer.
A structural alignment of the FDPs with known crystal structures shows that most of the enzymes that have been demonstrated to be exclusively oxygen reductases (namely, the FDPs from G. intestinalis and T. maritima), display these differences as compared with the E. coli NO-reducing enzyme (Fig. 2). In contrast, the FDPs that are not selective for either substrate exhibit different residues combinations at these positions: M. thermoacetica FDP has M. thermoacetica Tyr54 and Trp263, whereas D. gigas FDP has Lys52 and Trp263 (Fig. 2). The enzyme from M. marburgensis, an O2 selective FDP, may appear an exception, which instead of the tyrosine has a histidine (His267) in the respective sequence position; however, analysis of its three-dimensional structure reveals that M. marburgensis FDP Tyr25 may have the same role as G. intestinalis Tyr267 (22). Interestingly, in the other enzymes compared, this residue is a phenylalanine.
FIGURE 2.

Amino acid sequence alignment based on structural superposition of FDPs. The EhFdp1 amino acid sequence was aligned against the amino acid structural alignment built on the basis of the structures of the FDPs from G. intestinalis (PDB code 2Q9U, chain A), T. maritima (PDB code 1VME, chain A), M. marburgensis (PDB code 2OHI, chain A), E. coli FDP-D (PDB code 4D02), M. thermoacetica (PDB code 1YCF, chain C), and D. gigas (PDB code 1E5D, chain A). Gray shades indicate conserved residues; black shades correspond to diiron ligands; boxes indicate the residues in positions 1 and 2. The selectivity for O2 or NO is also indicated.
On the basis of this comparison, we decided to undertake a site-directed mutagenesis study of the E. histolytica EhFdp1. As the three-dimensional structure of this enzyme is not yet available, we generated a structural model employing I-TASSER (33, 34), using the structure of G. intestinalis FDP as a template (10). The obtained structural model also has the tyrosine and lysine found in the G. intestinalis enzyme in the same spatial location (Figs. 1B and 2). Therefore, EhFdp1 was used as a working model to mutate the residues at these positions, generating both single mutants (K53D and Y271S) and the K53D/Y271S double mutant.
Biochemical Properties of WT and Mutated EhFdp1
The EhFdp1 variants were successfully overexpressed in E. coli and purified to homogeneity. The WT enzyme and mutants were mainly isolated in their functional dimeric form, except for a minor fraction of the K53D/Y271S mutant in the monomeric state. The WT EhFdp1 and single mutants contained ∼2 Fe and 1 FMN per monomer; in contrast, the K53D/Y271S mutant showed approximately half of the expected cofactor load, although EPR data ruled out partially occupied diiron sites (see below).
The Flavin Moiety
As one of the mutated residues (Tyr271) is located in the vicinity of the flavin cofactor, possible effects of the mutations on the FMN moiety were assayed by UV-visible absorption spectroscopy and potentiometry. Like the WT protein (11), all the EhFdp1 variants were isolated in the oxidized state with UV-visible absorption bands with maxima at 377 and 456 nm, characteristic of the flavin moiety. The FMN reduction potentials were determined by anaerobic redox titrations (Fig. 3A). The spectrum of the fully reduced enzyme was subtracted from those obtained during the titration (inset to Fig. 3A); the data illustrate the appearance and subsequent disappearance of an optical species centered at 390 nm, characteristic of the anionic (red) semiquinone. The weak absorption band centered at ∼600 nm results from the used redox mediators, as deduced by performing control titrations in the absence of the protein. Consistently, these optical features displayed a redox potential dependence totally unrelated to that of the flavin moiety (not shown), and were thus excluded from the analysis. SVD analysis of the absorption changes measured in the 350–550 nm range yielded two significant components. The first two columns of the V matrix of the SVD output, scaled by their corresponding singular values, were fitted to a Nernst equation for two sequential one-electron steps (inset to Fig. 3B). According to Henry and Hofrichter (45), this allowed us to determine not only the reduction potentials for the transition of oxidized FMN to the semiquinone state and for its full reduction to hydroquinone, but also the absorption spectrum of FMN in the three states (Fig. 3B).
FIGURE 3.

Redox titration of FMN in WT EhFdp1. WT EhFdp1 (30 μm) in 50 mm Tris-HCl, 18% glycerol, pH 7.5, was anaerobically redox titrated, by stepwise addition of sodium dithionite, at 25 °C. Visible absorption spectra recorded along the titration were analyzed by SVD. Panel A, absolute spectra recorded in the course of the titration; inset, difference spectra obtained by subtracting the fully reduced spectrum to all other spectra. Panel B, optical components obtained by SVD analysis combined with curve fitting: EhFdp1 with oxidized (solid line), semiquinone (dashed line), and fully reduced (dotted line) FMN; inset, best fit of the first two columns of the V matrix obtained by SVD analysis, scaled by their corresponding singular values; data were fit using a Nernst equation for two sequential monoelectronic reduction steps, yielding E1 = −55 mV and E2 = −140 mV.
The same procedure was employed to follow the optical changes and determine the FMN reduction potentials in the EhFdp1 mutants. Results led us to conclude that the mutations have essentially no effect on the redox properties of the flavin moiety (see Table 1), or on its optical features (not shown). Accordingly, a similar amount of semiquinone proved to form in each protein variant (0.60 to 0.77 of total FMN), as estimated from the fitted reduction potentials.
TABLE 1.
Redox, spectroscopic, and functional properties of the EhFdp1 variants
| EvarianthFdp1 | E°(FMNox → FMNsq FMNsq → FMNred) | EPR g values | E°, (Fe3+Fe3+ → Fe3+Fe2+ Fe3+Fe2+ → Fe2+Fe2+) | kobs, NO reaction | NO Reductase activity | Vmax O2 |
|---|---|---|---|---|---|---|
| mV | mV | s−1 | μm NO s−1 μm protein | s−1 | ||
| WT | −55–140 | 1.95, 1.77 | + 170 + 132 | 0.12 ± 0.02 | 1.7 ± 0.4 | 400 ± 30 |
| K53D | −58–114 | 1.94, 1.78 | + 160 + 45 | 0.20 ± 0.01 | 3.4 ± 1.0 | 667 ± 29 |
| Y271S | −33–130 | 1.89, 1.86, 1.75 | + 165 + 59 | 0.94 ± 0.03 | 3.1 ± 0.9 | 1020 ± 12 |
| K53D/Y271S | −43–127 | 1.87, 1.75 | + 112 + 4 | 2.1 ± 0.1 | 6.0 ± 1.0 | 627 ± 41 |
The Diiron Center
The presence of the diiron center in the EhFdp1 variants was verified by performing anaerobic redox titrations monitored by EPR spectroscopy (Fig. 4). EPR data ruled out the presence of partially occupied iron sites, because in the oxidized protein resonances attributable to mononuclear high-spin ferric species (g ∼ 4.3) were not observed (data not shown). This indicates that even in the K53D/Y271S mutant, containing only 1 Fe/monomer, diiron centers with half-occupancy are not present. Consequently, for this variant, there are only two possible populations of each monomer: one with the fully loaded catalytic center and another population depleted of iron, thus inactive. The WT EhFdp1 exhibits in the mixed valence (Fe3+Fe2+) state an EPR spectrum similar to that observed for other FDPs, with g values below 2.0 (11, 20, 37, 46). In the course of the titration (Fig. 4A), the spectrum of the mixed valence species appeared and disappeared as the reduction potential decreased. The titration curves obtained by measuring the change in intensity of the resonances at g < 2.0 as a function of the redox potential were adjusted to a Nernst equation for two sequential one-electron steps (Fig. 4B and Table 1). The potentials of the WT EhFdp1 diiron center (E1 = +170 ± 20 mV and E2 = +132 ± 20 mV), particularly the value for the first transition, resemble those reported for the other protozoan FDPs (G. intestinalis FDP: E1 = +163 mV and E2 = +2 mV (46); T. vaginalis FDP: E1 = +190 mV and E2 = +50 mV (20)). Concerning the mutants, the most noticeable variation is the decrease in the second reduction potential (E2) for the double mutant. Although this could be consistent with an increase in the NO reductase activity (see below), because the standard reduction potential for the reduction of NO to N2O is +1,175 mV, it is unlikely that the observed negative shift will modulate the enzyme reactivity.
FIGURE 4.

EPR spectroscopy and redox titration of the diiron site. WT EhFdp1 (50 μm) in 50 mm Tris-HCl, 18% (v/v) glycerol, pH 7.5, was anaerobically redox titrated, by stepwise addition of sodium dithionite. EPR spectra were recorded at 7 K. Panel A, spectra recorded along the titration, with the corresponding redox potentials. Panel B, titration profile of the WT EhFdp1 mixed valence diiron center; dots represent normalized intensities (heights relative to the maximal values) measured at g = 1.95 and 1.77; full line represents the best adjustment using a Nernst equation for two sequential monoelectronic reduction steps, with E1 = +170 mV and E2 = +132 mV. Panel C, EPR spectra of the mixed valence diiron center in WT and mutated EhFdp1. Microwave frequency: 9.39 GHz; microwave power, 2 milliwatt; modulation amplitude, 1 millitesla.
For all mutants, resonances with g values below 2, characteristic of the diiron center in the mixed valence state, were observed (Fig. 4C). The EPR spectra for WT and K53D EhFdp1 are clearly rhombic, similar to one another and to those of the E. coli, G. intestinalis, and T. vaginalis FDPs (20, 37, 46). Major differences were observed in the shape and g values of the Y271S and K53D/Y271S mutants, which exhibit quasi-axial spectra, with gmax ∼ gmed ≫ gmin. The Y271S and K53D/Y271S proteins also exhibit an extra resonance with low intensity, also observable in partially reduced samples, probably due to an alternative conformation of the diiron center. Based on the observed spectral changes, we conclude that tyrosine substitution leads to a structural rearrangement in the diiron site vicinity.
Effect of Mutations on the Kinetics of EhFdp1 Oxidation by O2 and NO
The ability of the reduced EhFdp1 variants to react with O2 or NO was investigated by stopped-flow absorption spectroscopy at 5 °C. Reaction with each of the two substrates resulted in an absorbance increase, due to flavin oxidation mediated by the diiron centers (Fig. 5, A and B). Along the reaction, one single optical species was detected, corresponding to the difference of the spectrum of the oxidized form minus that of the reduced form (Fig. 5, A and B). Markedly different time courses were observed for the reaction of WT EhFdp1 with O2 or NO (Fig. 5C). The enzyme is indeed highly reactive with O2, with 80% of the protein being promptly oxidized within the first 10 ms and the remaining 20% being oxidized on a longer time scale (up to 10 s). The latter kinetic phase likely arises from a minor subpopulation of the enzyme lacking a functional diiron site. Despite the low temperature (5 °C), the fast phase of the reaction is too fast to be time-resolved even by stopped-flow spectroscopy. Under the tested experimental conditions, most of the protein reacts with O2 (∼120 μm after mixing) at a rate constant k′ > 200 s−1, consistent with a second-order rate constant k > 1 × 106 m−1 s−1. In contrast, NO (100 μm after mixing) reoxidized the enzyme only at ∼0.1 s−1, i.e. 3 orders of magnitude more slowly than O2 (Fig. 5), as reported for the G. intestinalis enzyme (10).
FIGURE 5.

Kinetics of WT EhFdp1 oxidation by O2 or NO. EhFdp1 (about 10 μm in FMN) in 50 mm Tris-HCl, 18% (v/v) glycerol, pH 7.5, was pre-reduced in anaerobic conditions by incubation with 300 μm NADH, 0.3 μm FlRd-Red, and 0.4 μm Rd-D in the presence of 640 nm catalase, and mixed in the stopped-flow apparatus with air-equilibrated buffer (∼240 μm O2) or 200 μm NO. T = 5 °C. Panel A, time-resolved spectra collected after mixing reduced EhFdp1 with air-equilibrated buffer. Thick solid line, oxidized EhFdp1; thick dotted line, reduced EhFdp1; thin lines, selected spectra collected at (bottom to top) 1 ms, 2 ms, 3ms, 4 ms, 5 ms, 20 ms, 100 ms, 0.5 s, and 1 s. Arrow indicates the direction of absorption changes. Panel B, spectra acquired during the reaction of reduced EhFdp1 with 200 μm NO. Thick solid line, oxidized EhFdp1; thick dotted line, reduced EhFdp1; thin lines, selected spectra collected at (bottom to top) 10 ms, 0.75 s, 2 s, 3 s, 4.5 s, 6 s, 8 s, 10 s, 13.7 s, 17.6 s, and 20 s. Arrow indicates the direction of absorption changes. Panel C, time course of the reactions of reduced EhFdp1 with O2 or NO, as obtained by SVD analysis of the data in panels A and B.
Under identical experimental conditions, the EhFdp1 mutants display differences in their reaction with O2 and NO (Fig. 6), among themselves and as compared with the WT protein. Similarly to the WT protein, all mutants exhibit a protein fraction very rapidly (<10 ms) oxidized by O2 and another one oxidized only on a much longer time scale (tens of seconds) (Fig. 6A). However, whereas the K53D mutant behaves quite similarly to the WT, the Y271S mutant shows a greater fraction (about 30% of the total enzyme) slowly oxidized by O2. This behavior is even more pronounced in the K53D/Y271S mutant, where the slowly oxidized fraction amounts to slightly over 50%. Therefore, within the limits of the stopped-flow time resolution, we conclude that the tested mutations, whereas affecting the amount of enzyme kinetically competent in the O2 reduction process, do not seem to significantly affect the initial rate of enzyme oxidation by O2, taking place within a few ms for all variants.
FIGURE 6.
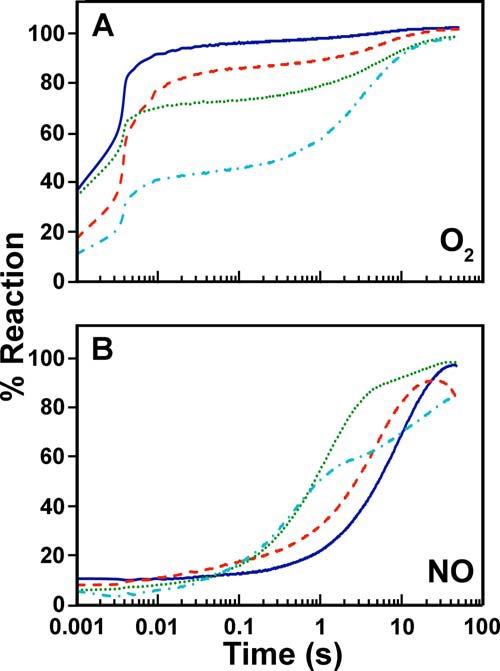
Reaction of reduced WT and mutated EhFdp1 with O2 and NO. Kinetic profile measured for each mutant in the reaction with O2 (panel A) or NO (panel B). Solid blue line, WT; dashed red line, K53D; dotted green line, Y271S; dash-dotted cyan line, K35D/Y271S. Experimental conditions as in the legend to Fig. 5.
Unlike with O2, the reaction of the EhFdp1 variants with NO is suitably time-resolved in stopped-flow experiments and, notably, it displays effects of the mutations fully consistent with the working hypothesis of this study. As shown in Fig. 6B and Table 1, all mutants are indeed oxidized by NO more quickly than the WT protein. The kinetic effect is modest (2-fold higher observed rate constant) in the K53D mutant, but significantly more pronounced (9-fold) in the Y271S, and even greater (20-fold) in the double mutant. Consistent with the results attained with O2, the K53D/Y271S mutant displayed the highest heterogeneity, among the tested variants.
In our stopped-flow experiments, we monitored the time course of iron-iron site-mediated flavin oxidation after the rapid mixing of reduced EhFdp1 with an excess of O2 or NO. Under these experimental conditions, we invariantly observed a single optical species, identical in shape in both kinetic phases and for all protein variants, corresponding to full oxidation of the flavin cofactor. Importantly, the optical intermediates recently reported by Caranto et al. (47) were not detected in our experiments, possibly due to faster electron transfer between the flavin and iron-iron site in E. histolytica EhFdp1 as compared with the T. maritima FDP. Furthermore, in our study no direct information was obtained by EPR or Mössbauer spectroscopy on the reactions taking place at the iron-iron site. This prevents us from correlating the observed time course of flavin oxidation with the chemistry occurring at the iron-iron site, namely with the formation of mono or dinitrosyl diiron sites (see Scheme 1). Nevertheless, our data suggest that the two kinetic phases observed are not related to impaired intramolecular electron transfer or to formation/dissociation of NO species at the catalytic site. Based on these results, a contribution of both mutated residues to substrate selectivity can be envisaged, with a major role of tyrosine 271 at position 2 in Fig. 1.
SCHEME 1.
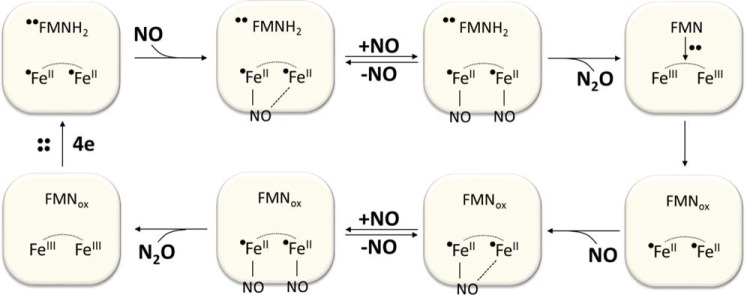
Mechanism for NO reduction at the diiron site as proposed by Caranto et al. (47).
Tyr271 Protects EhFdp1 in Turnover with O2
The effect of the mutations on EhFdp1 in turnover with O2 was investigated by high-resolution respirometry running multiple turnover assays in the presence of an excess of NADH and E. coli FlRd-Red/Rd-D acting as electron carriers. Oxygen consumption measurements were carried out in the presence of catalase and superoxide dismutase to protect the enzyme from reactive oxygen species, particularly H2O2, generated by the reaction of FlRd-Red with O2 (42, 43). Experiments were carried out at 25 °C in O2-equilibrated buffer to allow the enzymes (50 to 120 nm) to perform a high number (>8,000) of catalytic cycles with O2.
As shown in Fig. 7A, all EhFdp1 variants initially display an almost linear consumption profile, after which they are slowly inactivated. Notably, whereas initially displaying a higher consumption rate, the Y271S mutant is inactivated significantly more quickly (i.e. after much less turnover cycles) than the WT and K53D variants, which behave quite similarly to one another in that respect. The double mutant is even more prone to be inactivated in turnover with O2. Control experiments carried out in air-equilibrated buffer (∼240 μm O2) demonstrated that O2 consumption follows a linear time course (zero-order kinetics) down to quite low O2 concentrations (not shown). Altogether, these data suggest that mutation of Tyr271 results not only in a higher O2-reductase reactivity, but more importantly also in a higher propensity of the enzyme to be inactivated in turnover with O2.
FIGURE 7.
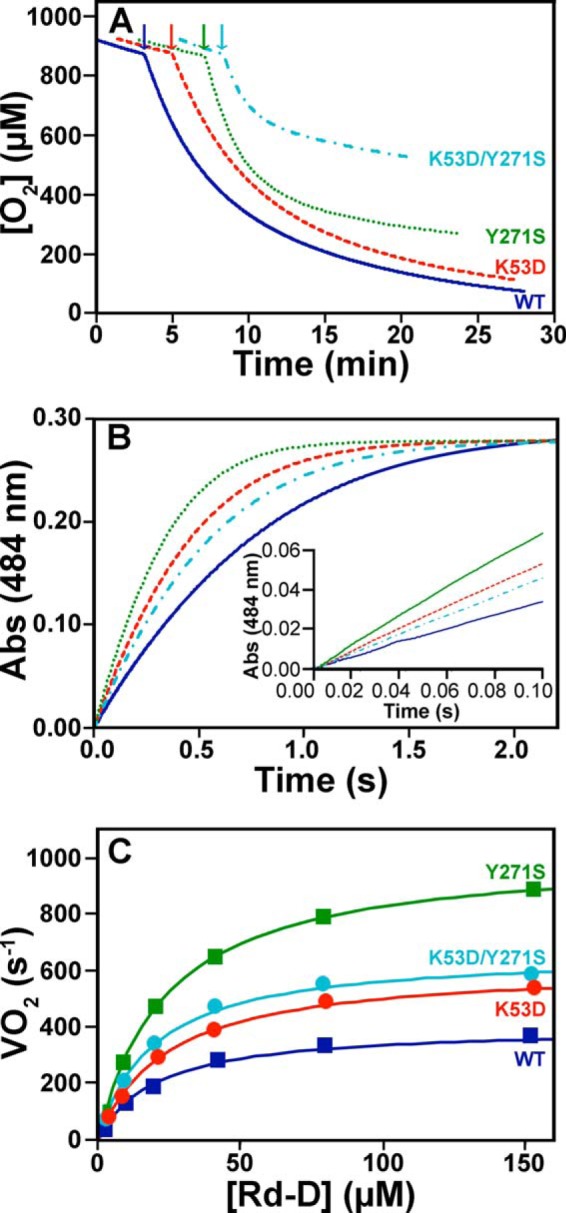
Reactivity of EhFdp1 variants with oxygen under multiple turnover conditions. Panel A, oxygraphic traces collected in O2-equilibrated buffer (50 mm Tris-HCl, 18% (v/v) glycerol, pH 7.5), in the presence of 5 mm NADH, 0.5 μm FlRd-Red, 2.5 μm Rd-D, 640 nm catalase, and 240 nm superoxide dismutase, following the addition of the EhFdp1 variants (50 to 120 nm). Panel B, Rd-D oxidation by EhFdp1 variants, with O2 as the terminal electron acceptor, as followed at 484 nm by stopped-flow absorption spectroscopy. Traces are shown for a single Rd-D concentration (40 μm). Rd-D was anaerobically pre-reduced with 400 μm NADH and 20 nm FlRd-Red in the presence of 1.3 μm catalase, and mixed at 25 °C with 100 nm EhFdp1 in air-equilibrated buffer. Data were fit with single exponential curves: solid blue line, WT; dashed red line, K53D; dotted green line, Y271S; dash-dotted cyan line, K53D/Y271S. Inset, zoom-in on the first 100 ms of the reaction for a better comparison of the initial rate of the reaction, among the tested EhFdp1 variants. Panel C, VO2 for WT and mutated EhFdp1 as measured at increasing concentrations of Rd-D.
To determine the Vmax of O2 consumption by WT and mutated EhFdp1, we carried out the following stopped-flow experiment. Rd-D, acting as the electron donor to EhFdp1, was pre-reduced anaerobically with NADH and catalytic amounts of FlRd-Red (in the presence of catalase), and then mixed at 25 °C with EhFdp1 at low concentration in air-equilibrated buffer. Rd-D oxidation was monitored at 484 nm; due to the relatively high concentrations of Rd-D (in the micromolar range) as compared with EhFdp1 and FlRd-Red (both in the nanamolar range), the latter enzymes did not interfere optically in these assays. In the absence of EhFdp1, direct oxidation of Rd-D by O2 was negligible, occurring only over tens of minutes (not shown). In contrast, EhFdp1 promptly oxidized Rd-D, following monoexponential curves (Fig. 7B). To attempt measuring the Vmax of O2 reduction by EhFdp1, we varied the Rd-D concentration in the experiment and calculated the rate of O2 consumption (VO2) from the initial rate of Rd-D oxidation. Fig. 7C reports the VO2 values measured at increasing Rd-D concentrations. Fitting these data to the Michaelis-Menten equation, we obtained Vmax values in the range of 400–1000 s−1 (Table 1), with the Y271S mutant displaying the highest Vmax among the tested variants. Similar apparent Km values for Rd-D (from 18.8 to 23.5 μm) were obtained for WT and mutated EhFdp1. Based on the high Vmax measured for WT EhFdp1 (400 s−1 for oxygen, corresponding to 1600 s−1 in terms of electrons, as O2 reduction to water requires 4 electrons), EhFdp1 displays a remarkable O2-reductase activity, comparable with that of respiratory heme-copper oxygen reductases (48, 49). It should be recalled that the measured Vmax may still be underestimated as Rd-D is not the physiological redox partner of EhFdp1. Notably, the Y271S mutant appears to be the most reactive variant toward O2, despite its higher propensity to be inactivated under multiple turnover conditions (Fig. 7A). Altogether, these results suggest that Tyr271 stabilizes EhFdp1 in turnover with O2, possibly preventing the formation of reactive intermediates leading to enzyme inactivation. This indicates that, in the reaction with O2, Tyr271 plays a role in ensuring both catalytic efficiency and preservation of protein activity in turnover.
Effect of Mutations on the NO-reductase Activity
We next attempted to employ the same strategies to assay the effects of mutations on the ability of EhFdp1 to metabolize NO. Consistent with stopped-flow measurements, all mutants proved to be more active than the WT protein, with the double mutant showing the highest NO reductase activity (Table 1). However, from a quantitative viewpoint, under turnover conditions the kinetic effect of each mutation (fold-increase relative to WT) was less than measured by stopped-flow spectroscopy for the reaction of the reduced enzyme with NO. As an example, as compared with the WT enzyme, the double mutant, whereas being oxidized by NO ∼20-fold faster, displayed a ∼3.5-fold higher NO reductase activity. This suggested a possible underestimation of the NO-reductase activity in amperometric measurements.
Therefore, we attempted to measure such activity in multiple turnover experiments by stopped-flow spectroscopy, as carried out with oxygen, i.e. testing the Rd-D oxidation mediated by catalytic amounts of EhFdp1, with NO as the terminal electron acceptor. However, while running the appropriate controls, we noticed that reduced Rd-D reacts promptly with NO over a time scale that interferes with the EhFdp1-mediated oxidation of Rd-D. Moreover, we observed that, by reacting with NO, Rd-D markedly loses its ability to act as a fast electron donor for EhFdp1. This prevented us from measuring the NO-reductase activity of WT and mutated EhFdp1 in stopped-flow multiple turnover experiments. However, it should be emphasized that in the amperometric assays both single mutations increased the NO reductase activity of the enzyme and the double mutant displays a cumulative increase in reactivity toward NO, enforcing the idea that both mutated residues contribute to substrate selectivity of FDPs.
Concluding Remarks
Based on the results herein presented, we conclude that the investigated amino acid residues contribute to modulate the substrate preference in flavodiiron proteins. Particularly, Tyr271 has a role in controlling the reactivity of EhFdp1 with O2 and preventing its inactivation in turnover. In respiratory heme-copper oxygen reductases, a tyrosine residue near the active site participates in the oxygen reduction cycle. Notably, this tyrosine residue is missing in respiratory NO reductases, which has been attributed as one of the factors contributing to substrate selectivity in this protein family (50). It appears that Tyr271 in EhFdp1 has a protective role in the reaction with O2, possibly preventing the formation of intermediates that otherwise could lead to enzyme inactivation. This implies that the remarkably high O2 reductase activity of the amoebic FDP results from a trade-off between catalytic efficiency and preservation of enzyme activity, where Tyr271 appears to play a pivotal role.
Acknowledgment
We acknowledge helpful discussions with all members of the involved research groups.
This work was supported by the Portuguese Fundação para a Ciência e Tecnologia project Grants PTDC/QUI-BIQ/111080/2009 and PTDC/SAU-MIC/111447/2009 (to J. B. V. and M. T.), Grants SFRH/BPD/94050/2013 (to C. V. R.), PEST-OE/EQB/LA0004/2013, PEST-OE/SAU/UI4013/2011 from the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (PNR-CNR Aging Program 2012–2014 (to A. G.), Fondo per gli Investimenti della Ricerca di Base Grants RBIN06E9Z8 and PRIN 20107Z8XBW_005 (to P. S.), and grants from the Fundação para a Ciência e Tecnologia-Consiglio Nazionale delle Ricerche Portugal-Italy bilateral project (to A. G. and M. T.), and a Federation of the European Biochemical Societies short-term fellowship (to J. B. V.).
- FDP
- flavodiiron protein
- Rd-D
- rubredoxin domain
- SVD
- singular value decomposition
- PDB
- Protein Data Bank.
REFERENCES
- 1. Saraiva L. M., Vicente J. B., Teixeira M. (2004) The role of the flavodiiron proteins in microbial nitric oxide detoxification. Adv. Microb. Physiol. 49, 77–129 [DOI] [PubMed] [Google Scholar]
- 2. Vicente J. B., Carrondo M. A., Teixeira M., Frazão C. (2008) Structural studies on flavodiiron proteins. Methods Enzymol. 437, 3–19 [DOI] [PubMed] [Google Scholar]
- 3. Vicente J. B., Justino M. C., Gonçalves V. L., Saraiva L. M., Teixeira M. (2008) Biochemical, spectroscopic, and thermodynamic properties of flavodiiron proteins. Methods Enzymol. 437, 21–45 [DOI] [PubMed] [Google Scholar]
- 4. Vicente J. B., Carrondo M. A., Teixeira M., Frazão C. (2011) Flavodiiron proteins: nitric oxide and/or oxygen reductases. in Encyclopedia of Inorganic and Bioinorganic Chemistry (Messerschmidt A., ed) pp. 1–20, John Wiley & Sons, Ltd., New York [Google Scholar]
- 5. Kurtz D. M. (2007) Flavo-diiron enzymes: nitric oxide or dioxygen reductases? Dalton Trans. 4115–4121 [Google Scholar]
- 6. Gonçalves V. L., Vicente J. B., Saraiva L. M., Teixeira M. (2011) Flavodiiron proteins and their role in cyanobacteria. in The Bioenergetic Processes of Cyanobacteria– from evolutionary singularity to ecological diversity (Obinger C., Peschek G. A., eds) pp. 631–656, Springer Verlag, New York [Google Scholar]
- 7. Justino M. C., Vicente J. B., Teixeira M., Saraiva L. M. (2005) New genes implicated in the protection of anaerobically grown Escherichia coli against nitric oxide. J. Biol. Chem. 280, 2636–2643 [DOI] [PubMed] [Google Scholar]
- 8. Silaghi-Dumitrescu R., Coulter E. D., Das A., Ljungdahl L. G., Jameson G. N., Huynh B. H., Kurtz D. M., Jr. (2003) A flavodiiron protein and high molecular weight rubredoxin from Moorella thermoacetica with nitric-oxide reductase activity. Biochemistry 42, 2806–2815 [DOI] [PubMed] [Google Scholar]
- 9. Silaghi-Dumitrescu R., Ng K. Y., Viswanathan R., Kurtz D. M. (2005) A flavo-diiron protein from Desulfovibrio vulgaris with oxidase and nitric-oxide reductase activities. Evidence for an in vivo nitric oxide scavenging function. Biochemistry 44, 3572–3579 [DOI] [PubMed] [Google Scholar]
- 10. Di Matteo A., Scandurra F. M., Testa F., Forte E., Sarti P., Brunori M., Giuffrè A. (2008) The O2-scavenging flavodiiron protein in the human parasite Giardia intestinalis. J. Biol. Chem. 283, 4061–4068 [DOI] [PubMed] [Google Scholar]
- 11. Vicente J. B., Tran V., Pinto L., Teixeira M., Singh U. (2012) A detoxifying oxygen reductase in the anaerobic protozoan Entamoeba histolytica. Eukaryot. Cell 11, 1112–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mills P. C., Richardson D. J., Hinton J. C., Spiro S. (2005) Detoxification of nitric oxide by the flavorubredoxin of Salmonella enterica serovar Typhimurium. Biochem. Soc. Trans. 33, 198–199 [DOI] [PubMed] [Google Scholar]
- 13. Baptista J. M., Justino M. C., Melo A. M., Teixeira M., Saraiva L. M. (2012) Oxidative stress modulates the nitric oxide defense promoted by Escherichia coli flavorubredoxin. J. Bacteriol. 194, 3611–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wink D. A., Mitchell J. B. (1998) Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 25, 434–456 [DOI] [PubMed] [Google Scholar]
- 15. Bogdan C. (2001) Nitric oxide and the immune response. Nat. Immunol. 2, 907–916 [DOI] [PubMed] [Google Scholar]
- 16. Thomas D. D., Flores-Santana W., Switzer C. H., Wink D. A., Ridnour L. A. (2010) Determinants of nitrc oxide chemistry: impact of cell signaling processes. in Nitric oxide-Biology and Pathobiology (Ignarro L. J., ed) pp. 3–25, Academic Press, Orlando, FL [Google Scholar]
- 17. Gonçalves V. L., Saraiva L. M., Teixeira M. (2011) Gene expression study of the flavodi-iron proteins from the cyanobacterium Synechocystis sp. PCC6803. Biochem. Soc. Trans. 39, 216–218 [DOI] [PubMed] [Google Scholar]
- 18. Allahverdiyeva Y., Ermakova M., Eisenhut M., Zhang P., Richaud P., Hagemann M., Cournac L., Aro E. M. (2011) Interplay between flavodiiron proteins and photorespiration in Synechocystis sp. PCC 6803. J. Biol. Chem. 286, 24007–24014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ermakova M., Battchikova N., Allahverdiyeva Y., Aro E. M. (2013) Novel heterocyst-specific flavodiiron proteins in Anabaena sp. PCC 7120. FEBS Lett. 587, 82–87 [DOI] [PubMed] [Google Scholar]
- 20. Smutná T., Gonçalves V. L., Saraiva L. M., Tachezy J., Teixeira M., Hrdy I. (2009) Flavodiiron protein from Trichomonas vaginalis hydrogenosomes: the terminal oxygen reductase. Eukaryot. Cell 8, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang P., Allahverdiyeva Y., Eisenhut M., Aro E. M. (2009) Flavodiiron proteins in oxygenic photosynthetic organisms: photoprotection of photosystem II by Flv2 and Flv4 in Synechocystis sp. PCC 6803. Plos One 4, e5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seedorf H., Hagemeier C. H., Shima S., Thauer R. K., Warkentin E., Ermler U. (2007) Structure of coenzyme F420H2 oxidase (FprA), a di-iron flavoprotein from methanogenic Archaea catalyzing the reduction of O2 to H2O. FEBS J. 274, 1588–1599 [DOI] [PubMed] [Google Scholar]
- 23. Frazão C., Silva G., Gomes C. M., Matias P., Coelho R., Sieker L., Macedo S., Liu M. Y., Oliveira S., Teixeira M., Xavier A. V., Rodrigues-Pousada C., Carrondo M. A., Le Gall J. (2000) Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat. Struct. Biol. 7, 1041–1045 [DOI] [PubMed] [Google Scholar]
- 24. Silaghi-Dumitrescu R., Kurtz D. M., Jr., Ljungdahl L. G., Lanzilotta W. N. (2005) X-ray crystal structures of Moorella thermoacetica FprA: novel diiron site structure and mechanistic insights into a scavenging nitric-oxide reductase. Biochemistry 44, 6492–6501 [DOI] [PubMed] [Google Scholar]
- 25. Fang H., Caranto J. D., Mendoza R., Taylor A. B., Hart P. J., Kurtz D. M. (2012) Histidine ligand variants of a flavo-diiron protein: effects on structure and activities. J. Biol. Inorg. Chem. 17, 1231–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carlton J. M., Hirt R. P., Silva J. C., Delcher A. L., Schatz M., Zhao Q., Wortman J. R., Bidwell S. L., Alsmark U. C., Besteiro S., Sicheritz-Ponten T., Noel C. J., Dacks J. B., Foster P. G., Simillion C., Van de Peer Y., Miranda-Saavedra D., Barton G. J., Westrop G. D., Müller S., Dessi D., Fiori P. L., Ren Q., Paulsen I., Zhang H., Bastida-Corcuera F. D., Simoes-Barbosa A., Brown M. T., Hayes R. D., Mukherjee M., Okumura C. Y., Schneider R., Smith A. J., Vanacova S., Villalvazo M., Haas B. J., Pertea M., Feldblyum T. V., Utterback T. R., Shu C. L., Osoegawa K., de Jong P. J., Hrdy I., Horvathova L., Zubacova Z., Dolezal P., Malik S. B., Logsdon J. M., Jr., Henze K., Gupta A., Wang C. C., Dunne R. L., Upcroft J. A., Upcroft P., White O., Salzberg S. L., Tang P., Chiu C. H., Lee Y. S., Embley T. M., Coombs G. H., Mottram J. C., Tachezy J., Fraser-Liggett C. M., Johnson P. J. (2007) Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 315, 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morrison H. G., McArthur A. G., Gillin F. D., Aley S. B., Adam R. D., Olsen G. J., Best A. A., Cande W. Z., Chen F., Cipriano M. J., Davids B. J., Dawson S. C., Elmendorf H. G., Hehl A. B., Holder M. E., Huse S. M., Kim U. U., Lasek-Nesselquist E., Manning G., Nigam A., Nixon J. E., Palm D., Passamaneck N. E., Prabhu A., Reich C. I., Reiner D. S., Samuelson J., Svard S. G., Sogin M. L. (2007) Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 317, 1921–1926 [DOI] [PubMed] [Google Scholar]
- 28. Loftus B., Anderson I., Davies R., Alsmark U. C., Samuelson J., Amedeo P., Roncaglia P., Berriman M., Hirt R. P., Mann B. J., Nozaki T., Suh B., Pop M., Duchene M., Ackers J., Tannich E., Leippe M., Hofer M., Bruchhaus I., Willhoeft U., Bhattacharya A., Chillingworth T., Churcher C., Hance Z., Harris B., Harris D., Jagels K., Moule S., Mungall K., Ormond D., Squares R., Whitehead S., Quail M. A., Rabbinowitsch E., Norbertczak H., Price C., Wang Z., Guillén N., Gilchrist C., Stroup S. E., Bhattacharya S., Lohia A., Foster P. G., Sicheritz-Ponten T., Weber C., Singh U., Mukherjee C., El-Sayed N. M., Petri W. A., Jr., Clark C. G., Embley T. M., Barrell B., Fraser C. M., Hall N. (2005) The genome of the protist parasite Entamoeba histolytica. Nature 433, 865–868 [DOI] [PubMed] [Google Scholar]
- 29. Rodrigues R., Vicente J. B., Félix R., Oliveira S., Teixeira M., Rodrigues-Pousada C. (2006) Desulfovibrio gigas flavodiiron protein affords protection against nitrosative stress in vivo. J. Bacteriol. 188, 2745–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gardner A. M., Helmick R. A., Gardner P. R. (2002) Flavorubredoxin, an inducible catalyst for nitric oxide reduction and detoxification Escherichia coli. J. Biol. Chem. 277, 8172–8177 [DOI] [PubMed] [Google Scholar]
- 31. Gomes C. M., Giuffrè A., Forte E., Vicente J. B., Saraiva L. M., Brunori M., Teixeira M. (2002) A novel type of nitric-oxide reductase: Escherichia coli flavorubredoxin. J. Biol. Chem. 277, 25273–25276 [DOI] [PubMed] [Google Scholar]
- 32. Mills P. C., Rowley G., Spiro S., Hinton J. C., Richardson D. J. (2008) A combination of cytochrome c nitrite reductase (NrfA) and flavorubredoxin (NorV) protects Salmonella enterica serovar Typhimurium against killing by NO in anoxic environments. Microbiology 154, 1218–1228 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protocols 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 36. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 37. Vicente J. B., Teixeira M. (2005) Redox and spectroscopic properties of the Escherichia coli nitric oxide-detoxifying system involving flavorubredoxin and its NADH-oxidizing redox partner. J. Biol. Chem. 280, 34599–34608 [DOI] [PubMed] [Google Scholar]
- 38. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 39. Susín S., Abián J., Sánchez-Baeza F., Peleato M. L., Abadía A., Gelpí E., Abadía J. (1993) Riboflavin 3′-sulfate and 5′-sulfate, 2 novel flavins accumulating in the roots of iron-deficient sugar-beet (β-vulgaris). J. Biol. Chem. 268, 20958–20965 [PubMed] [Google Scholar]
- 40. Fischer D. S., Price D. C. (1964) Simple serum iron method using new sensitive chromogen tripyridyl-S-triazine. Clin. Chem. 10, 21–31 [PubMed] [Google Scholar]
- 41. Stookey L. L. (1970) Ferrozine: a new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781 [Google Scholar]
- 42. Gomes C. M., Vicente J. B., Wasserfallen A., Teixeira M. (2000) Spectroscopic studies and characterization of a novel electron-transfer chain from Escherichia coli involving a flavorubredoxin and its flavoprotein reductase partner. Biochemistry 39, 16230–16237 [DOI] [PubMed] [Google Scholar]
- 43. Vicente J. B., Scandurra F. M., Forte E., Brunori M., Sarti P., Teixeira M., Giuffrè A. (2008) Kinetic characterization of the Escherichia coli nitric-oxide reductase flavorubredoxin. Methods Enzymol. 437, 47–62 [DOI] [PubMed] [Google Scholar]
- 44. Beckman J. S., Wink D. A., Crow J. P. (1996) Nitric oxide and peroxynitrite. in Methods in Nitric Oxide Research (Feelisch M., Stamler J. S., eds) pp. 61–71, John Wiley & Sons Ltd., New York [Google Scholar]
- 45. Henry E. R., Hofrichter J. (1992) Singular value decomposition: application to analysis of experimental data. Methods Enzymol. 210, 129–192 [Google Scholar]
- 46. Vicente J. B., Testa F., Mastronicola D., Forte E., Sarti P., Teixeira M., Giuffrè A. (2009) Redox properties of the oxygen-detoxifying flavodiiron protein from the human parasite Giardia intestinalis. Arch. Biochem. Biophys. 488, 9–13 [DOI] [PubMed] [Google Scholar]
- 47. Caranto J. D., Weitz A., Hendrich M. P., Kurtz D. M. (2014) The nitric oxide reductase mechanism of a flavo-diiron protein: identification of active-site intermediates and products. J. Am. Chem. Soc. 136, 7981–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pfitzner U., Odenwald A., Ostermann T., Weingard L., Ludwig B., Richter O. M. (1998) Cytochrome c oxidase (heme aa3) from Paracoccus denitrificans: analysis of mutations in putative proton channels of subunit I. J. Bioenerg. Biomembr. 30, 89–97 [DOI] [PubMed] [Google Scholar]
- 49. Hosler J. P., Espe M. P., Zhen Y., Babcock G. T., Ferguson-Miller S. (1995) Analysis of site-directed mutants locates a non-redox-active metal near the active site of cytochrome c oxidase of Rhodobacter sphaeroides. Biochemistry 34, 7586–7592 [DOI] [PubMed] [Google Scholar]
- 50. Ducluzeau A. L., van Lis R., Duval S., Schoepp-Cothenet B., Russell M. J., Nitschke W. (2009) Was nitric oxide the first deep electron sink? Trends Biochem. Sci. 34, 9–15 [DOI] [PubMed] [Google Scholar]