

Background: AT1Rs function as mechanosensors to activate β-arrestin-dependent signaling.
Results: Osmotic stretch induces active conformations of the AT1R and β-arrestin2 that are exclusively stabilized by β-arrestin-biased agonists.
Conclusion: Membrane stretch functions as an allosteric modulator to selectively enhance β-arrestin-biased signaling.
Significance: Membrane stretch induces a biased conformation of the AT1R similar to that of β-arrestin-biased agonists.
Keywords: Allosteric Regulation, Angiotensin II, Cell Signaling, Conformational Change, Mechanotransduction
Abstract
It has recently been appreciated that the angiotensin II type 1 receptor (AT1R), a prototypic member of the G protein-coupled receptor superfamily, also functions as a mechanosensor. Specifically, mechanical stretch activates the AT1R to promote downstream signaling mediated exclusively by the multifunctional scaffold protein, β-arrestin, in a manner consistent with previously identified β-arrestin-biased ligands. However, the ligand-independent mechanism by which mechanical stretch promotes β-arrestin-biased signaling remains unknown. Implicit in the concept of biased agonism (i.e. the ability of an agonist to activate a subset of receptor-mediated signaling pathways) is the notion that distinct active conformations of the receptor mediate differential activation of signaling pathways. Here we determined whether mechanical stretch stabilizes distinct β-arrestin-activating conformations of the AT1R by using β-arrestin2-biased agonists as conformational probes in pharmacological and biophysical assays. When tested at cells expressing the AT1R fused to β-arrestin (AT1R-β-arrestin2), we found that osmotic stretch increased the binding affinity and potency of the β-arrestin-biased agonist TRV120023, with no effect on the balanced agonist AngII. In addition, the effect of osmotic stretch on ERK activation was markedly augmented in cells expressing the AT1R-β-arrestin2 fusion compared with the wild type AT1R and completely blocked in cells expressing the AT1R-Gq fusion. Biophysical experiments with an intramolecular BRET β-arrestin2 biosensor revealed that osmotic stretch and TRV120023 activate AT1Rs to stabilize β-arrestin2 active conformations that differ from those stabilized by the AT1R activated by angiotensin II. Together, these data support a novel ligand-independent mechanism whereby mechanical stretch allosterically stabilizes specific β-arrestin-biased active conformations of the AT1R and has important implications for understanding pathophysiological AT1R signaling.
Introduction
The transduction of mechanical stimuli (i.e. mechanotransduction) by mechanosensitive cells mediates a variety of physiological processes such as tactile perception, proprioception, visceroception, hearing, and balance (1–3). Mechanotransduction is also thought to play an important role in pathophysiological processes such as vascular constriction (1, 4), cardiac hypertrophy (2), and neurosensory disorders (3, 5). Although the precise molecular entities that function as sensors are not completely understood, it is appreciated that a number of membrane proteins can activate intracellular signaling in response to mechanical force including ion channels, integrins, components of the cytoskeleton and some members of the heterotrimeric G protein-coupled receptor (GPCR)4 superfamily (1, 2, 5–7).
Of the GPCRs that have been identified as mechanosensors, the angiotensin II type 1 receptor (AT1R) remains one of the best characterized (2, 6, 8). The AT1R, like nearly all members of the GPCR superfamily, can transduce extracellular stimuli to activate intracellular signaling through both canonical G protein and noncanonical β-arrestin effector pathways (9, 10). Recently, it has been shown that the activation of intracellular signaling by mechanical stretch of the AT1R does not require the ligand angiotensin II (AngII) (6, 8, 11) but does require the recruitment and activation of the transducer β-arrestin (6). Thus, despite its apparent ligand independence, mechanical stretch activates the AT1R in a manner that is consistent with previously identified β-arrestin-biased ligands that stabilize a receptor conformation to preferentially activate a β-arrestin-mediated pathway (6, 12).
Implicit in the concept of biased agonism (i.e. the ability of an agonist to activate a subset of receptor-mediated signaling pathways) is the notion that ligands stabilize distinct active conformations of a GPCR, thereby promoting differential activation of signaling pathways (10, 13). In this context, it is intriguing to speculate that mechanical stretch induces active conformations of the AT1R that selectively promote β-arrestin signaling. Indeed, previous studies with several GPCRs, including the AT1R, suggest that mechanical stimuli alter receptor structure. Both rhodopsin (14, 15) and the B2 bradykinin receptor (16) have been shown to adopt a distinct active receptor conformation induced by mechanical stress. Through mutagenesis of the AT1R, it has been suggested that mechanical stretch induces a change in the conformation of the receptor, allowing it to couple mechanical stress to signaling (17). Although we have recently shown that mechanical stretch induces a conformation of β-arrestin similar to that induced by a biased ligand as measured by intramolecular bioluminescence resonance energy transfer (BRET) (6), direct evidence for a β-arrestin-biased AT1R conformation induced by mechanical stretch is lacking. This is due in large part to the considerable technical difficulties associated with measuring the effects of mechanical stress on GPCRs at the molecular level.
To determine whether mechanical stretch stabilizes distinct β-arrestin-activating conformations of the AT1R, we used biased agonists as novel conformational probes in pharmacological and biophysical assays. Critical to this approach were fusions between the AT1R and Gq (AT1R-Gq) or β-arrestin2 (AT1R-β-arrestin2), which were recently used to quantify the signaling bias of AT1R agonists (18). From these studies, we propose a ligand-independent mechanism whereby mechanical stretch allosterically stabilizes specific β-arrestin-activating conformations of the AT1R to engender β-arrestin-biased signaling.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK 293 cells stably expressing the following receptors: wild type AT1R (WT AT1R), AT1R-β-arrestin2 fusion protein, or AT1R-Gq fusion protein, were generated and maintained as previously described (18). HEK 293 cells stably expressing WT AT1R (1152 bp, 44 kDa), AT1R-β-arrestin2 fusion protein (2334 bp, 88 kDa), or AT1R-Gq (2280 bp, 87 kDa) fusion protein were grown in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 500 μg/ml neomycin at 37 °C in a humidified environment (5% CO2). Receptor densities of HEK 293 cells stably expressing WT AT1R, AT1R-β-arrestin fusion protein, or AT1R-Gq fusion protein are 400, 1400, and 1200 fmol/mg protein, respectively. The AT1R biased ligands used, TRV120023 (Sar-Arg-Val-Tyr-Lys-His-Pro-Ala-OH) and TRV120056 (Asp-Arg-Gly-Val-Tyr-Ile-His-Pro-Phe-OH), were developed and supplied by Trevena, Inc. (King of Prussia, PA).
Osmotic Stretch
Six-well flat bottom Corning Costar® cell culture plates (Sigma-Aldrich) were coated with 0.01% collagen (Sigma-Aldrich) for adherence. HEK 293 cells stably expressing WT AT1R, AT1R-β-arrestin fusion protein, or AT1R-Gq fusion protein were plated at a confluence of 60–70%. Cells were incubated for 24 h and then serum-starved for 16 h before osmotic stimulation. Osmotic stretch was applied by adding a defined volume of double-distilled H2O to the culture medium in each well of the 6-well plates. The cells were stimulated for 10 min 37 °C in a humidified environment (5% CO2). Control samples were maintained at the same condition with no application of osmotic stretch. Osmolality, measured as mOsm of solute/Kg of solvent (mOsm/kg), was measured using an Advanced InstrumentsTM 3250 Single Sample Osmometer (Thermo Fisher Scientific Inc., Waltham, MA) in the Duke University Clinical Laboratory.
Competition Radioligand Binding Assays with Whole Cells
Competition radioligand binding assays were performed on HEK 293 cells stably expressing WT AT1Rs, AT1R-β-arrestin2 fusion proteins, or AT1R-Gq fusion proteins after stimulation with isotonic or hypotonic conditions as described above. On the day of assay, cells were washed gently with ice-cold PBS without Ca2+ and Mg2+, covered with 5 ml of versene, and incubated on ice for 30 min. The cells were then gently harvested in 5 ml of cold MEM and spun down in a 15-ml conical tube at 500 rpm at 4 °C for 5 min. The pellet was gently resuspended in 1 ml of cold indicator-free minimal essential Eagle's medium (undiluted or diluted 1:1 with double-distilled H2O) and adjusted to achieve ∼1–2 μg of protein per μl of cells as determined from the Bradford assay. Competition binding assays with 50 μl of cells, 50 μl of serially diluted 5× test ligand in cold buffer, and 150 μl of 1.67× radioligand (125I-S1I8-AngII at 83.5 pm; 50 pm final concentration) were incubated at 4 °C for 4–5 h. Nonspecific binding was determined with 10 μm telmisartan. Bound radioactivity was collected on 0.3% PEI-treated GF/C filters (Brandel, Gaithersburg, MD) using cold wash buffer (50 mm Tris-HCl, pH 7.4). Bound radioactivity was quantified on a Packard Cobra gamma counter (GMI, Ramsey, MN).
Competition Radioligand Binding Assays with Detergent-solubilized AT1R
Human FLAG-tagged AT1R was stably expressed in a suspension adapted tetracycline-inducible HEK cell line (Invitrogen). AT1R expression was induced for 24 h by treatment with doxycycline (2 μg/ml) and sodium butyrate (5 mm). Cell lysate was solubilized in 0.5% lauryl maltose neopentyl glycol and subject to M1 anti-FLAG chromatography. Monomeric AT1R was isolated using size exclusion chromatography. Binding assays were performed as described above in MEM buffer or a 1:1 dilution of MEM and double-distilled H2O. The final concentration of AT1R was 0.4 ng/μl, and 125I-S1I8-AngII was used at a final concentration of 200 pm.
Immunoblotting
Cells were lysed in Nonidet P-40 lysis buffer containing 20 mm Tris (pH 7.4), 137 mm NaCl, 1% Nonidet P-40, 20% glycerol, 10 mm phenylmethylsulfonyl fluoride, 1 mm Na3VO4, 10 mm NaF, aprotinin (2.5 μg/ml), and leupeptin (2.5 μg/ml). Protein concentrations were assayed with Bio-Rad protein assay reagent (Bio-Rad), and 35 μg of protein was denatured by heating at 95 °C for 5 min before resolving by SDS-polyacrylamide gel (10%) electrophoresis. Immunoblotting for total ERK and phosphorylated ERK was performed as previously described (6). The following dilutions of primary antibody were used: total ERK (Upstate, Lake Placid, NY), 1:3000; phospho-ERK1/2 (Cell Signaling, Danvers, MA), 1:1000. Horseradish peroxidase-conjugated polyclonal donkey anti-rabbit IgG (Sigma-Aldrich) was employed as the secondary antibody. Protein bands were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences, Pittsburgh, PA). Individual bands were then quantified by densitometry with the Syngene Imager (Syngene USA, Frederick, MD) using GeneSnap software.
BRET Assay
BRET assays were performed as described previously (6, 19). HEK-293 cell stably expressing AT1Rs were transfected with the intramolecular BRET-based biosensor of β-arrestin2 and 24 h later were distributed onto collagen coated 96-well microplates. The BRET signal was determined as the ratio of the light emitted by YFP and the light emitted by Luc. BRET ratio was monitored at 10 min after ligand stimulation or osmotic stress. Values were corrected by subtracting the background BRET signals.
ERK Phosphorylation in Sodium-free Buffer
For the measurement of osmotic stretch-induced ERK signaling in buffer with or without sodium ions, HEK 293 stable expressing WT AT1R cells were grown in MEM on 10-cm polystyrol dishes and were split into 6-well plates. The cells were washed four times using sodium-free buffer as previously described (20), which contains 125 mmol/liter N-methyl-d-glucosamine hydrochloride, 25 mmol/liter Hepes/Tris (pH 7.4), 5.6 mmol/liter (+) glucose, 4.8 mmol/liter KCl, 1.2 mmol/liter KH2PO4, 1.2 mmol/liter CaCl2, and 1.2 mmol/liter MgSO4. After preincubation in sodium-free buffer for 20 min, the buffer was replaced with either sodium-free buffer or buffer containing sodium buffer (125 mmol/liter NaCl, 25 mmol/liter Hepes/NaOH, pH 7.4, 5.6 mmol/liter (+) glucose, 4.8 mmol/liter KCl, 1.2 mmol/liter KH2PO4, 1.2 mmol/liter CaCl2, and 1.2 mmol/liter MgSO4) (20). Stimulation with AngII, osmotic stretch, with or without the addition of telmisartan, was performed in MEM, sodium buffer, or sodium-free buffer for 10 min, and then the cell lysates were analyzed for ERK phosphorylation by immunoblotting.
Statistics
Competition binding curves were best fit by a one-site binding model using the nonlinear least squares regression program GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Differences in competition binding curves were tested using the F test, with a significance threshold of p < 0.05. Individual dose-response curves for ERK1/2 phosphorylation were fit to a three-parameter logistic model in Prism. Emax and LogEC50 average values for the data in Figs. 3 and 6 were calculated from the best fit of individual curves except for Fig. 6B where two of the dose-response curves did not fit a three-parameter logistic model, and therefore Emax and LogEC50 were calculated from the best fit of the average data. For each ligand the data were normalized to the fit bottom value determined for isotonic buffer, thus setting it equal to 1. The data are expressed as the means ± S.E. Statistical significance was determined with a one-way or two-way analysis of variance (ANOVA) to correct for multiple comparisons and either a Bonferroni or Holm-Sidak post hoc test using GraphPad Prism. A p value of <0.05 was considered significant.
FIGURE 3.
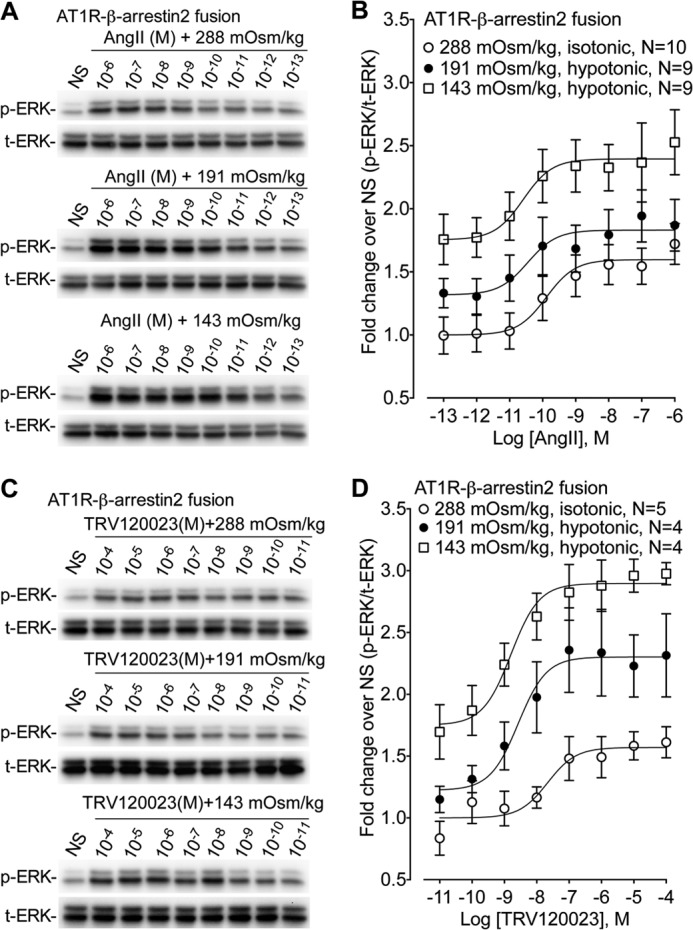
Osmotic stretch allosterically modulates AT1R signaling. The effect of the balanced agonist AngII and the β-arrestin-biased agonist (n = 9–10) on ERK phosphorylation was tested in cells stably expressing AT1R-β-arrestin2 fusion proteins under isotonic (288 mOsm/kg) and hypotonic osmotic stretch (191 and 143 mOsm/kg). A, representative immunoblots showing enhanced ERK phosphorylation in response to increasing hypotonic osmotic stretch over a range of AngII concentrations. NS, nonstimulation. B, dose-dependent increase in ERK phosphorylation with the balanced agonist AngII stimulation. Increasing hypotonicity augmented Emax but had no effect on EC50 (n = 9–10). Isotonic 288 mOsm/kg (Emax = 1.63 ± 0.15, LogEC50 = −9.84 ± 0.37 nm); hypotonic 191 mOsm/kg (Emax = 1.96 ± 0.20, LogEC50 = −9.36 ± 0.56 nm); hypotonic 143 mOsm/kg (Emax = 2.57 ± 0.27, LogEC50 = −9.78 ± 0.50 nm). Emax, p < 0.02, hypotonic 143 mOsm/kg versus isotonic 288 mOsm/kg (one-way ANOVA with Bonferroni correction). C, representative immunoblots showing enhanced ERK phosphorylation in response to increasing hypotonic osmotic stretch over a range of TRV120023 concentrations. NS, nonstimulation. D, dose-dependent increase in ERK phosphorylation with the β-arrestin-biased agonist TRV120023. Increasing hypotonicity significantly enhanced both Emax and EC50 (n = 4–5). Isotonic 288 mOsm/kg (Emax = 1.62 ± 0.07, LogEC50 = −7.24 ± 0.49 nm); hypotonic 191 mOsm/kg (Emax = 2.67 ± 0.36, LogEC50 = −8.37 ± 0.27 nm); hypotonic 143 mOsm/kg (Emax = 3.33 ± 0.19 ±, LogEC50 = −8.87 ± 0.18 nm). Emax, p < 0.001, hypotonic 143 mOsm/kg versus isotonic 288 mOsm/kg; p < 0.05 hypotonic 191 mOsm/kg versus isotonic 288 mOsm/kg. LogEC50, p < 0.05 hypotonic 143 mOsm/kg versus isotonic 288 mOsm/kg (one-way ANOVA with Bonferroni correction). The data represent the means ± S.E. NS, nonstimulation.
FIGURE 6.
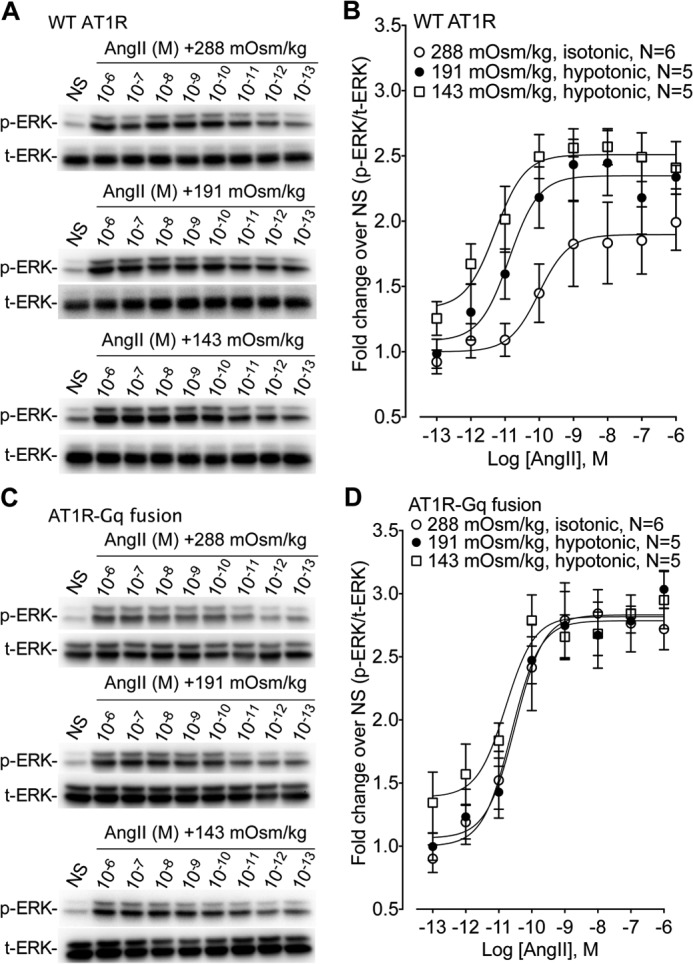
Dose-response characteristics of ERK1/2 phosphorylation for the WT AT1R and the AT1R-Gq fusion over a broad range of AngII concentrations. Shown is the effect of the balanced agonist AngII on ERK1/2 phosphorylation in cells stably expressing WT AT1R and AT1R-Gq fusion protein under isotonic (288 mOsm/kg) or hypotonic osmotic stretch (191 and 143 mOsm/kg) conditions. A, representative immunoblots showing enhanced ERK1/2 phosphorylation in cells expressing the WT AT1R in response to increasing hypotonic osmotic stretch over a range of AngII concentrations. B, osmotic stretch enhances ERK1/2 phosphorylation by increasing Emax without a change in the EC50 for the WT AT1R over a broad range of AngII concentrations. Isotonic 288 mOsm/kg (Emax = 1.90 ± 0.12, LogEC50 = −10.01 ± 0.45 nm); hypotonic 191 mOsm/kg (Emax = 2.35 ± 0.10, LogEC50 = −10.88 ± 0.33 nm); hypotonic 143 mOsm/kg (Emax = 2.51 ± 0.08, LogEC50 = −11.29 ± 0.30 nm) (n = 5–6 independent experiment at each AngII concentration). Emax, p < 0.005, hypotonic 143 mOsm/kg versus isotonic 288 mOsm/kg; Emax, p < 0.05, 191 mOsm/kg versus isotonic 288 mOsm/kg (one-way ANOVA with Bonferroni correction). There was no significant difference for LogEC50 between groups. C, representative immunoblots in cells stably expressing AT1R-Gq fusion proteins showing no enhancement of ERK1/2 phosphorylation in response to increasing hypotonic osmotic stretch over a range of AngII concentrations in cells. D, osmotic stretch does not enhance ERK1/2 phosphorylation in cells stably expressing AT1R-Gq fusion proteins in response to AngII stimulation. Isotonic 288 mOsm/kg (Emax = 2.84 ± 0.16, LogEC50 = −10.45 ± 0.31 nm); hypotonic 191 mOsm/kg (Emax = 2.84 ± 0.17, LogEC50 = −10.51 ± 0.16 nm); hypotonic 143 mOsm/kg (Emax = 2.83 ± 0.12, LogEC50 = −10.68 ± 0.20 nm) (n = 5–6 independent experiment at each AngII concentration). There was no significant difference for both Emax and LogEC50 between groups.
RESULTS
Mechanical Stretch Stabilizes AT1R Conformations That Favor the Binding of β-Arrestin2-biased Agonists
It is well established that agonists bind with higher affinity to active GPCR conformations. These conformations can be stabilized by mutations that confer constitutive activity (21) or by coupling to transduction proteins such as G proteins or β-arrestins that promote intracellular signaling (22–24). Recently, it has been demonstrated that AT1R-transducer fusion proteins can be used to interrogate these active conformations to precisely determine the signaling bias of AT1R agonists (18). To examine whether mechanical stretch stabilizes distinct β-arrestin2-activating conformations of the AT1R, we measured the binding affinities of balanced and β-arrestin2-biased agonists in intact cells expressing either the unfused WT AT1R (uncoupled) or AT1R-β-arrestin2 fusion protein (β-arrestin coupled) under normal tonicity (288 mOsm/kg) or hypotonic conditions (143 mOsm/kg) to induce membrane stretch (Fig. 1A). Compared with the WT AT1R, the balanced agonist AngII bound with 3.1-fold higher affinity to the AT1R-βarrestin2 fusion protein in isotonic media (Fig. 1B and Table 1). This was in agreement with the known allosteric enhancing effect of β-arrestin2 coupling on AngII affinity in whole cells (25) and membranes (18). We next found that osmotic stretch had no effect on AngII affinity for the WT AT1R (p = 0.1204 by F test) or the AT1R-βarrestin2 fusion protein (p = 0.9257 by F test). This yielded superimposed binding curves in Fig. 1B. When the β-arrestin2-biased agonist TRV120023 was tested, we found that it bound with 9.7-fold higher affinity to the AT1R-βarrestin2 fusion protein compared with the WT AT1R under isotonic conditions (Fig. 1C, filled black symbols, and Table 1). However, in contrast to the balanced ligand AngII, exposing cells to hypotonic osmotic stretch significantly increased the binding affinity of TRV120023 at both the WT AT1R (3.5-fold; Fig. 1C, filled black squares versus open red squares, p < 0001 by F test) and the AT1R-β-arrestin2 fusion protein (3.2-fold; Fig. 1C, filled black circles versus open red circles, p < 0.0001 by F test) (Fig. 1C). For the antagonist telmisartan, the superimposed binding curves in Fig. 1D indicated that its binding affinity was unaltered by β-arrestin2 coupling or changes in tonicity, consistent with previous reports (18, 25). Lastly, we tested the effects of osmotic stretch on the binding of the Gq-biased agonist TRV120056 and showed a pattern similar to AngII in which it bound with higher affinity to the AT1R-β-arrestin2 fusion protein but was unaffected by osmotic stretch (data not shown). This was consistent with reports that TRV120056 and the balanced agonist AngII activate β-arrestin2 to the same extent (12, 18). We were unable to measure the effects of osmotic stress on Gq coupling to the AT1R using the AT1R-Gq fusion protein in intact cells likely because of the high concentration of intracellular GTP, which uncouples G proteins from their cognate receptors in intact cellular systems (26). Taken together, these data suggest that osmotic stretch stabilizes a subset of β-arrestin2-specific AT1R conformations that are distinct from those stabilized by the balanced agonist AngII.
FIGURE 1.

Osmotic stretch stabilizes distinct β-arrestin2-activating conformations of the AT1R as measured by shifts in binding affinities. Competition binding isotherms of different ligands in HEK 293 cells stably expressing WT AT1R or AT1R-β-arrestin2 fusion proteins under isotonic (288 mOsm/kg) or hypotonic osmotic stretch (143 mOsm/kg). A, schematic diagrams of WT AT1R, AT1R-β-arrestin2 fusion, and AT1R-Gq fusion. B, competition binding isotherms for the balanced agonist AngII. No curve shift was observed under hypotonic osmotic stretch (143 mOsm/kg). C, competition binding isotherms for the β-arrestin-biased ligand TRV120023 showing that it bound with 9.7-fold greater affinity to the AT1R-βarrestin2 under isotonic condition, with a further 3.5-fold shift in binding affinity under hypotonic conditions. D, competition binding isotherms for the AngII antagonist telmisartan. Affinity shift was undetectable under hypotonic osmotic stretch. The data represent the means ± S.E. of three to nine independent experiments. Each experiment was performed in duplicate.
TABLE 1.
Ligand binding affinity at WT AT1R and AT1R-β-arrestin2 fusion
The data are expressed as means ± S.E.
| Ligand | pKi WT AT1R |
pKi AT1R-β-arrestin2 |
||
|---|---|---|---|---|
| Isotonic (288 mOsm/kg) | Hypotonic (143 mOsm/kg) | Isotonic (288 mOsm/kg) | Hypotonic (143 mOsm/kg) | |
| Angiotensin II | 8.29 ± 0.03 | 8.36 ± 0.03 | 8.78 ± 0.02a | 8.77 ± 0.03 |
| TRV120023 | 6.74 ± 0.05 | 7.28 ± 0.03b | 7.72 ± 0.03a | 8.23 ± 0.02b |
| Telmisartan | 8.68 ± 0.03 | 8.77 ± 0.03 | 8.66 ± 0.03 | 8.80 ± 0.03 |
a p < 0.05, AT1R-β-arrestin2 fusion versus WT AT1R under isotonic conditions indicating functional coupling between the receptor and fused β-arrestin2 transducer (F test).
b p < 0.05, hypotonic stretch (143 mOsm/kg) versus isotonic (288 mOsm/kg) buffer (F test).
To rule out the possibility that the observed shifts in β-arrestin2-biased agonist binding affinity were due to changes in buffer composition such as a dilution of Na+ cations (27, 28), we determined whether hypotonic media had any effect on ligand binding to detergent-purified AT1Rs. In this system, AT1Rs are exposed to the same hypotonic buffer but are unable to undergo the biophysical changes induced by osmotic stretch of intact cells. Hypotonic conditions elicited similar small increases in the binding affinities of both the balanced agonist AngII and the β-arrestin2-biased agonist TRV120023 (1.5- and 1.9-fold, respectively) (Fig. 2). Importantly, these results cannot account for the differential effects we observed in whole cells (Fig. 1, B and C) and are consistent with the lack of a requirement for Na+ ions in stretch-mediated ERK1/2 phosphorylation (see below).
FIGURE 2.
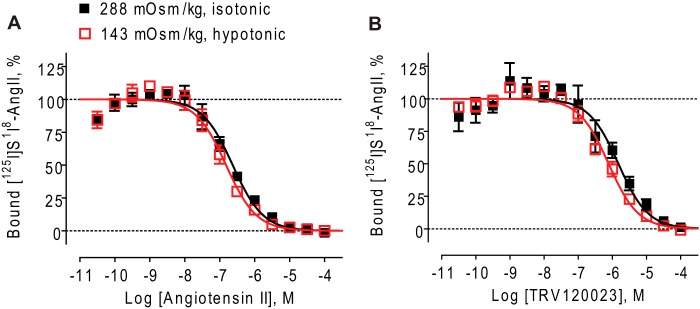
Minimal effect of hypotonic conditions on ligand binding affinities at detergent-purified human AT1Rs. Osmotic stretch has similar effects on ligand binding affinities at detergent-purified human AT1R receptor. A, shown is the competition binding curve of AngII at purified WT AT1Rs under isotonic (288 mOsm/kg) and hypotonic (143 mOsm/kg) buffer conditions. AngII affinity was increased by 1.5-fold in hypotonic buffer (p < 0.05 by F test). B, shown is the competition binding curve of TRV120023 at purified WT AT1Rs under isotonic and hypotonic buffer conditions. TRV120023 affinity was increased 1.9-fold in hypotonic buffer (p < 0.05 by F test). The data represent the means ± S.E. of four independent experiments performed in duplicate.
Osmotic Stretch Allosterically Modulates AT1R Signaling Leading to Enhanced Potency of a β-Arrestin2-biased Agonist
Osmotic stretch produced a pattern of affinity shifts for the β-arrestin2-biased agonist TRV120023 similar to that reported for agonist binding to a constitutively active mutant β2AR (21) (Fig. 1C). Because it is well documented that agonists signal with increased potency at constitutively active GPCRs, we next determined whether osmotic stretch modulates TRV120023 signaling. Here we measured the ability for AngII or the β-arrestin2-biased agonist TRV120023 to activate ERK1/2 in cells overexpressing the AT1R-βarrestin2 fusion protein under normal tonicity (288 mOsm/kg) or hypotonic conditions (191 and 143 mOsm/kg). We observed a dose-dependent increase in ERK phosphorylation with AngII stimulation, which was enhanced by osmotic stretch (Fig. 3, A and B). Although increasing hypotonicity significantly augmented AngII maximal signaling (Emax from 1.63 ± 0.15 to 2.57 ± 0.27, p < 0.02), it had no effect on AngII potency (LogEC50 from −9.84 ± 0.37 to −9.78 ± 0.50 nm, p = NS). This was consistent with our binding data showing no effect on AngII binding affinity (Fig. 1B and Table 1). Lastly, the increase in baseline ERK1/2 phosphorylation (e.g. at 10−13 m AngII) for 191 and 143 mOsm/kg most likely reflects the ligand-independent activation of AT1R by increasing hypotonicity.
Increasing hypotonicity had similar effects on both baseline ERK1/2 phosphorylation and TRV120023 maximal signaling (Emax from 1.62 ± 0.07 to 3.33 ± 0.19, p < 0.001) (Fig. 3D). However, unlike the balanced agonist AngII, exposing cells to osmotic stretch significantly increased the potency of the β-arrestin-biased agonist TRV120023 for ERK1/2 phosphorylation (LogEC50 from −7.24 ± 0.49 to −8.87 ± 0.18 nm, p < 0.05) (Fig. 3D). This is in good agreement with the ability for osmotic stretch to selectively enhance the binding affinity of TRV120023 (Fig. 1C), supporting our hypothesis that mechanical stretch stabilizes unique β-arrestin2-activating conformations of the AT1R.
Osmotic Stretch and β-Arrestin2-biased Agonists Activate AT1Rs to Stabilize β-Arrestin2 Conformations Distinct from AngII
Our data support a ligand-independent mechanism whereby osmotic stretch stabilizes unique β-arrestin2-activating conformations of the AT1R to promote β-arrestin2 signaling. To further test the effect of osmotic stretch on AT1R conformation, we used an intramolecular BRET β-arrestin2 biosensor (29) (Fig. 4A) that differentiates between conformations of β-arrestin2 stabilized by AngII or osmotic stretch (6) and is sensitive to conformations induced by a β-arrestin-biased agonist (19). Consistent with our previous report (6), activating the AT1R with AngII (1 μm) induced changes in the BRET signal that were of similar magnitude but opposite in direction from that induced by hypotonic osmotic stretch (143 mOsm/kg, p < 0.001; Fig. 4B). Importantly, activating the AT1R with the β-arrestin2-biased ligand TRV120023 (1 μm) produced a negative BRET ratio similar to that induced by osmotic stretch (p = NS versus hypotonic; Fig. 4B). We next tested whether the β-arrestin conformation induced by hypotonic conditions can be altered by the subsequent addition of the balanced agonist AngII. We measured the BRET ratio from normal tonicity (isotonic, 288 mOsm/kg) to hypotonicity (143 mOsm/kg) and then after the addition of AngII (1 μm). Control experiments were performed by adding isotonic solution (288 mOsm/kg) to the media followed by 1 μm AngII. We demonstrate that AngII is unable to overcome the effects of hypotonicity, suggesting that once the AT1R is stabilized in an allosterically modified conformation, AngII cannot shift the equilibrium to stabilize a different conformation (Fig. 4C). These biophysical data support our radioligand binding studies to indicate that membrane stretch stabilizes unique β-arrestin2-activating conformations of the AT1R.
FIGURE 4.
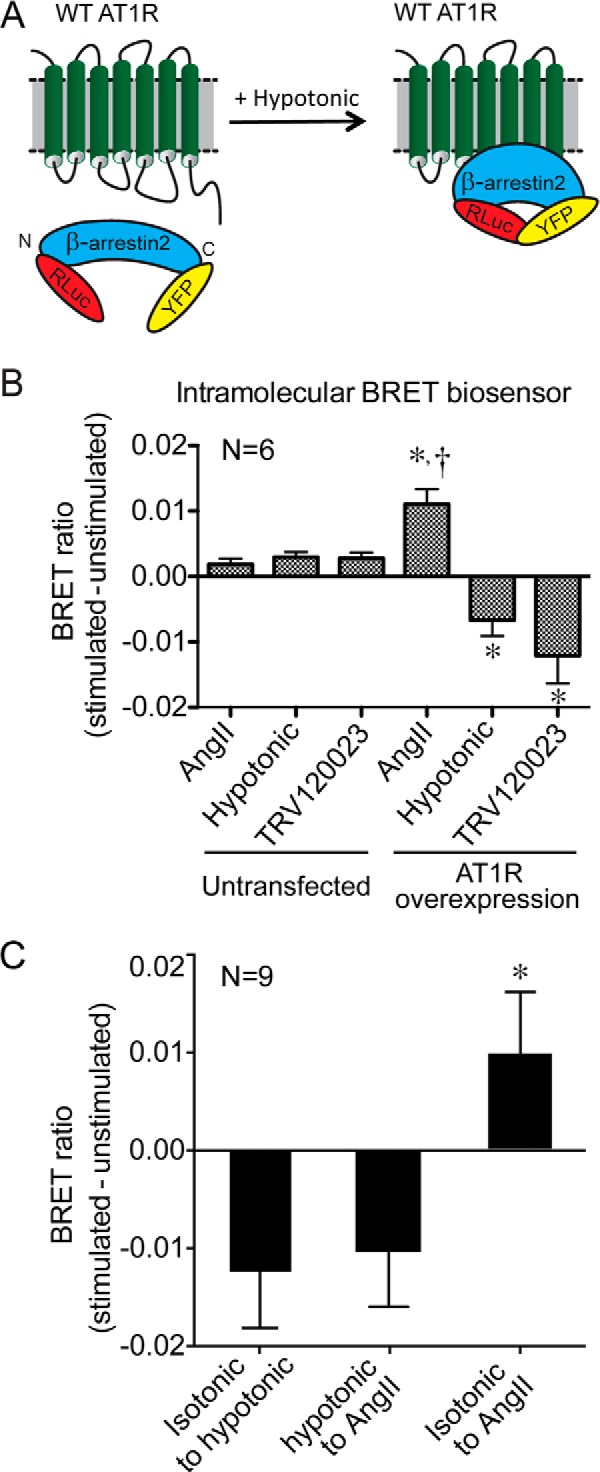
Osmotic stretch and β-arrestin2-biased agonists stabilize similar β-arrestin2 conformations. A, schematic of the intramolecular β-arrestin2 BRET biosensor used to monitor the effect of osmotic stretch on β-arrestin2-activating conformations of the AT1R. B, the balanced agonist AngII-induced changes in the BRET signal were of similar magnitude but in the opposite direction from that induced by osmotic stretch or a β-arrestin-biased agonist. Different conformations of β-arrestin2 were detected with the intramolecular BRET biosensor (Luc-β-arrestin-YFP) before and after AT1R-expressing cells were stimulated with AngII (1 μm) or TRV120023 (1 μm) or hypotonic osmotic stretch (143 mOsm/kg) (n = 6 independent experiments with 1 × 106 to 5 × 105 cells per experiment). *, p < 0.0001, AngII AT1R overexpression versus AngII untransfected; TRV120023 AT1R overexpression versus TRV120023 untransfected; or hypotonic osmotic stretch AT1R overexpression versus hypotonic osmotic stretch untransfected. †, p < 0.0001, AngII AT1R transfected versus either hypotonic osmotic stretch AT1R transfected or TRV120023 AT1R transfected. C, AT1R-overexpressing cells exposed to hypotonic conditions from isotonic conditions (isotonic → hypotonic) showed a negative deflection in the BRET signal. The addition of the balanced agonist AngII (1 μm) to AT1R-overexpressing cells under hypotonic conditions (hypotonic → AngII) continued to show a negative deflection in the BRET signal. In contrast, the addition of AngII (1 μm) to cells in isotonic media (isotonic → AngII) showed a positive deflection in the BRET signal (n = 9 independent experiments for each group). *, p < 0.05, isotonic → AngII versus either hypotonic → AngII or isotonic → hypotonic, one-way ANOVA with Holm-Sidak test. Hypotonic osmotic stretch (143 mOsm/kg).
Osmotic Stretch Promotes Active Conformations of the AT1R to Signal through β-Arrestin
The observation that osmotic stretch favors the binding and signaling of β-arrestin2-biased agonists is supported by our previous report that osmotic stretch promotes signaling in a β-arrestin2-dependent manner (6, 30). To more rigorously test this notion, we used the AT1R-β-arrestin2 and AT1R-Gq fusion proteins to achieve maximal transducer coupling and then measured ERK1/2 phosphorylation in HEK 293 cells stably expressing these receptor fusions before and after treatment with maximal hypotonicity (143 mOsm/kg) or AngII (1 μm) (Fig. 5, A and B). Compared with unstimulated cells, osmotic stretch significantly increased ERK1/2 phosphorylation by 2.3-fold in cells expressing the WT AT1R and by 3.8-fold in cells expressing the AT1R-β-arrestin2 fusion (p < 0.05; Fig. 5A). Notably, ERK1/2 phosphorylation in AT1R-β-arrestin2 cells exposed to osmotic stretch approached that of the full agonist AngII (4.3-fold). In contrast, osmotic stretch in cells expressing the AT1R-Gq fusion showed no significant increase in ERK1/2 phosphorylation over unstimulated conditions (Fig. 5B). This difference between the osmotic stretch-induced ERK1/2 response for the AT1R-β-arrestin2 (3.8-fold) and AT1R-Gq fusions (1.3-fold) is even more striking given the comparable expression levels and similar robust responses for AngII at both receptor fusions.
FIGURE 5.
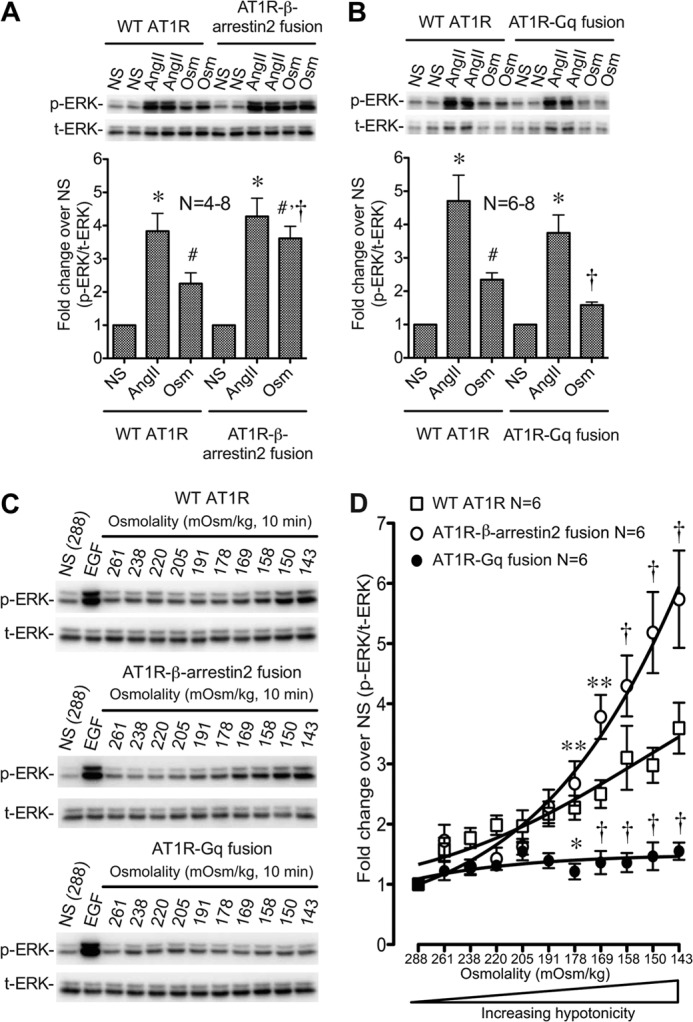
Osmotic stretch promotes active conformations of the AT1R to activate ERK1/2 through β-arrestin. Enhanced ERK1/2 signaling by osmotic stretch in cells stably expressing the AT1R-β-arrestin2 fusion proteins is shown. A, ERK signaling of AT1R-β-arrestin2 fusion following treatment with AngII (1 μm, 10 min) or osmotic stretch (143 mOsm/kg, 10 min). Increased ERK1/2 phosphorylation is observed for the AT1R-β-arrestin2 fusion compared with AT1R-WT under conditions of osmotic stretch. B, ERK1/2 signaling of AT1R-Gq fusion following treatment with AngII (1 μm, 10 min) or osmotic stretch (143 mOsm/kg, 10 min). No increase in ERK1/2 phosphorylation is observed in cells stably expressing AT1R-Gq fusion protein under osmotic stretch conditions. NS, nonstimulation; Osm, osmotic stretch. Summary data in A and B represent the means ± S.E. of 4–8 and 6–8 experiments, respectively. *, p < 0.01, treatment with AngII, compared with the control nontreatment in the same group; #, p < 0.05, treatment with osmotic stretch, compared with the control nontreatment in the same group; †, p < 0.05 osmotic stretch of the AT1R-β-arrestin2 fusion or AT1R-Gq fusion compared with osmotic stretch of the WT AT1R (one-way ANOVA). C, representative immunoblots of ERK1/2 phosphorylation in response to increasing osmotic stretch in cells stably expressing WT AT1R, AT1R-β-arrestin2 fusion, and AT1R-Gq fusion receptors. Cells were stimulated with increasing hypotonic osmotic stretch for 10 min. NS, nonstimulation; EGF, cells are stimulated by 10 ng/ml EGF for 10 min. D, ERK1/2 phosphorylation was significantly enhanced in AT1R-β-arrestin2 cells over a broad range of hypotonicity compared with WT AT1Rs and completely abrogated in HEK 293 stably expressing AT1R-Gq fusion proteins (n = 6 independent experiments for each receptor and each hypotonic condition). p < 0.0001 for interaction between all groups by two-way ANOVA. *, p < 0.05; **, p < 0.01; †, p < 0.0001 either AT1R-β-arrestin2 fusion or AT1R-Gq fusion versus WT AT1R by two-way ANOVA and Bonferroni correction for multiple comparisons. NS, nonstimulation (n = 6).
To further investigate the requirement for β-arrestin2 in mediating the response to osmotic stretch, we stimulated cells stably expressing WT AT1R, AT1R-β-arrestin2, or AT1R-Gq with a dose range of hypotonicity (288–143 mOsm/kg) (Fig. 5, C and D). ERK1/2 phosphorylation in response to the control ligand EGF was similar across all three cell lines (6.42-, 6.85-, and 6.47-fold for AT1R, AT1R-β-arrestin2, and AT1R-Gq, respectively). Compared with the WT AT1R, increases in ERK1/2 phosphorylation were significantly enhanced in AT1R-β-arrestin2 cells over a broad range of hypotonicity (Fig. 5D). However, despite the local excess of fused Gq, the ERK1/2 response in cells overexpressing the AT1R-Gq fusion was completely abrogated compared with both WT AT1R and AT1R-β-arrestin2-expressing cells over the entire range of osmolality (Fig. 5D). We next measured the inhibitory effects of the AT1R-Gq fusion protein by measuring ERK1/2 phosphorylation downstream of the WT AT1R and the AT1R-Gq fusion over a broad range of AngII concentrations. Consistent with our results at the AT1R-β-arrestin2 fusion protein (Fig. 3B), osmotic stretch significantly increased AngII Emax without significantly affecting AngII potency in WT AT1R cells (Fig. 6, A and B). No effect of hypotonicity on AngII Emax or EC50 was observed in cells expressing the AT1R-Gq fusion protein (Fig. 6, C and D). These data support our contention that in response to osmotic stretch the AT1R uses β-arrestin to induce transducer-specific signaling in the cell.
The Angiotensin Receptor Blocker Telmisartan Inhibits Osmotic Stretch-induced AT1R Signaling
We believe that osmotic stretch stabilizes a subset of β-arrestin2-specific active AT1R conformations that are distinct from those stabilized by the balanced agonist AngII. To confirm that stretch induces active conformations of the AT1R, we tested the ability for the inverse agonist telmisartan to inhibit ERK1/2 phosphorylation in cells treated with EGF (10 ng/ml), AngII (1 μm), or osmotic stretch (143 mOsm/kg). Consistent with the specific blockade of the AT1R, telmisartan (10 μm) had no effect on ERK1/2 phosphorylation induced by EGF activation of endogenous receptor tyrosine kinases (10 ng/ml; Fig. 7, A and B). By contrast, telmisartan completely blocked ERK1/2 phosphorylation induced by AngII and hypotonic osmotic stretch (143 mOsm/kg). Fig. 7C provides further evidence that stretch exerts its effect through active AT1R conformations. In these experiments, telmisartan inhibited stretch-induced ERK1/2 phosphorylation with a half-maximal effect (pIC50 = 7.54 ± 0.15) in close agreement with its affinity for the AT1R (pKi = 8.77 ± 0.03; Table 1). Together, these data support our hypothesis that mechanical stretch-induced signaling is indeed mediated through active conformations of the AT1R.
FIGURE 7.
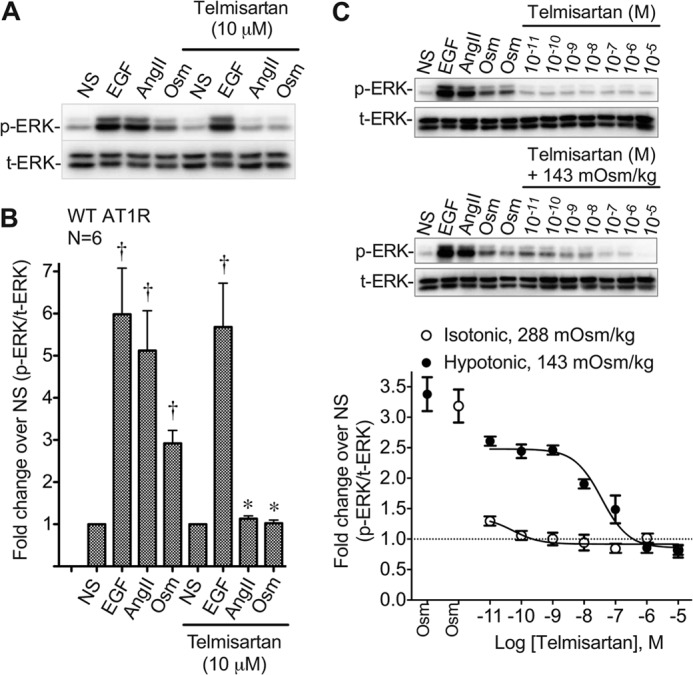
Telmisartan blocks osmotic stretch-induced AT1R signaling. Antagonist telmisartan blocks osmotic stretch induced ERK1/2 signaling in HEK 293 stably expressing AT1R receptor. A, representative immunoblot showing ERK1/2 phosphorylation in HEK 293 stably expressing WT AT1R in response to stimulation with AngII, EGF, osmotic stretch, with or without the addition of telmisartan (10 μm). NS, nonstimulation. B, quantification of ERK1/2 phosphorylation from HEK 293 stable expressing WT AT1R under stimulation with AngII, EGF, osmotic stretch, with or without the addition of telmisartan. †, p < 0.01 EGF, AngII, or Osm versus nonstimulation (NS); *, p < 0.05 AngII or Osm with telmisartan versus AngII or Osm without telmisartan by one-way ANOVA (n = 6 independent experiments). C, representative immunoblot of four experiments showing dose response of ERK1/2 phosphorylation from HEK 293 stable expressing WT AT1R under treatment with telmisartan and osmotic stretch at 143 mOsm/kg. Telmisartan-reduced ERK phosphorylation is expressed as a percentage of maximum osmotic stretch under different concentrations of telmisartan with or without osmotic stretch at 143 mOsm/kg. Telmisartan + hypotonic 143 mOsm/kg (Emax = 2.49 ± 0.07, LogIC50 = −7.54 ± 0.15 nm). p < 0.0001 for interaction between two groups by two-way ANOVA with Bonferroni correction for multiple comparisons. The data represent the means ± S.E. of four experiments. NS, nonstimulation; EGF, positive control treatment with EGF at 10 ng/ml for 10 min; AngII, 1 μm for 10 min; Osm, osmotic stretch at 143 mOsm/kg for 10 min.
Sodium Ions Are Not Required for Osmotic Stretch-induced ERK1/2 Phosphorylation
To further confirm that the enhancement of AT1R signaling by osmotic stretch is mediated through a subset of β-arrestin2-stabilized active AT1R conformations and not due to changes in Na+ cations (27), we tested the effect of osmotic stretch on ERK1/2 phosphorylation under normal and Na+-free culture conditions in intact cells. Following stimulation with either AngII or hypotonic osmotic stress, ERK1/2 phosphorylation increased to a similar extent in cells incubated in either MEM, Na+-containing buffer, or Na+-free buffer (Fig. 8). These data support our contention that the effect of osmotic stretch on AT1R signaling is most likely mediated through allosteric modulation of the receptor and not due to changes in the Na+ concentration the plasma membrane.
FIGURE 8.

Osmotic stretch-induced ERK phosphorylation in Na+-free buffer. A, representative immunoblot showing ERK phosphorylation in HEK 293 stably expressing WT AT1R in response to stimulation with AngII, osmotic stretch, with or without the addition of telmisartan (Tel), in MEM, sodium containing buffer, or sodium-free buffer. B, quantification of three experiments showing ERK phosphorylation from HEK 293 stable expressing WT AT1R under stimulation with AngII, osmotic stretch, with or without the addition of telmisartan, in MEM, sodium buffer, or sodium-free buffer. †, p < 0.05 Osm (143 mOsm/kg) versus AngII; *, p < 0.01 Osm (143 mOsm/kg) or AngII versus nonstimulation (288 mOsm/kg) or with telmisartan (one-way ANOVA). There was no significant difference for ERK phosphorylation under stimulation with AngII or osmotic stretch in MEM, sodium buffer, or sodium-free buffer. The data represent the means ± S.E. of three experiments. NS, nonstimulation; AngII, stimulation with AngII at 1 μm for 10 min; Osm, osmotic stretch at 143 mOsm/kg for 10 min.
DISCUSSION
We set out to investigate the molecular mechanism by which mechanical stretch modulates AT1R structure and function by using biased AT1R agonists as novel conformational probes in several pharmacological and biophysical approaches. When tested at AT1R-transducer proteins expressed in intact cells we show that osmotic stretch: 1) induces active conformations of the AT1R and β-arrestin2 that are exclusively stabilized by β-arrestin-biased agonists and 2) functions as an allosteric modulator to selectively enhance β-arrestin-biased ERK1/2 signaling. Our data provide some of the most compelling evidence to date that there is a biased conformation of the AT1R. We show that osmotic stretch acts through the AT1R to induce a conformation of β-arrestin2 similar to that induced by β-arrestin-biased agonists. Although the balanced agonist AngII stimulates the AT1R to couple to and activate G protein signaling, it also robustly recruits β-arrestin. Membrane stretch can also activate the AT1R, but in this case it only recruits and activates β-arrestin signaling, indicating that osmotic stretch induces conformations of the AT1R that activate signaling through β-arrestin but not through Gq. Our data provide molecular level support for the emerging concept that multiple conformations of the angiotensin receptor exist, each unique in its ability to engage transducers (i.e. either Gq, β-arrestin, or both), and that mechanical stretch stabilizes distinct β-arrestin2-activating conformations of the AT1R to activate selective signaling pathways.
Increased Binding Affinity and Potency of the β-Arrestin-biased Agonist TRV120023 by Stretch Provides Mechanistic Insight into AT1R Mechanotransduction
According to the conceptual framework of biased agonism, biased agonists signal disproportionately by stabilizing distinct active GPCR conformations that vary in their abilities to promote transducer coupling and activation (12, 13, 31). Recently, several β-arrestin-biased agonists have been identified that exclusively activate β-arrestin pathways downstream of the AT1R, presumably via stabilizing distinct receptor conformations that do not support Gq coupling and activation (18). In accordance with the allosteric effect linking the simultaneous binding of an agonist and transducer to topologically distinct sites on a receptor (21, 22, 32), β-arrestin-biased agonists such as TRV120023 were shown to bind with high affinity to β-arrestin2-activating conformations of the AT1R (18). Thus, β-arrestin2-biased agonists can function as conformational probes, binding β-arrestin2-activating conformations of the AT1R with high affinity. When used to monitor the conformational changes thought to accompany mechanical stretch of the AT1R (1), we found that osmotic stretch only enhanced the binding affinity of the β-arrestin2-biased agonist TRV120023 at both the WT AT1R and AT1R-β-arrestin2 fusion protein (Fig. 1C). Indeed, despite AngII being a full agonist for β-arrestin2-mediated signaling pathways (12), there was no increase in AngII affinity with osmotic stretch (Fig. 1B). Importantly, we did not observe changes in TRV120023 binding affinity using detergent-purified AT1Rs, suggesting that physical changes to the membrane are required for AT1R activation by stretch. Together with the exclusive ability for TRV120023 to bind preferentially to β-arrestin2-activating states of the AT1R, we contend that membrane stretch stabilizes distinct β-arrestin2-activating conformations of the AT1R.
Our biophysical studies using the intramolecular BRET β-arrestin2 biosensor support this line of reasoning because differences in the biosensor BRET signal between AT1Rs bound to AngII and those bound to TRV120023 indicate that AT1Rs can adopt different β-arrestin2-activating conformations depending on the stimulus. Importantly, the β-arrestin2 biosensor BRET signature in response to osmotic stretch and the β-arrestin2-biased agonist TRV120023 is distinct from that induced by AngII. This is consistent with previous work showing that membrane stretch may induce changes in the biophysical properties of the AT1R (17), which appear structurally distinct from changes typically associated with agonist activation (33). In fact, within the context of a multistate model of GPCR activation, it has been proposed that stretch stabilizes the AT1R in a different activated state, recently termed Rstretch (17).
It is interesting to speculate that a general mechanism could account for the β-arrestin-biased modulatory effects of stretch given that this effect is not unique to AT1Rs. Recently, stretch-induced activation of the apelin receptor was shown to favor β-arrestin pathways and inhibit G protein activation (7). It is currently thought that integral membrane proteins perceive stretch through different mechanisms. According to the “tethered” model, intracellular and/or extracellular anchoring to other proteins acts as a gating spring to impart mechanosensitivity to membrane proteins (1). Alternatively, the “membrane” model contends that an altered lateral pressure profile at the protein/phospholipid bilayer boundary directly translates membrane stretch into conformational changes (1). It remains a possibility that relaxing the AT1R through decreased lateral pressure promotes β-arrestin-activating conformations.
Although the precise mechanism of how the cell membrane transmits forces to modulate the conformation of a GPCR is not known, recent evidence for mechanosensitive ion channels has shown that the lipid bilayer is critically involved in opening and closing of these channels with membrane stretch (34). Proteins embedded in lipid bilayers are subject to strong localized forces that can be altered by membrane stretch resulting in an energetically more favorable conformation for the channel protein to assume (35). The change in the force profile at the lipid-channel interface may be the ultimate mechanoenergetic trigger for the ion channel to adopt an activated configuration (35). Although our studies do not address this concept directly, we propose that intrinsic forces between the lipid bilayer and the AT1R under conditions of membrane stretch promote the AT1R to adopt an energetically favorable conformation stabilized through its interaction with β-arrestin.
Mechanical Stretch Allosterically Modulates Angiotensin II Type 1 Receptors
The activity of GPCRs can be modulated by a diverse spectrum of drugs ranging from full agonists to partial agonists, antagonists, and inverse agonists by their binding to the orthosteric pocket of the receptor. It is now appreciated that ligands can also bind to sites on the GPCR that are physically distinct from the orthosteric pocket, termed allosteric, and modulate the signaling properties of GPCRs (36). Because both allosteric and orthosteric sites are structurally linked, allosteric agonists have the capacity to modulate the binding and/or signaling of orthosteric ligands (36–38). For instance, Zn2+ acts as a positive allosteric modulator at the β2-adrenergic receptor to increase agonist affinity through the bridging of the cytoplasmic extensions of TM5 and TM6 and binding to His-269, Cys-265, and Glu-225 (39). Although allosteric modulators bind to distinct regions of the receptor, the recent crystal structure of active muscarinic acetylcholine receptor bound to an allosteric modulator noted that the actual conformational change induced by the binding of an allosteric modulator is quite mild compared with that induced by the orthosteric agonist alone (40). More generally, given the large number of conformational states available to a GPCR, and the notion that allosteric modulators modulate orthosteric ligand efficacy, our data are consistent with the concept that membrane stretch is functioning to allosterically modulate or induce β-arrestin-biased active conformations and signaling of the AT1R.
Molecular Mechanism for Stretch-mediated β-Arrestin Signaling
It is now well established that β-arrestin not only serves to desensitize G protein signaling but also leads to signaling in its own right by activating a variety of intracellular signaling pathways (10, 41–43). Indeed, as we have recently shown, β-arrestin-dependent signaling by the AT1R can be activated by mechanical stretch and does not require AngII (6, 8). However, the precise molecular nature of this AT1R-β-arrestin interaction under conditions of membrane stretch is not well defined. Here we propose that mechanical stretch activates AT1Rs in a manner consistent with constitutive activation of a GPCR. Constitutively active GPCRs have the following characteristics (21, 44–47): 1) their activation is ligand-independent, 2) this ligand-independent activation is inhibited by antagonists which preferentially stabilize inactive conformations (i.e. inverse agonists), 3) agonists bind to these receptors with increased affinities, and 4) the conformational constraints that suppress spontaneous signaling are reduced in these receptors, thereby allowing agonists to signal with increased potency. Previous studies have reported that stretch-activated AT1Rs exhibit some of these characteristics including ligand independence (6, 8, 17) and the ability for inverse agonists such as candesartan (8, 17) and losartan (6) to block this stretch-mediated activation. Although we show in this study that osmotic stretch activates ERK1/2 in the absence of exogenous AngII and is inhibited by the inverse agonist telmisartan (Fig. 7), the strongest support for stretch-mediated constitutive activation of the AT1R is the observation that osmotic stretch increases the binding affinity of the β-arrestin-biased agonist TRV120023 for both the WT AT1R (i.e. uncoupled) and AT1R-β-arrestin2 fusion protein (i.e. β-arrestin2-coupled) (Fig. 1). Here, the pattern of affinity shifts produced by osmotic stretch were similar to those observed for agonist binding to the constitutively active mutant β2AR (21) and suggest to us that osmotic stretch removes the conformational constraints repressing basal AT1R activity. Such reductions in the energy barrier to receptor activation are known to have functional (i.e. signaling) consequences. Indeed, we show that osmotic stretch significantly increased the potency of the biased agonist TRV120023 for activating ERK in cells expressing the AT1R-β-arrestin fusion protein (Fig. 3D), without affecting potency for the balanced agonist AngII (Fig. 3B) and is consistent with the increase in agonist potency observed for a constitutive active receptor (21, 46). Lastly, in addition to the biophysical forces applied to the lipid membrane under stretch conditions, the composition of the membrane microdomain may influence trafficking and signal transduction of the receptor with mechanical stretch (48). Given that cell membrane proteins and lipid bilayers form compartmentalized domains (rafts/caveolae) with different biophysical properties, mechanical stretch may differentially influence the conformational state of AT1Rs residing in these compartments (49).
Based on our data, we speculate that the unique AT1R conformations induced by osmotic stretch recruit a unique subset of GRKs to result in a specific phosphorylation pattern or “bar code” on the C-terminal tail of the receptor. At the level of the transducer, these different phosphorylation bar codes stabilize distinct, active conformations of β-arrestin transducers to promote unique signaling profiles (50, 51). This is supported by our earlier study showing that osmotic stretch induced ERK activation was dependent only on GRK5/6 (6) and studies using the β2AR showing that β-arrestin function was dependent on the phosphorylation pattern or bar code of the receptor (50, 51). Future studies using mass spectrometry phosphoproteomic-based approaches will determine whether osmotic stretch induces unique phosphorylation patterns of the AT1R compared with AngII.
Recent high resolution crystal structure information for the δ-opioid receptor has revealed the presence of a sodium ion centrally located in its architecture and its role in mediating allosteric control of opioid receptor function (27). We considered the possibility that our use of hypotonic conditions to induce membrane stretch could be effecting allostery of the receptor by altering the local sodium ion concentration. To address this, we used two strategies: 1) whether hypotonic media had any effect on ligand binding to detergent-purified AT1Rs and 2) whether sodium-free buffer altered the level of ERK phosphorylation with osmotic stretch in intact cells. In both cases, we show that changes in the sodium concentration do not alter the behavior of the AT1R either by changing its ligand binding affinity or activation of β-arrestin-mediated ERK signaling.
The physiological importance of stretch-activated AT1R as it relates to human physiology has been explored in recent years. Recent work has shown that ligand-independent stretch-activated AT1Rs are a mechanism for control of myogenic constriction in vascular smooth muscle cells (52) and in the vessel wall of mesenteric and renal resistance arteries (11). Similar data have recently been generated for shear stress-induced AT1R activation, which can activate both G protein-dependent and -independent signals (53). Mechanoactivation of AT1Rs under conditions of pressure overload may contribute to cardiac hypertrophy (8) and the stimulation of myocyte prosurvival signaling pathways (6, 30). Lastly, AT1Rs appear to be involved in the osmoregulation of thirst and salt appetite (54), which may be mediated, in part, by β-arrestin because salt intake was stimulated by the β-arrestin-biased AngII analog Sar(1), Ile(4),Ile(8)-AngII (SII) (55).
In conclusion, we adopted a novel strategy to provide molecular level insight into the conformational changes driving β-arrestin-biased AT1R mechanotransduction. We show that osmotic stretch selectively increases the affinity and potency of a β-arrestin-biased agonist, as well as stabilizes β-arrestin2 conformations that are distinct from those stabilized by AngII. These findings support a ligand-independent mechanism whereby mechanical stretch functions as an allosteric modulator to stabilize specific β-arrestin-activating conformations of the AT1R to engender β-arrestin-biased signaling. That mechanical stretch induces a conformational state of the AT1R to promote high affinity binding of the transducer β-arrestin indicates that biased ligands, which recognize this complex, may be effective in enhancing protective β-arrestin-mediated signaling in the heart under conditions of hemodynamic overload (6, 30).
Acknowledgments
We thank Roma Agrawal for reading the manuscript and Dr. Arun Shukla for assistance with the BRET biosensor experiments. Dr. Dean P. Staus kindly provided purified AT1R for control experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants HL56687 and HL75443 (to H. A. R.), HL16037 and HL70631 (to R. J. L.), and T32 HL007101 to (W. T.).
- GPCR
- G protein-coupled receptor
- AT1R
- angiotensin II type 1 receptor
- AngII
- angiotensin II
- BRET
- bioluminescence resonance energy transfer
- ANOVA
- analysis of variance
- MEM
- minimum essential medium.
REFERENCES
- 1. Storch U., Mederos y Schnitzler M., Gudermann T. (2012) G protein-mediated stretch reception. Am. J. Physiol. Heart Circ. Physiol. 302, H1241–H1249 [DOI] [PubMed] [Google Scholar]
- 2. Zablocki D., Sadoshima J. (2013) Solving the cardiac hypertrophy riddle: The angiotensin II-mechanical stress connection. Circ. Res. 113, 1192–1195 [DOI] [PubMed] [Google Scholar]
- 3. Chalfie M. (2009) Neurosensory mechanotransduction. Nat. Rev. Mol. Cell Biol. 10, 44–52 [DOI] [PubMed] [Google Scholar]
- 4. Osol G., Brekke J. F., McElroy-Yaggy K., Gokina N. I. (2002) Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am. J. Physiol. Heart Circ. Physiol. 283, H2260–H2267 [DOI] [PubMed] [Google Scholar]
- 5. Mederos y Schnitzler M., Storch U., Gudermann T. (2011) AT1 receptors as mechanosensors. Curr. Opin. Pharmacol. 11, 112–116 [DOI] [PubMed] [Google Scholar]
- 6. Rakesh K., Yoo B., Kim I. M., Salazar N., Kim K. S., Rockman H. A. (2010) β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 3, ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scimia M. C., Hurtado C., Ray S., Metzler S., Wei K., Wang J., Woods C. E., Purcell N. H., Catalucci D., Akasaka T., Bueno O. F., Vlasuk G. P., Kaliman P., Bodmer R., Smith L. H., Ashley E., Mercola M., Brown J. H., Ruiz-Lozano P. (2012) APJ acts as a dual receptor in cardiac hypertrophy. Nature 488, 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou Y., Akazawa H., Qin Y., Sano M., Takano H., Minamino T., Makita N., Iwanaga K., Zhu W., Kudoh S., Toko H., Tamura K., Kihara M., Nagai T., Fukamizu A., Umemura S., Iiri T., Fujita T., Komuro I. (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 6, 499–506 [DOI] [PubMed] [Google Scholar]
- 9. Rajagopal S., Rajagopal K., Lefkowitz R. J. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schleifenbaum J., Kassmann M., Szijártó I. A., Hercule H. C., Tano J. Y., Weinert S., Heidenreich M., Pathan A. R., Anistan Y. M., Alenina N., Rusch N. J., Bader M., Jentsch T. J., Gollasch M. (2014) Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ. Res. 115, 263–272 [DOI] [PubMed] [Google Scholar]
- 12. Rajagopal S., Ahn S., Rominger D. H., Gowen-MacDonald W., Lam C. M., Dewire S. M., Violin J. D., Lefkowitz R. J. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 80, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kahsai A. W., Xiao K., Rajagopal S., Ahn S., Shukla A. K., Sun J., Oas T. G., Lefkowitz R. J. (2011) Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat. Chem. Biol. 7, 692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okada T., Sugihara M., Bondar A. N., Elstner M., Entel P., Buss V. (2004) The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J. Mol. Biol. 342, 571–583 [DOI] [PubMed] [Google Scholar]
- 15. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 16. Chachisvilis M., Zhang Y. L., Frangos J. A. (2006) G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 103, 15463–15468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yasuda N., Miura S., Akazawa H., Tanaka T., Qin Y., Kiya Y., Imaizumi S., Fujino M., Ito K., Zou Y., Fukuhara S., Kunimoto S., Fukuzaki K., Sato T., Ge J., Mochizuki N., Nakaya H., Saku K., Komuro I. (2008) Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep. 9, 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strachan R. T., Sun J. P., Rominger D. H., Violin J. D., Ahn S., Rojas Bie Thomsen A., Zhu X., Kleist A., Costa T., Lefkowitz R. J. (2014) Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem. 289, 14211–14224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shukla A. K., Violin J. D., Whalen E. J., Gesty-Palmer D., Shenoy S. K., Lefkowitz R. J. (2008) Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 9988–9993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fork C., Bauer T., Golz S., Geerts A., Weiland J., Del Turco D., Schömig E., Gründemann D. (2011) OAT2 catalyses efflux of glutamate and uptake of orotic acid. Biochem. J. 436, 305–312 [DOI] [PubMed] [Google Scholar]
- 21. Samama P., Cotecchia S., Costa T., Lefkowitz R. J. (1993) A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 268, 4625–4636 [PubMed] [Google Scholar]
- 22. De Lean A., Stadel J. M., Lefkowitz R. J. (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J. Biol. Chem. 255, 7108–7117 [PubMed] [Google Scholar]
- 23. Gurevich V. V., Pals-Rylaarsdam R., Benovic J. L., Hosey M. M., Onorato J. J. (1997) Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J. Biol. Chem. 272, 28849–28852 [DOI] [PubMed] [Google Scholar]
- 24. Seifert R., Wenzel-Seifert K., Kobilka B. K. (1999) GPCR-Gα fusion proteins: molecular analysis of receptor-G-protein coupling. Trends Pharmacol. Sci. 20, 383–389 [DOI] [PubMed] [Google Scholar]
- 25. Sanni S. J., Hansen J. T., Bonde M. M., Speerschneider T., Christensen G. L., Munk S., Gammeltoft S., Hansen J. L. (2010) β-Arrestin 1 and 2 stabilize the angiotensin II type I receptor in distinct high-affinity conformations. Br. J. Pharmacol. 161, 150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerwins P., Nordstedt C., Fredholm B. B. (1990) Characterization of adenosine A1 receptors in intact DDT1 MF-2 smooth muscle cells. Mol. Pharmacol. 38, 660–666 [PubMed] [Google Scholar]
- 27. Fenalti G., Giguere P. M., Katritch V., Huang X. P., Thompson A. A., Cherezov V., Roth B. L., Stevens R. C. (2014) Molecular control of δ-opioid receptor signalling. Nature 506, 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katritch V., Fenalti G., Abola E. E., Roth B. L., Cherezov V., Stevens R. C. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Charest P. G., Terrillon S., Bouvier M. (2005) Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep. 6, 334–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim K. S., Abraham D., Williams B., Violin J. D., Mao L., Rockman H. A. (2012) β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 303, H1001–H1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deupi X., Kobilka B. (2007) Activation of G protein-coupled receptors. Adv. Protein Chem. 74, 137–166 [DOI] [PubMed] [Google Scholar]
- 32. Onaran H. O., Costa T. (2012) Where have all the active receptor states gone? Nat. Chem. Biol. 8, 674–677 [DOI] [PubMed] [Google Scholar]
- 33. Kobilka B. K. (2011) Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 32, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nilius B., Honoré E. (2012) Sensing pressure with ion channels. Trends Neurosci. 35, 477–486 [DOI] [PubMed] [Google Scholar]
- 35. Kung C. (2005) A possible unifying principle for mechanosensation. Nature 436, 647–654 [DOI] [PubMed] [Google Scholar]
- 36. May L. T., Leach K., Sexton P. M., Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51 [DOI] [PubMed] [Google Scholar]
- 37. Christopoulos A., Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 54, 323–374 [DOI] [PubMed] [Google Scholar]
- 38. Keov P., Sexton P. M., Christopoulos A. (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60, 24–35 [DOI] [PubMed] [Google Scholar]
- 39. Swaminath G., Lee T. W., Kobilka B. (2003) Identification of an allosteric binding site for Zn2+ on the β2 adrenergic receptor. J. Biol. Chem. 278, 352–356 [DOI] [PubMed] [Google Scholar]
- 40. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., Garcia K. C., Wess J., Kobilka B. K. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Violin J. D., DeWire S. M., Yamashita D., Rominger D. H., Nguyen L., Schiller K., Whalen E. J., Gowen M., Lark M. W. (2010) Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 335, 572–579 [DOI] [PubMed] [Google Scholar]
- 42. Zimmerman B., Beautrait A., Aguila B., Charles R., Escher E., Claing A., Bouvier M., Laporte S. A. (2012) Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal. 5, ra33. [DOI] [PubMed] [Google Scholar]
- 43. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., Rockman H. A. (2007) β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cotecchia S., Exum S., Caron M. G., Lefkowitz R. J. (1990) Regions of the α1-adrenergic receptor involved in coupling to phosphatidylinositol hydrolysis and enhanced sensitivity of biological function. Proc. Natl. Acad. Sci. U.S.A. 87, 2896–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gether U., Ballesteros J. A., Seifert R., Sanders-Bush E., Weinstein H., Kobilka B. K. (1997) Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J. Biol. Chem. 272, 2587–2590 [DOI] [PubMed] [Google Scholar]
- 46. Smit M. J., Vischer H. F., Bakker R. A., Jongejan A., Timmerman H., Pardo L., Leurs R. (2007) Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu. Rev. Pharmacol. Toxicol. 47, 53–87 [DOI] [PubMed] [Google Scholar]
- 47. Bond R. A., Leff P., Johnson T. D., Milano C. A., Rockman H. A., McMinn T. R., Apparsundaram S., Hyek M. F., Kenakin T. P., Allen L. F. (1995) Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the β2-adrenoceptor. Nature 374, 272–276 [DOI] [PubMed] [Google Scholar]
- 48. Nair K. S., Balasubramanian N., Slepak V. Z. (2002) Signal-dependent translocation of transducin, RGS9–1-Gβ5L complex, and arrestin to detergent-resistant membrane rafts in photoreceptors. Curr. Biol. 12, 421–425 [DOI] [PubMed] [Google Scholar]
- 49. Barnett-Norris J., Lynch D., Reggio P. H. (2005) Lipids, lipid rafts and caveolae: their importance for GPCR signaling and their centrality to the endocannabinoid system. Life Sci. 77, 1625–1639 [DOI] [PubMed] [Google Scholar]
- 50. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wisler J. W., Xiao K., Thomsen A. R., Lefkowitz R. J. (2014) Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mederos y Schnitzler M., Storch U., Meibers S., Nurwakagari P., Breit A., Essin K., Gollasch M., Gudermann T. (2008) Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 27, 3092–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barauna V. G., Magalhaes F. C., Campos L. C., Reis R. I., Kunapuli S. P., Costa-Neto C. M., Miyakawa A. A., Krieger J. E. (2013) Shear stress-induced Ang II AT1 receptor activation: G-protein dependent and independent mechanisms. Biochem. Biophys. Res. Commun. 434, 647–652 [DOI] [PubMed] [Google Scholar]
- 54. Johnson A. K., Cunningham J. T., Thunhorst R. L. (1996) Integrative role of the lamina terminalis in the regulation of cardiovascular and body fluid homeostasis. Clin. Exp. Pharmacol. Physiol. 23, 183–191 [DOI] [PubMed] [Google Scholar]
- 55. Daniels D., Yee D. K., Faulconbridge L. F., Fluharty S. J. (2005) Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology 146, 5552–5560 [DOI] [PubMed] [Google Scholar]