

Background: Stalled replication forks are foci for genomic instability.
Results: Both RecG and RuvAB can regress stalled forks; however, RuvAB completely unwinds the nascent DNA, whereas RuvC cleaves the Holliday junctions formed by RecG.
Conclusion: RecG and RuvAB activities are distinct.
Significance: Replication fork regression is a major step in processing stalled forks.
Keywords: DNA Enzyme, DNA Recombination, DNA Repair, DNA Replication, Genomic Instability
Abstract
The orderly progression of replication forks formed at the origin of replication in Escherichia coli is challenged by encounters with template damage, slow moving RNA polymerases, and frozen DNA-protein complexes that stall the fork. These stalled forks are foci for genomic instability and must be reactivated. Many models of replication fork reactivation invoke nascent strand regression as an intermediate in the processing of the stalled fork. We have investigated the replication fork regression activity of RecG and RuvAB, two proteins commonly thought to be involved in the process, using a reconstituted DNA replication system where the replisome is stalled by collision with leading-strand template damage. We find that both RecG and RuvAB can regress the stalled fork in the presence of the replisome and SSB; however, RuvAB generates a completely unwound product consisting of the paired nascent leading and lagging strands, whereas RuvC cleaves the Holliday junction generated by RecG-catalyzed fork regression. We also find that RecG stimulates RuvAB-catalyzed regression, presumably because it is more efficient at generating the initial Holliday junction from the stalled fork.
Introduction
Accurate transmission of the genetic information requires complete duplication of the chromosomal DNA content of the cell during each cellular growth cycle. However, the simple idea that replication forks would form at origins of DNA replication and proceed without impairment to copy the chromosomes in the cell has not held. The first suggestion that replication forks might be inactivated at the site of a strand break was put forward by Hanawalt (1), in considering the consequences of UV damage to the DNA. Skalka (2) subsequently suggested that pre-existing nicks in the template could cause replication fork inactivation and that the inactivated fork might be repaired by recombinational properties. Our finding that inactivation of the PriA replication protein constitutively induced the SOS response (3) led us to suggest that SOS induction was a result of the inability to restart replication at replication forks that had been inactivated by endogenous roadblocks on the DNA template (4). Subsequent studies by many laboratories have elaborated the genetic pathways involved in the processing and repair of stalled replication forks (for reviews, see Refs. 5–11). A central feature of many of the models described is one in which the nascent strands at the stalled fork have been unwound from the respective parental template strand and paired together. This annealing of the nascent DNA causes the rewinding of the parental strands and regression of the growth point of the replication fork (Fig. 1), a process variously termed either replication fork regression or reversal (RFR).3
FIGURE 1.

Reaction scheme and possible outcomes. i, template DNA. Black line, lagging-strand template; green line, leading-strand template; thin red arrow, direction of replication. The strand that is methylated is marked CH3. Replication is initiated on the supercoiled template (ii) in the absence of a topoisomerase, leading to the accumulation of an early replication intermediate where replication has been stalled because of the accumulation of positive supercoils (iii). The replication forks in the early intermediate are released by the addition of EcoRI, and labeled precursor is added at the same time. Stalled forks are accumulated by stopping DNA synthesis by the addition of ddNTPs and digesting the DNA with PvuI (iv). If the stalled forks are regressed by the addition of RecG and RuvAB, the regressed fork may be cut by the HJ resolvase RuvC (v) to give two classes of products (vi and vii), termed CP1 and CP2, that are roughly the size of full-length EcoRI-PvuI DNA and stall DNA, respectively, but differ in their strand composition (thick red arrow, nascent leading-strand DNA; thick blue arrows, nascent lagging-strand DNA). Alternatively, RuvC may not cut the regressed fork, and the nascent leading and lagging strands may be unwound from their respective template strands to give a complete NDD the same size as the stall product (viii), which will be labeled, and the duplex template DNA (ix), which will not be labeled.
RFR generates a four-stranded DNA structure similar to the Holliday junction (HJ) (see Fig. 1) formed during recombination between two DNA molecules. RFR was first proposed as a means of providing parental duplex DNA at forks stalled by template damage that flanked the damage site in order to allow repair of the damage by either nucleotide or base excision repair (12, 13). In a topologically constrained molecule, positive supercoiling will provide a strong driving force for spontaneous RFR (14, 15). With respect to rescue of the stalled fork (5–11), the HJ formed can be cleaved by the HJ resolvase, RuvC (16), resulting in rescue of replication by homologous recombination-directed DNA replication, whereby a recombinant joint molecule is formed between the two sister chromosomes, and the replisome is reloaded by a PriA-directed pathway downstream of the damage. In cases where the 5′-end of the last Okazaki fragment is further downstream than the 3′-end of a stalled nascent leading strand, a template strand switch can occur in the regressed intermediate, extending the nascent leading strand, which, upon resetting of the fork back to its original position, will result in bypass of the lesion. Similarly, regression can allow access of various repair proteins, correction of the lesion, and origin-independent restart of replication after the regressed fork is reset to its original position.
There are three proteins that have been suggested to be the agents that catalyze RFR: RecG, a 3′ → 5′ DNA helicase (17) that also branch migrates HJs (18, 19); the HJ branch migration protein RuvAB (19); and the strand exchange protein RecA (20–22). Conclusions about which protein acts when vary, depending on the type of damage that is used to arrest the replication fork. RuvAB-catalyzed RFR has been invoked in the rescue of replication forks stalled by either denaturation of temperature-sensitive components of the DNA polymerase III holoenzyme (Pol III HE), the cellular replicase (23) (the α and ψ subunits (24)), or in mutants lacking the Rep DNA helicase (24), which causes slower replication fork progression (25) because of reduced ability to clear proteins from in front of the replication fork (26). RecG-catalyzed fork regression has been suggested to occur in UV-irradiated cells after replication forks stall, primarily because they encounter an RNA polymerase that has stalled at a leading-strand cyclopyrimidine dimer (CPD) (27, 28), whereas RecA-catalyzed regression is proposed to occur after stalling forks by denaturation of a thermosensitive replication fork DNA helicase, DnaB (29, 30), and also possibly after UV irradiation (31).
RecG has been shown to catalyze RFR using small oligonucleotide substrates (15, 18, 27, 32, 33), a large M13-based substrate (34), replication forks stalled by positive supercoiling in a reconstituted DNA replication system (15), and hairpin substrates by single molecule analysis (35). Similarly, RuvAB fork reversal has also been demonstrated using small oligonucleotide substrates (36–38). The large M13-based substrate has also been used to model RecA-catalyzed RFR (39). Although all of these studies have yielded valuable information on the specifics of DNA substrate utilization by these proteins, they did not model the action of these proteins at a bona fide replication fork stalled by template damage. We have developed a reconstituted replication system that allows examination of the consequences of the collision between a replisome and leading-strand template damage (40, 41). Using this system, we have assessed the activity of RecG, RuvAB, and RecA to catalyze RFR. We find that whereas both RecG and RuvAB can catalyze RFR on stalled forks where replication proteins are still resident, only the HJs formed by RecG are substrates for cleavage by RuvC; RuvAB RFR results in the complete unwinding of the nascent DNA from the template strands and the winding of the nascent DNA strands with each other to form a complete nascent strand duplex DNA. Furthermore, we find that RecG can stimulate RuvAB-catalyzed RFR, as has been suggested previously (27). Our studies with RecA are reported in the accompanying article (42).
EXPERIMENTAL PROCEDURES
DNA Templates, Recombination, and Replication Proteins
The standard DNA template was prepared from pJY1M13 single-stranded DNA (ssDNA) as described using the oligonucleotide 5′-GAATAATGGAAGGG(TT)AGAACCTACCAT-3′ (where (TT) represents the positon of the CPD) as the primer as described previously (40). The 5903 tetrahydrofuran (5903THF) template was prepared using 5′-GAATAATGGAAGGGXTAGAACCTACCAT-3′ (where (X) represents the position of the THF synthetic abasic site) as the primer. Fully methylated and hemimethylated DNA substrates were prepared by digesting replicative form DNA and template DNA with the PstI and PvuI restriction enzymes, respectively. Unmethylated DNA was prepared by PCR using replicative form DNA as a template and 5′-AGGGCAATCAGCTCG-3′ and 5′-CTGTTGGGAAGGGC-3′ as the primers to give a DNA fragment identical to the fully methylated and hemimethylated PstI-PvuI DNA fragments. Escherichia coli replication proteins (DnaA, DnaB, DnaC, DnaG, HU, the single-stranded DNA-binding protein (SSB), Tus, Pol III* (the Pol III HE lacking the β subunit (43)), and the β subunit of the Pol III HE) were prepared as described (40). RecG and RuvAB were prepared as described (44). RecG K302A was the gift of Peter McGlynn and Robert Lloyd (University of Nottingham, Nottingham, UK). RuvC was prepared by modification of a published procedure (45). BL21(DE3)pLysS cells carrying the RuvC overexpression vector pGS775 (a gift of Robert Lloyd) were induced by the addition of isopropyl 1-thio-β-d-galactopyranoside to cultures for 3 h. Cells were harvested and resuspended, a lysate was prepared by sonication, and RuvC was precipitated by dialysis at pH 8.0 and 0.1 m KCl. This pellet was redissolved at 0.5 m KCl and passed through a column of hydroxylapatite to which the RuvC did not bind. RuvC was further purified by column chromatography on phosphocellulose. E. coli DNA polymerase I, DNA ligase, and restriction enzymes were from New England Biolabs. RNase H was as described (46).
DNA Replication and Fork Reversal Assays
DNA replication reaction mixtures (30–80 μl) to generate stalled replication forks containing 2 nm DNA template, 50 mm HEPES-KOH (pH 8.0), 10 mm Mg(OAc)2, 75 mm potassium glutamate, 200 μm CTP, UTP, and GTP, 1 mm ATP, 40 μm dNTPs, 10 mm DTT, 100 μg/ml BSA (New England Biolabs), 140 nm DnaA, 200 nm DnaB (monomer), 180 nm DnaC, 250 nm DnaG, 12.5 nm HU (dimer), 20 nm Pol III*, 30 nm β (dimer), 8 nm Tus, 250 nm SSB (tetramer) were incubated at 37 °C for 3 min to form early replication intermediates. EcoRI-HF and [α-32P]dATP were then added to 0.6 unit/μl and 15 nm, respectively, and the incubation continued for 1 min at 37 °C. Two volumes of a STOP buffer containing 50 mm HEPES-KOH (pH 8.0), 10 mm Mg(OAc)2, 75 mm potassium glutamate, 10 mm DTT, 100 μg/ml BSA, 200 μm 2′,3′-dideoxyribonucleoside 5′-triphosphates (ddNTPs), and 0.6 units/μl PvuI I (when indicated) was then added, and the incubation continued at 37 °C for 10 min to form the stalled forks. Replication fork reversal assays were performed with 15 μl of this stalled fork reaction mixture and the indicated concentrations of RecG, RuvC, and RuvAB (premixed on ice) for 10 min at 37 °C. EDTA was added to 30 mm to terminate the reactions. Reaction products were analyzed by electrophoresis through either 0.8% neutral agarose gels or 0.6% denaturing alkaline agarose gels as described (40). Gels were dried, exposed to PhosphorImager screens, and then autoradiographed. RuvC cleavage products were quantified using ImageGauge software (Fuji). The amounts of products generated are given as fractions of the total radioactivity in the lane.
RESULTS
An Assay for Replication Fork Reversal Using Bona Fide Replication Forks Arrested by Template Damage
To investigate how RFR might be generated at a bona fide stalled replication fork, we used a replication system that we developed to examine the consequences of collision of the replisome with template damage. The template is a 10.4-kbp supercoiled M13 plasmid DNA carrying oriC and either a CPD or synthetic THF abasic site at a specific site in the leading-strand template 5.3 kbp clockwise from the origin (Fig. 1). The plasmid template also contains a pair of Ter sites 0.8 kbp counterclockwise from the origin oriented to arrest both the clockwise- and counterclockwise-moving replication forks. Hence, when this template is replicated in the presence of Tus, the counterclockwise-moving fork is arrested at the Ter site (47), whereas the clockwise-moving fork encounters the leading-strand template damage. We have previously reported using this replication system that the replisome pauses transiently at the template damage, generating a stalled nascent leading strand 5.3 kb in length and then skips over the lesion by restarting leading-strand replication via a primase-directed leading-strand priming event downstream of the lesion (40). Restarted products vary in length between 3.5 and 4.2 kb. While the leading-strand polymerase is paused by the template damage, DnaB-catalyzed template unwinding and lagging-strand synthesis proceed downstream slowly (41).
For assay of RFR, we used a version of the replication assay that results in synchronization of the reaction. Replication on the CPD template is initiated in the absence of a topoisomerase, resulting in the accumulation of positive supercoils that pause the replication forks in an early replication intermediate. Topological constraint is released by the rapid digestion of the template DNA with EcoRI, at which time [α-32P]dATP is also added to label the replication products (Fig. 1). Subsequent analysis is after digestion with PvuI so that only the products of the clockwise-moving fork are observed on the gels. Native agarose gel electrophoresis shows the rapid appearance post-EcoRI cleavage of a slow moving band representing the stalled replication fork that is gradually processed to full-length duplex DNA (Fig. 2, A and C). Denaturing agarose gel electrophoresis showed that initially, at 1 min post-EcoRI cleavage, the leading-strand stall product and Okazaki fragments could be observed, whereas by 6 min, restart products have also become prominent (Fig. 2, B and D). The addition of neither RecG nor RuvAB had any obvious effect on the replication products generated (Fig. 2).
FIGURE 2.

Neither RecG nor RuvAB affects DNA replication directly. Replication reaction mixtures (20 μl) were as described under “Experimental Procedures.” The indicated concentrations of either RecG (A and B) or RuvAB (C and D) were added at the same time as EcoRI and [α-32P]dATP. Aliquots (5 μl) were removed at the indicated times post-EcoRI addition, and the DNA replication reaction was terminated by the addition of two volumes of STOP buffer. After an additional 10 min of incubation to digest the DNA products with PvuI, EDTA was added, and the reaction products were analyzed by either neutral gel electrophoresis (A and C) or denaturing alkaline gel electrophoresis (B and D). SF, stalled forks; FL, full-length EcoRI-PvuI DNA product; NDD, nascent duplex DNA; RS, restart products; OF, Okazaki fragments.
In order to observe RFR, stalled replication forks were accumulated by a 1-min incubation post-EcoRI cleavage, at which time an excess of ddNTPs was added to prevent further DNA synthesis. Under these conditions, we expect the Pol III HE to remain present at the fork. Pol III HE stalled by deprivation of two nucleotides is quite stable as it cycles between digestion of the nascent DNA and polymerization (48), and digestion of a DNA strand with a 3′-terminal dideoxymononucleotide by the 3′ → 5′ exonuclease of the Pol III HE is slowed only by a factor of 6 compared with digestion of a DNA strand terminated with a 3′-deoxymononucleotide (49); thus, the cycle of polymerization/exonuclease digestion is unlikely to be perturbed significantly. Indeed, in experiments using a rolling circle DNA template, McInerney and O'Donnell (50) found that leading-strand synthesis could resume after blockages to the leading-strand polymerase (ddNTP addition) and the helicase (ATP depletion) were eliminated by diluting the reaction mixture. Furthermore, it was clear that DnaB was still present on these templates under these conditions because if we terminated DNA synthesis by the addition of the ddNTPs and then followed any subsequent reaction, products appeared representing the fully unwound, partially replicated leading- and lagging-strand sister duplexes (41) (Fig. 3). These products appeared at 2 min post-PvuI cleavage, indicating that DnaB-catalyzed template unwinding proceeded at about 35 bp/s after replication fork arrest, which is the expected rate (51).
FIGURE 3.

DnaB remains on the template DNA during the RFR incubation period. A replication reaction mixture (80 μl) was incubated as described in the legend to Fig. 2. After the 10-min incubation to digest the DNA with PvuI, the ATP concentration was restored to 1 mm, and the incubation was continued. Aliquots (15 μl) were withdrawn at the indicated times, the reactions were terminated by the addition of EDTA, and the products were analyzed by native gel electrophoresis (lanes 4–11) (II). For comparison, the products of a replication reaction that was terminated directly at the indicated times post-EcoRI cleavage are shown in lanes 1–3 (I). UC, uncoupled products (40, 41).
To detect RFR, we included the RuvC HJ resolvase in the reaction mixtures. RuvC digestion of a regressed stalled fork can occur in two different orientations (item v in Fig. 1), which will result in two distinct cleavage products of different size (CP1 and CP2, items vi and vii in Fig. 1); however, each cleavage product will be a mixture of cleaved leading- and lagging-strand sister duplexes. The sequence of the plasmid template was such that there were four RuvC recognition sequences (52) within 100 bp of the site of template damage, so we expected CP1 and CP2 to be nearly the same size as the full-length duplex and stall product, respectively. This expectation is supported by the data presented in Fig. 7. Of course, once the SF is cleaved by RuvC, any further RFR is prevented; thus, multiple cleavage products will not be observed.
FIGURE 7.
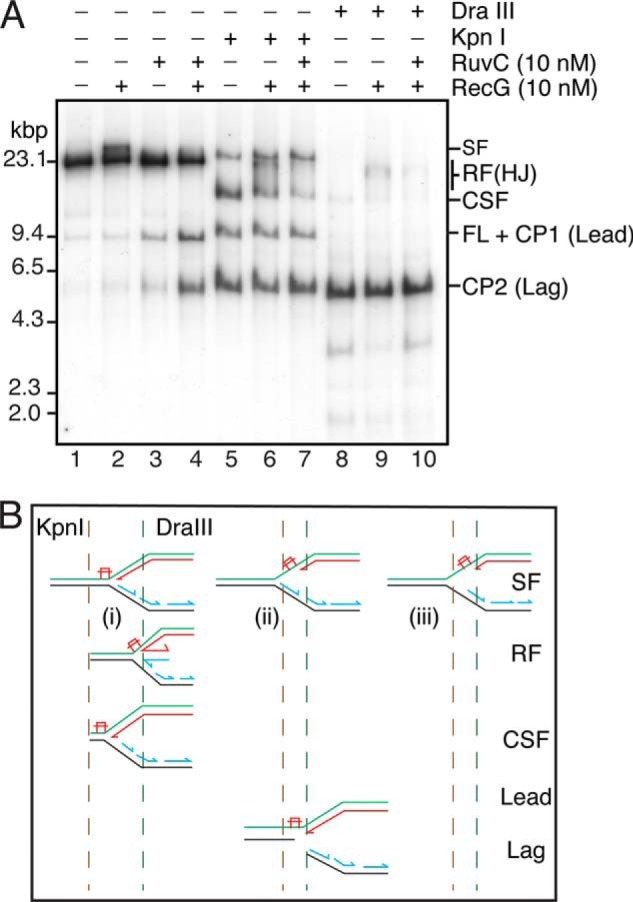
RecG forms HJs from the stalled forks in the absence of RuvC. A, standard RFR reaction mixtures containing the 5903THF template and the indicated concentrations of RecG and RuvC were incubated for 10 min at 37 °C. DraIII (5 units) and KpnI (5 units) were then added as indicated, and the incubation continued for 10 min at 37 °C. The products were then analyzed by native gel electrophoresis. B, schematic of possible products of KpnI digestion. A stalled fork where neither the nascent leading nor lagging strand has progressed past the template lesion (i) will be cut by KpnI to give a CSF. A stalled fork where the nascent leading strand remains stalled at the lesion and lagging-strand synthesis has continued (ii) will be cut by KpnI to give separate products from the leading- and lagging-strand sister duplexes (Lead and Lag, respectively). If parental DNA unwinding proceeds in the absence of either leading- or lagging-strand synthesis, the stalled fork will not be cut by KpnI (iii). Regressed forks (RF) cut by KpnI will give products that migrate between the CSF and the SF on the gel. DraIII will cut all possible products; however, forks that have regressed past the DraIII site will retain the HJ and therefore migrate more slowly than the CSF.
The addition of RuvC alone to the stalled replication forks had little effect, although some cleavage products were detectable at extended incubation (Fig. 4A). This cleavage derives from the known activity of RuvC in cleaving forks that have a gap in the nascent leading strand and a complete nascent lagging strand (53). We have found that the presence of SSB inhibits this type of cleavage by RuvC (42). The addition of both RecG and RuvC resulted in the accumulation after a lag of both CP1 and CP2 with time (Fig. 4, A and B), suggesting that RecG was catalyzing RFR and RuvC was processing the HJ formed. The lag in the kinetics most likely reflects the initial steps in RecG organizing the nascent strands in the SF for RFR. Surprisingly, the addition of RuvAB alone generated a product that had a mobility similar to that of CP2, which we call nascent DNA duplex (NDD) (Fig. 4C). Generation of NDD required the presence of both RuvA and RuvB (Fig. 4D). Higher concentrations of RuvC did not generate a cleavage product (data not shown), and, using an assay described in the accompanying article (42), where RuvC cleavage of a model oligonucleotide fork structure with a nascent lagging strand is measured, RuvAB did not inhibit cleavage of the forked structure by RuvC (data not shown). Because NDD was a labeled DNA produced by a branch migration protein and was roughly the same size as CP2 and the stall product, we considered that it was generated by complete unwinding of the nascent DNA from the template strands (item viii in Fig. 1). As shown below, this proved to be the case.
FIGURE 4.

Both RecG and RuvAB generate products from stalled forks consistent with regression. A, RecG stimulates cleavage of stalled forks by RuvC. RFR reaction mixtures (120 μl) containing either 10 nm RuvC or 10 nm RuvC and 10 nm RecG were incubated at 37 °C. Aliquots (15 μl) were removed at the indicated times, and the reaction products were analyzed by native gel electrophoresis. CP1, cleavage product 1; CP2, cleavage product 2. A representative gel is shown. B, quantification of the kinetics of CP2 production by RuvC either in the presence or absence of RecG. Mean and S.D. (error bars) are shown for three experiments. C, RuvAB generates a product from stalled forks in the absence of RuvC. Standard RFR reactions containing the indicated concentrations of RecG, RuvC, and RuvAB were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis. D, formation of NDD requires both RuvA and RuvB. Standard RFR reactions containing the indicated concentrations of RecG, RuvC, RuvA, RuvB, and RuvAB were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis.
Product Formation by RecG and RuvAB Requires a Stalled Replication Fork
We investigated whether replication fork stalling was indeed required for the formation of products by RecG and RuvAB. We addressed this question in two ways. First, we compared the results of incubating RecG-RuvC and RuvAB under standard conditions with templates that either contained a CPD or were undamaged. In each case, products were only generated by RecG-RuvC (Fig. 5A) and RuvAB (Fig. 5C) with the CPD template. Second, we used the CPD template but incubated the reaction for 6 min post-EcoRI cleavage before adding the ddNTPs. At this time, most of the stalled fork is processed to full-length duplex (Fig. 2). Under these conditions, product formation by both RecG-RuvC and RuvAB is reduced significantly compared with using stalled replication forks accumulated for 1 min (Fig. 5, B and D). We conclude that the products produced by the RecG-RuvC combination and RuvAB arise from processing of a stalled replication fork and are not the product of the action of these proteins on the template DNA itself.
FIGURE 5.

Both template damage and the presence of stalled forks are required for the generation of RFR products by RecG-RuvC and RuvAB. A, template damage is required for RecG-RuvC production of RFR products. Standard RFR reactions prepared using either undamaged template or CPD template containing the indicated concentrations of RecG and RuvC were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis. B, stalled forks are required for RecG-RuvC production of RFR products. RFR reaction mixtures derived from replication reactions incubated for either 1 min or 6 min post-EcoRI addition containing the indicated concentrations of RecG and RuvC were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis. C, template damage is required for RuvAB production of RFR products. Standard RFR reactions prepared using either undamaged template or CPD template containing the indicated concentrations of RuvB and RuvAB were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis. D, stalled forks are required for RuvAB production of RFR products. RFR reaction mixtures derived from replication reactions incubated for either 1 min or 6 min post-EcoRI addition containing the indicated concentrations of RecG, RuvC, and RuvAB were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis.
RecG Forms HJs from the Stalled Forks
RFR is proposed to proceed via the generation of a HJ. The requirement for RuvC to be present in order to observe any cleavage products with RecG suggested that this was the case. To examine the issue more directly, we added increasing concentrations of RuvA, which is known to inhibit cleavage of HJs by RuvC (54), to reactions that contained RecG and RuvC. RuvA was a very potent inhibitor of CP1 and CP2 formation by RecG and RuvC (Fig. 6). Even 3 nm RuvA (0.75 nm tetramer) completely eliminated product formation, indicating that a HJ is formed in the RecG-RuvC reaction. At first glance the effectiveness of RuvA seems remarkable; however, it is worth noting that because of the manner in which we generate the substrate, we estimate that 10–20% carry stalled forks. In addition, the concentration of total DNA template in the RFR reaction mixture is diluted to 0.67 nm.
FIGURE 6.

RuvA inhibits the generation of RFR products by RecG-RuvC. Standard RFR reaction mixtures containing the indicated concentrations of RecG, RuvC, and RuvA (increasing by a factor of 2 in lanes 5–10) were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis.
It remained possible that RuvC was involved in stabilizing the HJs that were inferred to be present based on the inhibition of product formation by RuvA. We therefore used a physical assay to detect HJ formation by RecG. The KpnI restriction enzyme cleaves the template DNA once 65 bp downstream from the site of template damage. Under conditions where cleavage products were generated by RecG-RuvC, we also treated the products with KpnI (Fig. 7). As described earlier, we have demonstrated that template unwinding and lagging-strand synthesis continues slowly downstream of the stalled leading-strand polymerase. In the reaction scheme we are using here, DnaB-catalyzed template unwinding continues (Fig. 3) even after adding the ddNTPs to arrest DNA synthesis. A mixture of stalled forks that have varied lengths of Okazaki fragment downstream of the stall point followed by unwound template will therefore be generated after the addition of the ddNTPs. Thus, several different products could be observed (Fig. 7B), depending on whether the KpnI site was duplex or single-stranded on the leading- and lagging-strand sisters. Note that not all of the stalled fork will be digested by KpnI because some of the templates will have been unwound by DnaB in the absence of any DNA synthesis, making both the leading- and lagging-strand sister duplexes resistant to cleavage. Also note that if replication progresses before the ddNTPs are added, the products labeled Lead and Lag in Fig. 7B will also arise.
In the presence of RecG, treatment with KpnI generated a smear of products between the position of the cleaved stalled fork (CSF) and the undigested stalled fork (SF) (Fig. 7A, lane 6) that were not present in the absence of RecG (Fig. 7A, lane 5) and that were removed by the inclusion of RuvC in the reaction (Fig. 7A, lane 7). These results argue that the smear of products represents branched DNA molecules generated by RecG that were resolved into duplex DNAs by the HJ resolvase RuvC. These data also suggest that RecG acts, for the most part, on forks that were arrested in close proximity to the damage (i.e. before the KpnI site) because only the CSF gets depleted in the presence of RecG (Fig. 7A, compare lane 6 with lane 5). If RecG was acting on forks that had progressed further downstream before the addition of the ddNTPs, a reduction in the amount of the lead and lag cleavage products would also be expected.
We used treatment with another restriction enzyme, DraIII, which cuts the template 183 bp upstream of the site of template damage to assess the extent of RFR. Similar to the results with KpnI digestion, branched molecules were formed by RecG (Fig. 7A, compare lanes 8 and 9) that were eliminated by RuvC treatment (Fig. 7A, lane 10). The fact that these branched molecules could still be observed after DraIII treatment indicated that at least some of the stalled replication forks had been regressed about 180 bp by RecG.
RuvAB Branch Migrates the Nascent DNA Strands Off of the Template DNA
The requirements for generating a RuvAB-dependent product from the stalled forks (Fig. 4C) suggested that, unlike RecG-RuvC, RuvAB was unwinding the nascent DNA completely from the template DNA. Such a product would be possible if a branch migration reaction occurred in the direction of oriC from a HJ either formed or stabilized by RuvAB. If this were the case, the product would be free of the template. In our reaction scheme, EcoRI digestion of the template DNA is used to release topological constraint, and PvuI digestion is used to isolate the stalled clockwise-moving fork from the counterclockwise-moving fork (Fig. 1). As illustrated in Fig. 8A, the RecG-RuvC product is released for analysis by RuvC and PvuI cleavage. In the absence of PvuI cleavage, because regression is not complete to the origin, no products should be observed. On the other hand, because RuvAB-catalyzed branch migration and unwinding of the nascent DNA proceeds from the stall point back to the origin, the product should be free of the template and not require PvuI digestion to be observed. This proved to be the case.
FIGURE 8.
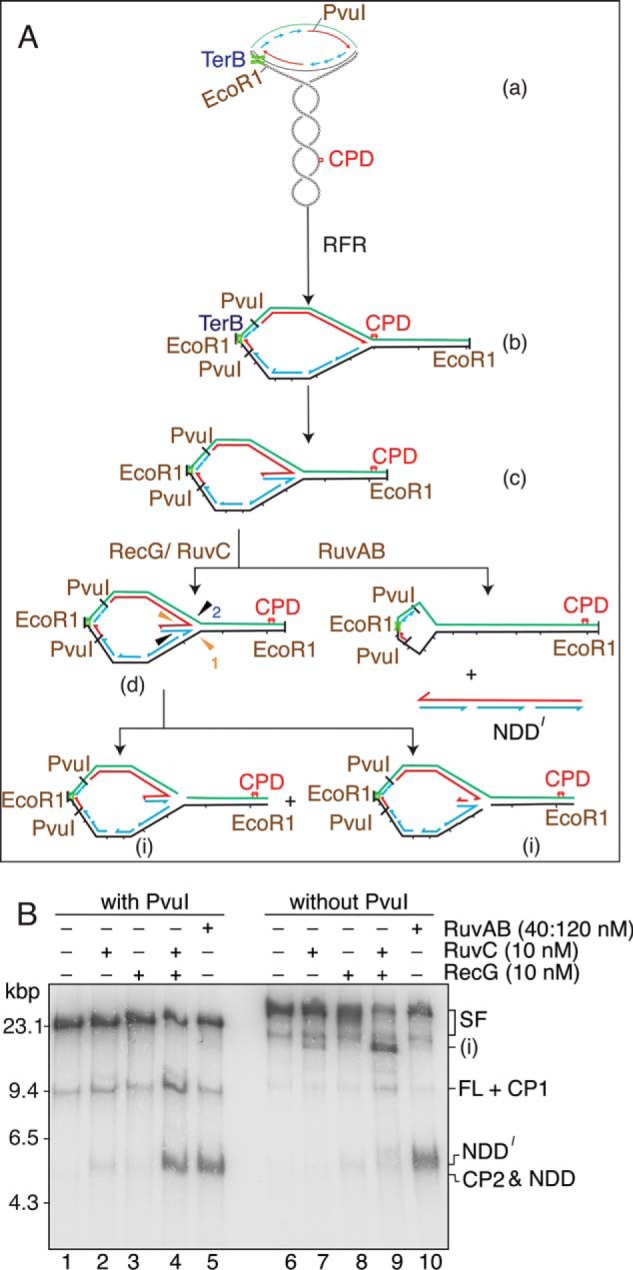
RuvAB regresses the nascent DNA completely from the template strands. A, schematic of the reaction. The early replication intermediate (a) is digested with EcoRI but not PvuI. The stalled forks (both the counterclockwise moving fork that is stalled at the Ter site and the clockwise-moving fork that is stalled by the template damage) therefore generate a large replication bubble in the EcoRI-digested template (b). After RFR by either RecG or RuvAB, the size of this bubble is reduced (c). Note that the counterclockwise moving fork may also be regressed. RuvC can cleave the regressed fork in two different orientations (d), generating a large Y-like structure (i). RuvAB regresses the nascent DNA completely off of the template strands, generating a free nascent strand duplex (NDD′) that is a little longer than NDD because it encompasses the region from the stall point back to where the fork initiated at oriC. B, standard RFR reaction mixtures containing the indicated proteins and either digested with PvuI (with PvuI) or not treated (without PvuI) were incubated for 10 min at 37 °C and analyzed by native gel electrophoresis.
Products generated from stalled forks by RecG-RuvC and RuvAB, both individually and in combination, were compared in the presence and absence of PvuI digestion (Fig. 8B). Treatment of stalled forks in the presence of PvuI cleavage gave the expected products. In the absence of PvuI digestion, cleavage of stalled forks with RuvC alone generated two products (Fig. 8B). Product i corresponds to cleavage of the template damage stalled fork on the lagging-strand template (as discussed above) and at the counterclockwise-moving stalled fork at the Ter site. The addition of RecG resulted in a significant increase in product i because now the damage-stalled fork has been regressed. In the presence of RuvAB alone, NDD′ (a slightly longer version of NDD because of the lack of PvuI cleavage) was generated as predicted, indicating that the nascent DNA has been completely unwound from the template. Formally speaking, however, these results did not indicate that NDD was double-stranded. To do so, we assessed its methylation state using digestion by DpnI and MboI, restriction enzymes that differ in their sensitivity to Dam methylation.
We first tested the activity of the two restriction enzymes on the KpnI-PvuI DNA fragment that was either fully methylated (prepared from the replicative form DNA), hemimethylated (prepared from the template DNA), or unmethylated (prepared by PCR from the replicative form DNA). MboI could not digest fully methylated DNA (Fig. 9A), whereas it was partially active on hemimethylated DNA (Fig. 9B) and could digest unmethylated DNA completely at 0.1 unit of enzyme (Fig. 9C; we had identical results with DpnII (data not shown)). DpnI did not digest unmethylated DNA (Fig. 9C) but did digest fully methylated DNA (Fig. 9A) and, surprisingly, hemimethylated DNA at concentrations in excess of that required to digest methylated DNA (Fig. 9C). The action of these enzymes on CP2, generated by RecG-RuvC, and NDD, generated by RuvAB, was then compared.
FIGURE 9.

Digestion of differentially methylated DNA by DpnI and MboI. Duplex DNA fragments (5.3 kbp) that were either methylated on both strands (A), hemimethylated (B), or unmethylated (C), prepared as described under “Experimental Procedures,” were treated with the indicated amounts of DpnI and MboI for 10 min at 37 °C in replication reaction buffer. The products of digestion were analyzed by native agarose gel electrophoresis.
Neither 1 nor 5 units of DpnI digested CP2 (Fig. 10, A (compare lanes 2–4) and B). This result is consistent with the predicted methylation state of CP2, which is a combination of hemimethylated and unmethylated DNA (the template DNA is hemimethylated because it is prepared in vitro from single-stranded phage DNA; see Fig. 1), depending on how the HJ is resolved. One-tenth unit of MboI digested about half of the CP2 (Fig. 10, A (compare lanes 2 and 5) and B), suggesting that HJ resolution was about equal in either direction. On the other hand, whereas 1 unit of DpnI did not digest NDD (Fig. 10, A (compare lanes 7 and 8) and B), NDD was very sensitive to MboI, with about half being digested at 0.01 unit and 90% at 1 unit (Fig. 10, A (compare lanes 9 and 10 with lane 7) and B). Although, because of the activity of MboI on hemimethylated DNA (Fig. 9), we cannot conclude that NDD is exclusively unmethylated, the contrast with the sensitivity of CP2 to MboI strongly suggests that NDD is unmethylated, as would be predicted for nascent leading- and lagging-strands of DNA that had been paired together by a RuvAB-catalyzed branch migration reaction.
FIGURE 10.

The RuvAB RFR product is a nascent strand duplex. A, the indicated restriction enzymes (1 unit of DpnI, 0.1 unit of MboI in lanes 5 and 10 and 0.01 unit in lane 9) were added to regression products formed by either RecG + RuvC or RuvAB in standard RFR reaction mixtures, and the incubations continued for 10 min at 37 °C. The DNA products were then analyzed by native gel electrophoresis. B, quantification of the fraction of CP2 or NDD that was resistant to digestion by the indicated restriction enzyme. The plot shows the mean and S.D. from two experiments. A representative gel is shown in A.
RecG Stimulates RuvAB-catalyzed Regression of Stalled Forks
We used the generation of NDD to assess whether RecG and RuvAB action at the stalled forks could be synergized (Fig. 11). When the action of RecG is analyzed in this manner in the absence of RuvC, no products are generated that are free of the template strands (i.e. NDD) as there are when RuvAB activity is examined. The combination of the two enzymes resulted in a clear stimulation by RecG of RuvAB-catalyzed regression (Fig. 11, A (compare lanes 3 and 4) and B). This stimulation was not observed when the RecG K230A variant, which does not have DNA helicase activity (32), was used in place of the wild-type RecG (Fig 11, A (compare lanes 3 and 6) and B), indicating that the stimulation was dependent on the ability of RecG to unwind DNA. Thus, this stimulation probably results from RecG being more efficient than RuvAB at generating the initial HJ from the stalled fork, as has been proposed by McGlynn and Lloyd (36).
FIGURE 11.

RecG stimulates RuvAB regression. A, standard RFR reaction mixtures containing the indicated concentrations of RecG, RecG K302A, and RuvAB were incubated for 10 min at 37 °C and analyzed by native gel electrophoresis. A representative gel is shown. B, quantification of the amount of NDD generated. The plot shows the mean and S.D. (error bars) from three experiments.
DISCUSSION
Nature of the Stalled Fork
We have examined the RFR activity of RecG and RuvAB, two helicase/branch migration enzymes commonly invoked as agents that regress stalled replication forks during the process of replication fork repair or restart (8–11), using bona fide replication forks stalled by leading-strand template damage in a reconstituted replication system in vitro, a scenario that should be a reflection of events in vivo. In our replication system, replication forks formed at oriC collide with and stall transiently at leading-strand template damage (a CPD) (40). During the stall, the replication fork helicase, DnaB, and the lagging-strand polymerase proceed slowly downstream of the damage, continuing Okazaki fragment synthesis while still being associated with the stalled leading-strand polymerase (41). Once a new primer made by the primase on the leading-strand template downstream of the damage is captured by the stalled leading-strand polymerase, leading-strand synthesis will resume with the net effect of the damage being skipped over with a gap left behind.
In order to observe RFR, we arrested DNA synthesis by adding ddNTPs early in the reaction so that stalled forks accumulated, but there was little restart of leading-strand synthesis. Thus, as used herein, the form of the stalled replication forks is one where there is likely to be small, but not extensive, gaps in the nascent leading strand with a completed lagging-strand sister opposite (Fig. 1). We can also say definitively that DnaB is still associated with the template DNA (Fig. 3) and that, based on the known stability of paused, cycling Pol III HE (48–50), the replicase is likely to be as well. Because SSB is present in the reaction mixture, we also expect that the leading-strand template will be bound to SSB on the leading-strand sister. Because we did not include the enzymes required for processing of Okazaki fragments, the lagging-strand sister will contain nicks and probably small gaps as well. Using these stalled forks, we found that whereas RecG and RuvAB could both catalyze RFR, the products generated in the presence of RuvC were different.
Action of RecG
RecG-catalyzed RFR was detected by assaying conversion of the stalled fork to a substrate that could be cleaved by RuvC. We do note, however, that RuvC activity on the stalled fork itself was evident but was suppressed by the presence of SSB (42). Thus, it should not be a given, despite the precision activity of RuvC toward resolution of HJs (53, 55, 56), that any detectable RuvC cleavage of a DNA is indicative of the presence of a HJ. However, we could detect DNA fragments with decreased mobility that were derived from the region about the stalled fork, whose appearance was dependent on treatment of the stalled fork DNA with RecG, and that were eliminated by treatment with RuvC (Fig. 7). These results are consistent with the argument that RecG bound the stalled fork, regressed it into a HJ, and branch migrated the HJ at least as far as the first few RuvC recognition sequences. Our results are also consistent with the known affinity of RecG for a fork configured as the one generated herein (27, 33, 57–59).
RecG branch migration of the HJ formed did not seem extensive, because CP2 was essentially the size expected for a duplex DNA spanning the distance between the template damage site and the PvuI site that was composed of the stalled nascent leading strand and the leading-strand template. There are 175 RuvC recognition sites between the template damage site and the oriC. Extensive branch migration followed by RuvC cleavage would therefore have generated a smear of faster moving products extending from the position of CP2 on the neutral agarose gels, which was not observed (Fig. 4).
Action of RuvAB
RuvAB catalyzed RFR, generating a product that was shown to be the result of complete unwinding of the nascent leading and lagging strands from the template and their rewinding about each other, yielding a complete nascent duplex DNA. Generation of such a product can only occur by a branch migration reaction from a HJ formed by RuvAB at the stalled fork. This observation is counter to previous arguments that RuvAB is deficient in its ability to convert a stalled fork into a HJ (36, 59). We considered that the difference might result from the presence of the replication proteins at the stalled fork somehow endowing RuvAB with this activity; however, RuvAB has identical activity on deproteinized stalled forks (data not shown). We do not know the exact disposition of the 3′-ends of the Okazaki fragments that terminate closest to the site of template damage, so we cannot speak to the absolute structure of the stalled fork. Perhaps either the size of the gap in the nascent leading strand or the presence of consecutive Okazaki fragments on the lagging-strand sister is a factor. These elements are not reflected by the small oligonucleotide substrates used in previous studies (36, 59).
Remarkably, RuvC appeared unable to coordinate with RuvAB to cleave the HJ during this extensive branch migration reaction. This observation runs counter to the common view of RuvABC being a resolvasome (60), which is supported by considerable data; the phenotypes of mutations in ruvA, ruvB, and ruvC are similar (61, 62). Monoclonal antibodies against RuvA, RuvB, or RuvC inhibit resolution of a HJ in a reconstituted branch migration system (60). RuvB and RuvC interact physically (60). RuvA and RuvC can bind simultaneously to a HJ, forming a complex (54). RuvB can stimulate the binding to and resolution of a HJ by RuvC, and RuvC can stimulate RuvB-catalyzed branch migration in the absence of RuvA (63). RuvAB also clearly stimulated RuvC-catalyzed HJ resolution over a 310-bp homologous region in a fashion that was dependent on the DNA helicase activity of RuvB (64).
However, extensive branch migration reactions, such as the one described herein, have rarely been examined. In a four-strand exchange reaction catalyzed by RecA, branch migration and RuvC HJ cleavage were assayed over a 3-kbp region. There was little difference in the kinetics of HJ resolution in the presence of RecA and RuvAB compared with RecA and RuvABC (60). Similarly, with this same system, the distribution and extent of cleavage at specific sites by RuvC was not much different with RecA and RuvC in the reaction compared with RecA and RuvABC (65). Such observations led Eggleston et al. (60) to suggest the existence of two complexes: a RuvAB complex capable of extensive branch migration, and a RuvABC complex, capable of both branch migration and HJ resolution. Indeed, two tetramers of RuvA bound to a HJ exclude binding of RuvC (66) and are required for efficient branch migration (38).
Perhaps the reaction that we have observed is too processive to allow access of RuvC to the HJ, or there is another unknown factor that was not present in our reactions or an unknown event required to convert a RuvAB complex to a RuvABC complex that did not occur in our reactions. The apparent cooperation between RecG and RuvC that we describe may simply be a function of a reduced processivity of RecG in an RFR reaction, as has been reported by Cox (34). However, it is interesting that a synthetic lethal screen designed to uncover an alternative to RuvC that would act as the HJ resolvase coupled to RecG-catalyzed branch migration yielded only mutations in polA, dam, and uvrD (67).
Synergistic Action of RecG and RuvAB
It has been suggested that RecG is the primary initiator of RFR and that RuvAB can only branch migrate a HJ once it is formed at a stalled fork by another agent (36). However, assay systems that used only small oligonucleotide substrates cannot demonstrate cooperation between these two proteins because the final product of the reaction would be identical with RecG in either the presence or absence of RuvAB. Because we could detect the completely unwound nascent duplex (NDD) branch migrated off of the template by RuvAB and RecG did not form such a product, we were able to show that, in fact, RecG is able to stimulate RuvAB branch migration, presumably by providing increased amounts of the starting material, the HJ. This observation is consistent with the argument that the starting material for RuvAB is a HJ (36).
Access to Stalled Replication Forks
The RFR reactions we describe are probably occurring with the replisome proteins present at the stalled fork. The 3′-ends of both the nascent leading and lagging strands must be available to freely rotate about the template strands in order for RFR to occur. It seems unlikely that either DnaB or the lagging-strand polymerase would be an impediment to RFR. These enzymes continue to migrate downstream away from the stalled leading-strand polymerase (41). Thus, as long as the Okazaki fragment opposite the stalled leading-strand polymerase is not sealed to the next one downstream before the RFR reaction initiates, its 3′-end will be available. The interesting question is what happens on the leading strand. We envision two possibilities. It is now clear that DNA polymerases can freely exchange on the β clamp (68). If this exchange reaction reflects the repeated association/dissociation of the leading-strand polymerase while it is stalled, then it is possible that a competition for the 3′-end of the nascent, stalled leading strand could be established between the α subunit of the Pol III HE and an RFR protein. Alternatively, the 3′-end of the nascent, stalled leading strand could be freed because the leading-strand polymerase cycles forward to a new primer downstream on the leading-strand template (40), in which case RFR might actually be postreplicative. Either way, the β clamp so freed would just be pushed backward in the branch migration reaction and would not be an obstacle.
This work was supported, in whole or in part, by National Institutes of Health Grant GM34557 (to K. J. M.).
- RFR
- replication fork regression
- HJ
- Holliday junction
- Pol III HE
- DNA polymerase III holoenzyme
- THF
- tetrahydrofuran
- CPD
- cyclopyrimidine dimer
- SSB
- E. coli single-stranded DNA-binding protein
- ddNTP
- 2′,3′-dideoxynucleoside 5′-triphosphate
- CP1 and CP2
- RuvC cleavage product 1 and 2, respectively
- NDD
- nascent DNA duplex
- CSF
- cleaved stalled fork
- SF
- stalled fork.
REFERENCES
- 1. Hanawalt P. C. (1966) The U.V. sensitivity of bacteria: its relation to the DNA replication cycle. Photochem. Photobiol. 5, 1–12 [PubMed] [Google Scholar]
- 2. Skalka A. (1974) in Mechanisms of Recombination (Grell R. F., ed) pp. 421–432, Plenum Press, New York [Google Scholar]
- 3. Nurse P., Zavitz K. H., Marians K. J. (1991) Inactivation of the Escherichia coli priA DNA replication protein induces the SOS response. J. Bacteriol. 173, 6686–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zavitz K. H., Marians K. J. (1991) Dissecting the functional role of PriA protein-catalysed primosome assembly in Escherichia coli DNA replication. Mol. Microbiol. 5, 2869–2873 [DOI] [PubMed] [Google Scholar]
- 5. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 6. Gabbai C. B., Marians K. J. (2010) Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair 9, 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yeeles J. T., Poli J., Marians K. J., Pasero P. (2013) Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 5, a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGlynn P., Lloyd R. G. (2002) Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 3, 859–870 [DOI] [PubMed] [Google Scholar]
- 9. Atkinson J., McGlynn P. (2009) Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 37, 3475–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michel B., Grompone G., Florès M. J., Bidnenko V. (2004) Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 101, 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R. (2007) Recombination proteins and rescue of arrested replication forks. DNA Repair 6, 967–980 [DOI] [PubMed] [Google Scholar]
- 12. Higgins N. P., Kato K., Strauss B. (1976) A model for replication repair in mammalian cells. J. Mol. Biol. 101, 417–425 [DOI] [PubMed] [Google Scholar]
- 13. Fujiwara Y., Tatsumi M. (1976) Replicative bypass repair of ultraviolet damage to DNA of mammalian cells: caffeine sensitive and caffeine resistant mechanisms. Mutat. Res. 37, 91–110 [DOI] [PubMed] [Google Scholar]
- 14. Postow L., Ullsperger C., Keller R. W., Bustamante C., Vologodskii A. V., Cozzarelli N. R. (2001) Positive torsional strain causes the formation of a four-way junction at replication forks. J. Biol. Chem. 276, 2790–2796 [DOI] [PubMed] [Google Scholar]
- 15. McGlynn P., Lloyd R. G., Marians K. J. (2001) Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. U.S.A. 98, 8235–8240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Connolly B., Parsons C. A., Benson F. E., Dunderdale H. J., Sharples G. J., Lloyd R. G., West S. C. (1991) Resolution of Holliday junctions in vitro requires the Escherichia coli ruvC gene product. Proc. Natl. Acad. Sci. U.S.A. 88, 6063–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitby M. C., Vincent S. D., Lloyd R. G. (1994) Branch migration of Holliday junctions: identification of RecG protein as a junction specific DNA helicase. EMBO J. 13, 5220–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whitby M. C., Ryder L., Lloyd R. G. (1993) Reverse branch migration of Holliday junctions by RecG protein: a new mechanism for resolution of intermediates in recombination and DNA repair. Cell 75, 341–350 [DOI] [PubMed] [Google Scholar]
- 19. Tsaneva I. R., Müller B., West S. C. (1992) ATP-dependent branch migration of Holliday junctions promoted by the RuvA and RuvB proteins of E. coli. Cell 69, 1171–1180 [DOI] [PubMed] [Google Scholar]
- 20. Cassuto E., West S. C., Mursalim J., Conlon S., Howard-Flanders P. (1980) Initiation of genetic recombination: homologous pairing between duplex DNA molecules promoted by recA protein. Proc. Natl. Acad. Sci. U.S.A. 77, 3962–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McEntee K., Weinstock G. M., Lehman I. R. (1979) Initiation of general recombination catalyzed in vitro by the recA protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 2615–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shibata T., Cunningham R. P., DasGupta C., Radding C. M. (1979) Homologous pairing in genetic recombination: complexes of recA protein and DNA. Proc. Natl. Acad. Sci. U.S.A. 76, 5100–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marians K. J. (1992) Prokaryotic DNA replication. Annu. Rev. Biochem. 61, 673–719 [DOI] [PubMed] [Google Scholar]
- 24. Baharoglu Z., Petranovic M., Flores M. J., Michel B. (2006) RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J. 25, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lane H. E., Denhardt D. T. (1975) The rep mutation. IV. Slower movement of replication forks in Escherichia coli rep strains. J. Mol. Biol. 97, 99–112 [DOI] [PubMed] [Google Scholar]
- 26. Guy C. P., Atkinson J., Gupta M. K., Mahdi A. A., Gwynn E. J., Rudolph C. J., Moon P. B., van Knippenberg I. C., Cadman C. J., Dillingham M. S., Lloyd R. G., McGlynn P. (2009) Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell 36, 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McGlynn P., Lloyd R. G. (2000) Modulation of RNA polymerase by(p) ppGpp reveals a RecG-dependent mechanism for replication fork progression Cell 101, 35–45 [DOI] [PubMed] [Google Scholar]
- 28. Gregg A. V., McGlynn P., Jaktaji R. P., Lloyd R. G. (2002) Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell 9, 241–251 [DOI] [PubMed] [Google Scholar]
- 29. LeBowitz J. H., McMacken R. (1986) The Escherichia coli dnaB replication protein is a DNA helicase. J. Biol. Chem. 261, 4738–4748 [PubMed] [Google Scholar]
- 30. Seigneur M., Ehrlich S. D., Michel B. (2000) RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol. Microbiol. 38, 565–574 [DOI] [PubMed] [Google Scholar]
- 31. Courcelle J., Donaldson J. R., Chow K. H., Courcelle C. T. (2003) DNA damage-induced replication fork regression and processing in Escherichia coli. Science 299, 1064–1067 [DOI] [PubMed] [Google Scholar]
- 32. McGlynn P., Mahdi A. A., Lloyd R. G. (2000) Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res. 28, 2324–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McGlynn P., Lloyd R. G. (2001) Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. U.S.A. 98, 8227–8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robu M. E., Inman R. B., Cox M. M. (2004) Situational repair of replication forks: roles of RecG and RecA proteins. J. Biol. Chem. 279, 10973–10981 [DOI] [PubMed] [Google Scholar]
- 35. Manosas M., Perumal S. K., Bianco P., Ritort F., Benkovic S. J., Croquette V. (2013) RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat. Commun. 4, 2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGlynn P., Lloyd R. G. (2001) Action of RuvAB at replication fork structures. J. Biol. Chem. 276, 41938–41944 [DOI] [PubMed] [Google Scholar]
- 37. Baharoglu Z., Bradley A. S., Le Masson M., Tsaneva I., Michel B. (2008) ruvA Mutants that resolve Holliday junctions but do not reverse replication forks. PLoS Genet. 4, e1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bradley A. S., Baharoglu Z., Niewiarowski A., Michel B., Tsaneva I. R. (2011) Formation of a stable RuvA protein double tetramer is required for efficient branch migration in vitro and for replication fork reversal in vivo. J. Biol. Chem. 286, 22372–22383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robu M. E., Inman R. B., Cox M. M. (2001) RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yeeles J. T. P., Marians K. J. (2011) The Escherichia coli replisome is inherently DNA damage tolerant. Science 334, 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yeeles J. T., Marians K. J. (2013) Dynamics of leading-strand lesion skipping by the replisome. Mol. Cell 52, 855–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta S., Yeeles J. T. P., Marians K. J. (August 19, 2014) Regression of replication forks stalled by leading-strand template damage: II. RecA-catalyzed regression is inhibited by SSB. J. Biol. Chem. 10.1074/jbc.M114.587907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wickner W., Schekman R., Geider K., Kornberg A. (1973) A new form of DNA polymerase 3 and a copolymerase replicate a long, single-stranded primer-template. Proc. Natl. Acad. Sci. U.S.A. 70, 1764–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suski C., Marians K. J. (2008) Resolution of converging replication forks by RecQ and topoisomerase III. Mol. Cell 30, 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dunderdale H. J., Sharples G. J., Lloyd R. G., West S. C. (1994) Cloning, overexpression, purification, and characterization of the Escherichia coli RuvC Holliday junction resolvase. J. Biol. Chem. 269, 5187–5194 [PubMed] [Google Scholar]
- 46. Minden J. S., Marians K. J. (1985) Replication of pBR322 DNA in vitro with purified proteins. Requirement for topoisomerase I in the maintenance of template specificity. J. Biol. Chem. 260, 9316–9325 [PubMed] [Google Scholar]
- 47. Hill T. M., Marians K. J. (1990) Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 87, 2481–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Donnell M., Studwell P. S. (1990) Total reconstitution of DNA polymerase III holoenzyme reveals dual accessory protein clamps. J. Biol. Chem. 265, 1179–1187 [PubMed] [Google Scholar]
- 49. Griep M. A., Reems J. A., Franden M. A., McHenry C. S. (1990) Reduction of the potent DNA polymerase III holoenzyme 3′–5′ exonuclease activity by template-primer analogues. Biochemistry 29, 9006–9014 [DOI] [PubMed] [Google Scholar]
- 50. McInerney P., O'Donnell M. (2007) Replisome fate upon encountering a leading strand block and clearance from DNA by recombination proteins. J. Biol. Chem. 282, 25903–25916 [DOI] [PubMed] [Google Scholar]
- 51. Kim S., Dallmann H. G., McHenry C. S., Marians K. J. (1996) Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell 84, 643–650 [DOI] [PubMed] [Google Scholar]
- 52. Shah R., Bennett R. J., West S. C. (1994) Genetic recombination in E. coli: RuvC protein cleaves Holliday junctions at resolution hotspots in vitro. Cell 79, 853–864 [DOI] [PubMed] [Google Scholar]
- 53. Benson F. E., West S. C. (1994) Substrate specificity of the Escherichia coli RuvC protein. Resolution of three- and four-stranded recombination intermediates. J. Biol. Chem. 269, 5195–5201 [PubMed] [Google Scholar]
- 54. Whitby M. C., Bolt E. L., Chan S. N., Lloyd R. G. (1996) Interactions between RuvA and RuvC at Holliday junctions: inhibition of junction cleavage and formation of a RuvA-RuvC-DNA complex. J. Mol. Biol. 264, 878–890 [DOI] [PubMed] [Google Scholar]
- 55. Dunderdale H. J., Benson F. E., Parsons C. A., Sharples G. J., Lloyd R. G., West S. C. (1991) Formation and resolution of recombination intermediates by E. coli RecA and RuvC proteins. Nature 354, 506–510 [DOI] [PubMed] [Google Scholar]
- 56. Bennett R. J., Dunderdale H. J., West S. C. (1993) Resolution of Holliday junctions by RuvC resolvase: cleavage specificity and DNA distortion. Cell 74, 1021–1031 [DOI] [PubMed] [Google Scholar]
- 57. Whitby M. C., Lloyd R. G. (1998) Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J. Biol. Chem. 273, 19729–19739 [DOI] [PubMed] [Google Scholar]
- 58. Singleton M. R., Scaife S., Wigley D. B. (2001) Structural analysis of DNA replication fork reversal by RecG. Cell 107, 79–89 [DOI] [PubMed] [Google Scholar]
- 59. Abd Wahab S., Choi M., Bianco P. R. (2013) Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J. Biol. Chem. 288, 26397–26409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eggleston A. K., Mitchell A. H., West S. C. (1997) In vitro reconstitution of the late steps of genetic recombination in E. coli. Cell 89, 607–617 [DOI] [PubMed] [Google Scholar]
- 61. Sharples G. J., Benson F. E., Illing G. T., Lloyd R. G. (1990) Molecular and functional analysis of the ruv region of Escherichia coli K-12 reveals three genes involved in DNA repair and recombination. Mol. Gen. Genet. 221, 219–226 [DOI] [PubMed] [Google Scholar]
- 62. Lloyd R. G., Benson F. E., Shurvinton C. E. (1984) Effect of ruv mutations on recombination and DNA repair in Escherichia coli K12. Mol. Gen. Genet. 194, 303–309 [DOI] [PubMed] [Google Scholar]
- 63. van Gool A. J., Shah R., Mézard C., West S. C. (1998) Functional interactions between the holliday junction resolvase and the branch migration motor of Escherichia coli. EMBO J. 17, 1838–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zerbib D., Mézard C., George H., West S. C. (1998) Coordinated actions of RuvABC in Holliday junction processing. J. Mol. Biol. 281, 621–630 [DOI] [PubMed] [Google Scholar]
- 65. Eggleston A. K., West S. C. (2000) Cleavage of holliday junctions by the Escherichia coli RuvABC complex. J. Biol. Chem. 275, 26467–26476 [DOI] [PubMed] [Google Scholar]
- 66. Dickman M. J., Ingleston S. M., Sedelnikova S. E., Rafferty J. B., Lloyd R. G., Grasby J. A., Hornby D. P. (2002) The RuvABC resolvasome. Eur. J. Biochem. 269, 5492–5501 [DOI] [PubMed] [Google Scholar]
- 67. Zhang J., Mahdi A. A., Briggs G. S., Lloyd R. G. (2010) Promoting and avoiding recombination: contrasting activities of the Escherichia coli RuvABC Holliday junction resolvase and RecG DNA translocase. Genetics 185, 23–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Johnson A., O'Donnell M. (2005) Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 74, 283–315 [DOI] [PubMed] [Google Scholar]