

Background: Stalled replication forks are foci for genomic instability.
Results: RecA can only regress a stalled fork in the absence of SSB.
Conclusion: Regression by RecA in vivo is likely to require additional activities.
Significance: Replication fork regression is a major step in processing stalled forks.
Keywords: DNA Enzyme, DNA Recombination, DNA Repair, DNA Replication, Genomic Instability
Abstract
Stalled replication forks are sites of chromosome breakage and the formation of toxic recombination intermediates that undermine genomic stability. Thus, replication fork repair and reactivation are essential processes. Among the many models of replication fork reactivation is one that invokes fork regression catalyzed by the strand exchange protein RecA as an intermediate in the processing of the stalled fork. We have investigated the replication fork regression activity of RecA using a reconstituted DNA replication system where the replisome is stalled by collision with leading-strand template damage. We find that RecA is unable to regress the stalled fork in the presence of the replisome and SSB. If the replication proteins are removed from the stalled fork, RecA will catalyze net regression as long as the Okazaki fragments are sealed. RecA-generated Holliday junctions can be detected by RuvC cleavage, although this is not a robust reaction. On the other hand, extensive branch migration by RecA, where a completely unwound product consisting of the paired nascent leading and lagging strands is produced, is observed under conditions where RuvC activity is suppressed. This branch migration reaction is inhibited by SSB, possibly accounting for the failure of RecA to generate products in the presence of the replication proteins. Interestingly, we find that the RecA-RuvC reaction is supported to differing extents, depending on the template damage; templates carrying a cyclopyrimidine dimer elicit more RecA-RuvC product than those carrying a synthetic abasic site. This difference could be ascribed to a higher affinity of RecA binding to DNAs carrying a thymidine dimer than to those with an abasic site.
Introduction
Replication forks will stall when they encounter damage in the leading-strand template. These stalled forks become a target for genome-destabilizing events. Thus, it becomes imperative to repair the stalled fork by removing or bypassing the damage, allowing DNA replication to proceed. A common intermediate in many models of replication fork repair and reactivation is one in which the nascent DNA at a stalled replication fork has been unwound from the template strands and paired to each other, resulting in the generation of a Holliday junction (HJ)3 and the rewinding of the template strands (reviewed in Refs. 1–7). The rewinding of the template strands causes the regression of the replication fork, a process known as replication fork regression or reversal (RFR).
Three proteins have been suggested to be the agents of RFR in vivo: the 3′ → 5′ DNA helicase/branch migration protein RecG (8); the HJ branch migration protein RuvAB (9); and the strand exchange protein itself, RecA (10–12). We have used a DNA replication system reconstituted with purified proteins and a DNA template containing specifically located leading-strand template damage (13) to stall replication forks and examine their susceptibility to regression by these proteins. Our studies with RecG and RuvAB are reported in the accompanying article (14); here, we describe our studies with RecA.
The role of homologous recombination (HR) in rescuing replication forks that collapse because of encountering a nick in the template strand is well established (1, 15–18). This process was termed “recombination-dependent replication” by Kogoma and co-workers (19–21), based on the observation that cell viability could be maintained in Escherichia coli when oriC function was suppressed via two pathways termed induced-stable DNA replication and constitutive-stable DNA replication that initiated from D-loops and R-loops, respectively, both of which required RecA and the restart primosomal protein PriA.
A role for RecA in catalyzing RFR seemed natural, given its ability to branch migrate the HJ formed by the initial strand exchange (22, 23). RecA RFR has been proposed to occur in vivo after denaturation of a thermosensitive DnaB replication fork helicase (24) and after UV irradiation (25). Using a mimic of a fork stalled by a leading-strand template lesion composed of a gapped M13 circle with a homologous double-stranded DNA tail attached to the 5′-end of the gap, Robu et al. (26) showed that RecA could catalyze a regression reaction that was stimulated by the single-stranded DNA-binding protein (SSB).
We have investigated RecA RFR using bona fide replication forks stalled at leading-strand template damage. We find that although RecA can catalyze RFR, it cannot do so in the presence of the replication proteins, presumably because SSB prevents initiation of the formation of a RecA filament, suggesting that Rec RFR in vivo may need to be assisted by additional proteins. Using deproteinized stalled forks, RecA RFR can be observed either by RuvC cleavage of the HJ formed or by the appearance of a completely branch migrated nascent strand duplex. RecA filament formation appeared to be affected by the nature of the template damage, with a cyclopyrimidine dimer (CPD) eliciting a greater extent of regression than a synthetic abasic site (tetrahydrofuran (THF)).
EXPERIMENTAL PROCEDURES
Replication Fork Reversal Assays
All reagents and procedures were as detailed in the accompanying article (14) with the following exceptions. To seal Okazaki fragments, DNA polymerase I (New England Biolabs), RNase H, E. coli DNA ligase, and NAD+ were added to the replication reaction mixtures to final concentrations of 2 nm (0.0005 units/μl), 0.2 nm, 0.08 units/μl, and 100 μm, respectively. Deproteinized stalled forks were prepared by incubating the stalled forks formed in the replication reaction mixtures (130 μl) after PvuI digestion for 20 min at 37 °C with 0.6% SDS and 0.2 mg/ml proteinase K in the presence of 30 mm EDTA. Stalled forks were recovered after extraction with phenol/chloroform and ethanol precipitation, resuspended in 100 μl of 50 mm HEPES-KOH (pH 8.0), and dialyzed against the resuspension buffer by spotting the sample onto a Millipore filter (type VSWP, 0.25 μm) floating on 50 ml of dialysis buffer in a Petri dish. Dialyzed, deproteinized stalled forks (now about 120 μl) were then mixed with two volumes of a buffer containing the components of the replication reaction mixture to give 50 mm HEPES-KOH (pH 8.0), 10 or 3 mm Mg(OAc)2 (as indicated), 75 mm potassium glutamate, 200 μm CTP, UTP, and GTP, 1 mm ATP, 40 μm dNTPs, 10 mm DTT, and 100 μg/ml BSA (New England Biolabs). RFR reaction mixtures containing 15 μl of this preparation of deproteinized stalled forks, 3 μm RecA, and 10 nm RuvC, as indicated, were incubated at 37 °C for 20 min. Reactions were terminated by the addition of EDTA to 30 μm and analyzed by native agarose gel electrophoresis as described (14). The following oligonucleotides were used to prepare the additional templates used in the experiments described in Fig. 8: 119CPD, 5′-CATTAAAGGTGAA(TT)ATCACCGTCACCG-3′ (where (TT) marks the site of the CPD); 119THF, 5′- CATTAAAGGTGAATXATCACCGTCACCG-3′ (where X marks the site of the THF lesion). SSB W54S was the gift of Tim Lohman (Washington University, St. Louis, MO).
FIGURE 8.

CPD templates support a greater extent of RecA RFR than THF templates. A and B, standard RFR reaction mixtures with deproteinized stalled forks formed from either the 225CPD template (the standard template) or the 5903THF template (a template with a THF in the same position as the CPD on the 225 template) (A) or the 119CPD and 119THF templates (a pair of templates where the CPD and THF are in identical positions, 3.27 kbp from the PvuI site) (B) at 10 mm Mg(OAc)2 containing 10 nm RuvC and 3 μm RecA, as indicated, were incubated for 20 min at 37 °C, and the DNA products were analyzed by native gel electrophoresis. C, quantification of the ratio of product formation, RecA-RuvC/RuvC, on the various template DNAs. D and E, SSB-stimulated RuvC cleavage is identical on the CPD and THF templates. Standard RFR reaction mixtures with deproteinized stalled forks formed from either the 225CPD template or the 5903THF template at 10 mm Mg(OAc)2 containing 10 nm RuvC, 3 μm RecA, and 1 μm SSB, as indicated, were incubated for 20 min at 37 °C, and the DNA products were analyzed by native gel electrophoresis. F, quantification of the amount of either CP2 and NDD formed in the experiments shown in panels D and E. The mean and S.D. (error bars) of three experiments is shown in C and F. Representative gels are shown in A, B, D, and E.
RuvC Cleavage of a Model Fork Substrate and HJ
An oligonucleotide model stalled fork composed of a 60-bp duplex region, 38-nucleotide nonhomologous tails (1b-98, top; 3L-98, bottom), and a 38-nucleotide oligonucleotide representing the nascent lagging strand (27) was prepared by annealing the three oligonucleotides together (with 3L-98 labeled at the 5′-end by polynucleotide kinase (New England Biolabs) and [γ-32P]ATP). The fork substrate was then gel-purified. RuvC cleavage reaction mixtures containing 50 mm Tris-HCl (pH 8.0 at 37 °C), 10 mm MgCl2, 1 mm DTT, 100 μg/ml BSA, 2 nm oligonucleotide substrate, and the indicated concentrations of RuvC were incubated at 37 °C for 10 min, and the reaction was then terminated by the addition of 30 mm EDTA. For RuvC HJ cleavage, the HJ1 synthetic HJ of Shah et al. (28) was used in reaction mixtures containing 10 nm RuvC that were incubated for 10 min at 37 °C. Reactions were terminated as above. Cleavage products were analyzed by electrophoresis at 5 V/cm for 4 h through 8% polyacrylamide gels (29:1, acrylamide/bisacrylamide) using 50 mm Tris borate (pH 8.3), 1 mm EDTA as the electrophoresis buffer. Gels were dried, exposed to PhosphorImager screens, and autoradiographed. Cleavage was quantified using ImageGauge software (Fuji).
RecA Binding to Model Oligonucleotide Substrates
Oligonucleotide substrates were prepared by annealing either 5′-[32P]ACGCTGTCTG(TT)AACATACTTCGTATTGAGGAGTCTAA-3′ (CG17CPD) or 5′-[32P]ACGCTGTCTGXTAACATACTTCGTATTGAGGAGTCTAA-3′ (CG17THF) to 5′-TTAGACTCCTCAATACGAAGTATGTTA-3′ (CG21) to form the CPD and THF substrates, respectively. The undamaged substrate was prepared by annealing 5′-[32P]ACGCTGTCTGCTAACATACTTCGTATTGAGGAGTCTAA-3′ (CG16) to CG21. All substrates were gel-purified. RecA DNA-binding reaction mixtures containing 50 mm HEPES-KOH (pH 8.0), either 10 or 3 mm MgCl2, as indicated, 25 mm ATPγS, 10 mm DTT, 1 mg/ml BSA (New England Biolabs), 0.1% Triton X-100, 2 nm oligonucleotide substrate, and the indicated concentrations of RecA were incubated at 37 °C for 10 min and then loaded directly onto 10% polyacrylamide gels (60:1, acrylamide/bisacrylamide) and electrophoresed at 2.5 V/cm at 4 °C for 13 h using 6.7 mm Tris-HCl (pH 8.0), 3.3 mm NaOAc, 1 mm EDTA as the electrophoresis buffer. Gels were dried, exposed to PhosphorImager screens, and autoradiographed. DNA binding was quantified using ImageGauge software (Fuji).
RESULTS
RecA Has Little Effect on Stalled Forks When the Replisome and SSB Are Present
We used a replication system that we developed previously (13) to generate stalled forks where the replisome is still present. Replication from oriC on a circular plasmid DNA with site-specific damage in the leading-strand template 5.3 kb clockwise from the origin was synchronized by initiating in the absence of a topoisomerase to allow the accumulation of early replication intermediates. The forks were released by rapid digestion with EcoRI, at which time labeled precursor was also added. After a 1-min incubation, stalled forks accumulated, and an excess of 2′,3′-dideoxyribonucleoside 5′-triphosphate (ddNTPs) were added to arrest DNA synthesis. The counterclockwise moving fork was arrested after 0.8 kpb by a Tus-Ter complex. At this point, DnaB and the lagging-strand polymerase continue moving slowly downstream of the stall point (29, 30), whereas the leading-strand polymerase either remains stalled at the lesion (30) or could, in principle, eventually cycle forward to a new primer made on the leading-strand template even in the absence of accompanying lagging-strand synthesis (13). Thus, the nascent DNA at the stalled fork takes the form of the stalled nascent leading strand positioned just 5′ of the damage with Okazaki fragments of varying lengths opposite and a nascent leading-strand gap of varying size downstream. Digestion with PvuI, which cuts close to and on the clockwise side of the origin, releases the clockwise-moving stalled fork for analysis (see Fig. 1 in the accompanying article (14)). We first determined whether RecA had any effect on replication.
FIGURE 1.

RecA does not affect DNA replication directly. Replication reaction mixtures (20 μl) were as described under “Experimental Procedures” except that the SSB concentration (monomer) was as indicated. RecA (3 μm) was added where indicated at the same time as the EcoRI and [α-32P]dATP. Aliquots (5 μl) were removed at the indicated times post-EcoRI addition, and the DNA replication reaction was terminated by the addition of two volumes of STOP buffer. After an additional 10 min of incubation to digest the DNA products with PvuI, EDTA was added, and the reaction products were analyzed by either neutral gel electrophoresis (A) or denaturing alkaline gel electrophoresis (B). SF, stalled forks; FL, the full-length EcoRI-PvuI DNA product; NDD, nascent DNA duplex; RS, restart products; OF, Okazaki fragments.
The addition of RecA to ongoing replication reactions (i.e. no ddNTPs have been added to accumulate stalled forks, and label is present continuously post-EcoRI cleavage) had little effect on either the kinetics of generation of stalled forks or their processing to full-length material via leading-strand replication restart (13) (Fig. 1). At optimal concentrations of SSB (1 μm), stalled fork processing proceeded with kinetics similar to those we have reported previously (13, 29), even when RecA was present in a 3-fold excess (Fig. 1A, compare lanes 10–12 with lanes 1–3). In order to determine whether an even greater excess of RecA over SSB might affect the reaction, we reduced the concentration of SSB to 0.1 μm, a level that supports replication but slows the processing of stalled forks to full-length duplex (Fig. 1A, compare lanes 4–6 with lanes 1–3). However, even at this 30-fold excess of RecA to SSB, we did not observe any effect of RecA on replication (Fig. 1A, compare lanes 7–9 with lanes 4–6). Analysis by denaturing gel electrophoresis showed the appearance of the leading-strand restart products as expected in all cases (Fig. 1B). Our observations seem counter to those recently reported by Indiani et al. (31), who, using a rolling-circle replication system, demonstrated that RecA slowed the replisome. Possible reasons for this difference will be considered under “Discussion.”
As described in the accompanying article (14), cleavage by the HJ resolvase RuvC (32) can be used to detect RFR. We generated stalled forks and tested whether the combination of RecA and RuvC would generate cleavage products (see Fig. 1 in Ref. 14 for a description of the possible cleavage products: CP1, which has a mobility identical to the full-length EcoRI-PvuI fragment; CP2, which runs at about 5.3 kbp; and NDD, the completely unwound and repaired nascent DNA, which also runs at about 5.3 kbp). No products were observed (Fig. 2A), although the control of RecG-RuvC gave the expected cleavage (14).
FIGURE 2.

Formation of a RFR product by RecA in the presence of the replisome is stimulated by sealing the Okazaki fragments. Stalled forks formed in either the absence (without Ligase; A) or presence (with Ligase; B) of DNA polymerase I, RNase H, and DNA ligase to seal the Okazaki fragments were incubated in standard RFR reaction mixtures (14) containing 10 nm RuvC, 10 nm RecG, and 0.75–6 μm RecA (in increments of 2-fold; lanes 4–7), as indicated, for 20 min at 37 °C. DNA products were analyzed by native gel electrophoresis. C, quantification of the “with ligase” experiment (average of two experiments). Error bars, S.D.
Our replication reactions are conducted in the absence of the Okazaki fragment-processing enzymes DNA polymerase I, RNase H, and DNA ligase so that we can observe leading- and lagging-strand products separately on the denaturing agarose gels used for analysis. We considered that having a lagging-strand sister duplex with nicks or small gaps between fragments might inhibit a RecA RFR reaction, so we included these enzymes in the reaction at concentrations where all of the Okazaki fragments are sealed (33) and again tested whether the RecA-RuvC combination could generate cleavage products (Fig. 2B). However, although there was a slight increase in background cleavage by RuvC alone, we did not observe any increase in cleavage products in the presence of RecA. We conclude that RecA, unlike RecG and RuvAB (14), is unable to access the stalled fork in a manner that allows RFR when the replisome and SSB are also present.
SSB Inhibits RecA-catalyzed RFR
Based on the observations of Robu et al. (26), we expected that RecA would be able to regress the stalled fork if the replication proteins were removed. This expectation was tested using stalled forks recovered from the replication reactions by treatment with phenol/chloroform followed by ethanol precipitation. These deproteinized stalled forks, prepared in either the presence or absence of the Okazaki fragment-processing enzymes (with Ligase and without Ligase in Fig. 3), were used in reaction mixtures otherwise identical to the replication and RFR assays described above except that no replication proteins were present.
FIGURE 3.

Formation of RecA RFR products with deproteinized stalled forks requires sealed Okazaki fragments. Standard RFR reaction mixtures with deproteinized stalled forks formed either in the absence (without Ligase) or presence (with Ligase) of DNA polymerase I, RNase H, and DNA ligase, containing RecG (10 nm), RuvC (10 nm), or RecA (3 μm), as indicated, were incubated for 20 min at 37 °C. DNA products were analyzed by native gel electrophoresis. The histogram shows quantification of the amount of either CP2 or NDD produced (average of three experiments). Error bars, S.D.
Using these deproteinized stalled forks as substrates, we found that RecA could, as expected, generate CP2; however, sealing of the Okazaki fragments appeared to be required (Fig. 3, compare lane 5 with lane 10). Possible reasons for this effect are considered under “Discussion.” We also detected a RecA product in the absence of RuvC, which, as we will discuss below, is NDD, the product of a branch migration reaction that unwinds the nascent DNA strands from the template strands completely and pairs them to each other (14).
In order for RecA to regress the fork via a strand-exchange reaction, it has to bind to the single-stranded gap in the leading-strand duplex just downstream of the 3′-end of the stalled nascent leading strand. The extension of the RecA filament onto the double-stranded DNA formed by the leading-strand template and the nascent leading strand enables strand invasion of the lagging-strand sister duplex DNA by the nascent leading strand. The HJ so formed can then be branch migrated away from the point of fork stalling back toward the origin (Fig. 4).
FIGURE 4.
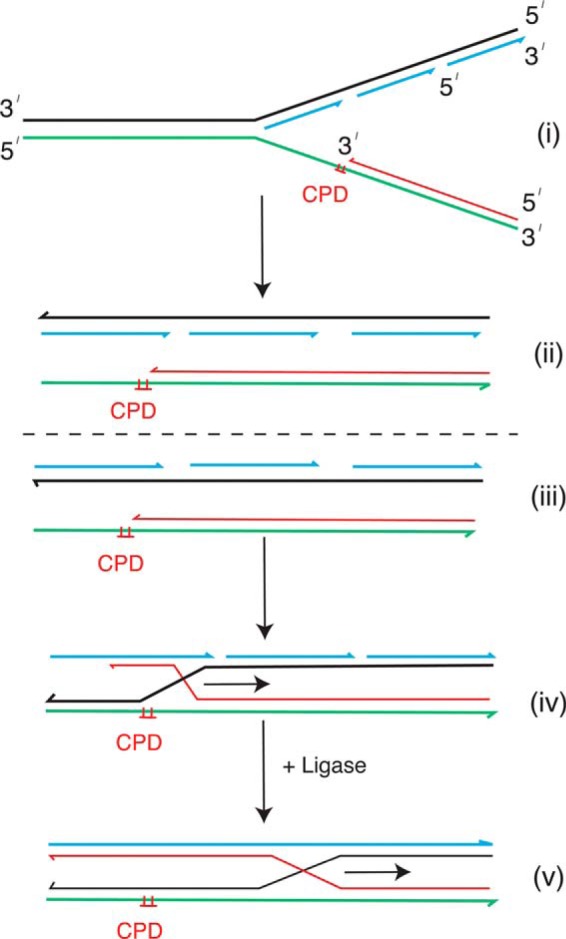
Schematic of the RecA RFR reaction. i, the stalled fork; ii, lagging-strand and leading-strand sisters aligned; iii, reorientation of the lagging-strand sister; iv, recombinant joint molecule formed by invasion of the nascent leading strand from the leading-strand sister into the lagging-strand sister; v, unsealed Okazaki fragments and gaps between them could prevent branch migration from proceeding. When the Okazaki fragments are sealed (v), this possibility is eliminated, and the branch migration reaction should proceed. The arrow denotes the direction of branch migration. Black lines, lagging-strand template; green lines, leading-strand template; blue lines, nascent lagging-strand DNA; red lines, nascent leading-strand DNA.
We considered it likely that it was SSB that was inhibiting RecA RFR in the presence of the replication proteins. SSB stimulates the RecA-promoted branch migration reaction (34, 35), most likely because it helps eliminate secondary structure in the ssDNA (36). Binding of SSB and RecA to ssDNA is competitive. In the absence of ATP, SSB prevents RecA from binding to ssDNA. In the presence of the non-hydrolyzable ATP analog, ATPγS, RecA wins the competition. And in the presence of ATP, both bound forms are found, with the equilibrium position being set by the temperature and Mg2+ concentration (37). Thus, access of RecA to a ssDNA that is completely coated with SSB can be restricted. We titrated SSB into RFR reactions with the deproteinized stalled forks and RecA-RuvC. Surprisingly, SSB did not appear to inhibit formation of CP2, although the nature of the band on the gel became more diffuse (Fig. 5A). However, it was apparent that high concentrations of SSB could stimulate RuvC cleavage as well, even in the absence of RecA (Fig. 5, A and B, compare lanes 9 with lanes 2). This SSB-stimulated RuvC cleavage appeared to be derived from the formation of HJs, because the RuvC-directed cleavage of replication forks modeled with oligonucleotides having a completed nascent lagging strand opposite a nascent leading-strand gap (38) (such as the stalled forks used herein) is inhibited by SSB (Fig. 5C). We suspect that increasing concentrations of SSB can destabilize the duplex formed by the stalled nascent leading strand and the leading-strand template, encouraging strand invasion into the lagging-strand sister duplex (Fig. 4). Nevertheless, however this reaction was achieved, we could not assess the reason that RecA did not catalyze RFR in the presence of the replication proteins in this manner.
FIGURE 5.

SSB stimulates RuvC cleavage of deproteinized stalled forks. A, standard RFR reaction mixtures with deproteinized stalled forks formed in the presence of DNA polymerase I, RNase H, and DNA ligase containing 0.25–2 μm SSB (increasing in 2-fold increments in lanes 5–8) were incubated at 37 °C for 5 min. RuvC and RecA were then added as indicated to 10 nm and 3 μm, respectively, and the incubation continued for 20 min at 37 °C. The DNA products were analyzed by native gel electrophoresis. B, quantification of the amount of CP2 and NDD formed (as a fraction of the total DNA products). The mean and S.D. (error bars) from three experiments is shown. A representative gel is shown in A. C, SSB inhibits RuvC cleavage of a model stalled fork, where the nascent lagging strand is ahead of the nascent leading strand. A model stalled fork was formed, as described under “Experimental Procedures,” that had a duplex lagging-strand sister arm and a single-stranded leading-strand sister arm. Reaction mixtures containing the model stalled fork with the nascent lagging-strand labeled at the 5′-end with 32P, 1 μm SSB, and 10 nm RuvC, as indicated, were incubated at 37 °C for 10 min. The reactions were terminated by the addition of EDTA to 30 mm, and the products were analyzed by electrophoresis through an 8% polyacrylamide gel (29:1, acrylamide/bisacrylamide).
We therefore sought to answer this question by assessing the effect of SSB on the production of the completely unwound NDD produced by RecA-promoted branch migration in the absence of RuvC. We found that RecA generation of NDD was improved at lower Mg2+ concentrations (Fig. 6A), which suppressed RuvC activity (Fig. 6B). That RecA was generating the fully unwound product under these conditions is shown by the fact the NDD product was resistant to digestion by the DpnI restriction endonuclease, which will cut unmethylated and hemimethylated DNA (Fig. 6C, compare lanes 5 and 6), whereas it was sensitive to digestion with the MboI restriction endonuclease, which will not cut fully methylated DNA but does digest hemimethylated and unmethylated DNA (Fig. 6D, compare lanes 5 and 6). This behavior was identical to that of NDD generated by RuvAB (Fig. 6, C and D, compare lanes 3 and 4), as established in the accompanying article (14). (The relative activities of DpnI and MboI on differentially methylated substrates can be found in Fig. 9 of the accompanying article (14).) Formation of NDD by RecA using deproteinized stalled forks as the substrate was inhibited by increasing concentrations of SSB (Fig. 6E). We used a variant SSB, SSB W54S, which binds dT35 with one-five hundredth the avidity of the wild type (39), to determine whether the ssDNA binding activity of SSB was required for this inhibition. This proved to be the case (Fig. 6F). Thus, we conclude that the failure of RecA to express any type of RFR activity on stalled forks in the presence of the replication proteins is, at least in part, because of SSB occupying the leading-strand gap, where RecA has to bind to initiate strand exchange (Fig. 4).
FIGURE 6.

RecA branch migrates the nascent strands off of the template strands of deproteinized stalled forks at lower magnesium concentrations. A, RecA generates a RFR product in the absence of RuvC. Standard RFR reaction mixtures at 3 mm Mg(OAc)2 either in the presence or absence of 10 nm RuvC, as indicated, and in the presence of RecA (from 0.75 to 6 μm, increasing by 2-fold in lanes 2–5 and 7–10) were incubated for 20 min at 37 °C, and the DNA products were analyzed by native gel electrophoresis. B, dependence of RuvC cleavage on Mg2+ concentration. RuvC was incubated with a synthetic HJ (HJ1 (28)) as described under “Experimental Procedures” at the indicated concentrations of Mg(OAc)2. Cleavage products were analyzed by electrophoresis through a 8% polyacrylamide gel. C and D, the RecA RFR product is a nascent strand DNA duplex. Standard RFR reaction mixtures at 3 mm Mg(OAc)2 containing 3 μm RecA, RuvAB (40–120 nm), or no RFR proteins were incubated for 20 min at 37 °C. Either DpnI (1 unit) (C) or MboI (0.1 unit) (D) was then added, and the incubation continued for 10 min. DNA products were then analyzed by native agarose gel electrophoresis. E and F, SSB inhibits formation of the RecA RFR product. Standard RFR reaction mixtures at 3 mm Mg(OAc)2 containing 3 μm RecA and either SSB (E) or SSB W54S (F) (25–500 nm, increasing by 2-fold in lanes 3–8), as indicated, were incubated for 20 min at 37 °C, and the DNA products were analyzed by native gel electrophoresis.
FIGURE 9.
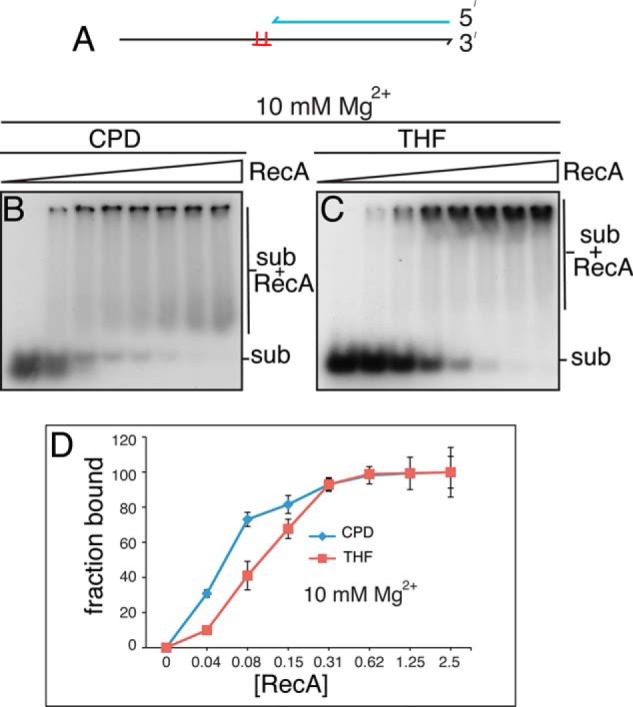
RecA displays preferential binding to model oligonucleotide substrates carrying a CPD compared with a THF. A, schematic of the oligonucleotide substrates. DNA binding reaction mixtures containing either the CPD or THF oligonucleotide substrates at 10 mm Mg(OAc)2 (B and C, respectively) and increasing amounts of RecA (no RecA, and 0.04–2.5 μm RecA, increasing by a factor of 2-fold from left to right) were incubated at 37 °C for 10 min and then analyzed by electrophoresis through native polyacrylamide gels as described under “Experimental Procedures.” D, DNA binding curves. Shown are the mean and S.D. (error bars) from three experiments. Representative gels are shown. Sub, substrate.
Effect of RecA on RecG-RuvAB NDD Product Formation
In the accompanying article (14), we showed that RuvAB also could generate the NDD product from SFs in a reaction that was stimulated by RecG. To begin to investigate the competition of different modes of RFR, we compared the kinetics of RecA- and RuvAB-catalyzed RFR using deproteinized SFs as the substrate (Fig. 7A). Because RecA NDD formation requires the formation of a RecA filament, we expected that the RuvAB-catalyzed reaction would be faster. This proved to be the case. Significant amounts of RuvAB NDD product had accumulated even by 5 min of incubation, whereas only some faint RecA NDD product accumulation could be seen at 10 min of incubation (Fig. 7A).
FIGURE 7.

RecA does not interfere with RuvAB RFR. A, comparison of the kinetics of RecA and RuvAB RFR. RFR reaction mixtures (100 μl) containing deproteinized SFs and either 3 μm RecA or RuvAB (40–120 nm) were incubated at 37 °C. Aliquots (15 μl) were removed at the indicated times, the reactions were stopped by the addition of 30 mm EDTA, and the products were analyzed by native gel electrophoresis. B, RecA does not interfere with RuvAB RFR. RFR reaction mixtures containing the replisome and SSB, as described in the accompanying article (14), containing RecA (3 μm), RecG (10 nm), and RuvAB (40–120 nm), as indicated, were incubated for 10 min at 37 °C, and the products were analyzed by native gel electrophoresis.
It is the case, however, that RecA, RuvAB, RecG, and SSB could all be present coincidentally at a SF in vivo. We therefore asked whether RecA interfered with RecG-stimulated RuvAB NDD formation in the presence of the replisome and SSB in standard RFR reaction mixtures, as described in the accompanying article (14). No inhibition was observed (Fig. 7B), suggesting that at an SF in vivo, RecG and RuvAB are the likely first actors, not RecA.
RecA RFR Activity Is Modulated by the Type of Template Lesion
During the course of our studies, we noted that there was a small, but significant, difference in the extent of RecA RFR product formation, depending on the nature of the site-specific template damage used. This effect is illustrated in Fig. 8. The standard template used to generate stalled forks throughout the studies reported in this and the accompanying paper (14) is one carrying a CPD lesion. This is template 225CPD in Fig. 8A. There was little RecA-RuvC product formation over the RuvC alone background when a template carrying a THF (5903THF) in the identical position was used, whereas RecA did stimulate RuvC cleavage of stalled forks when the 225CPD template was used (Fig. 8, A and C). We asked whether this effect might be sequence context-dependent by preparing CPD and THF templates where the lesion was in a different position on the template. Product formation with CPD templates was always better (varying between 0.5- and 2.5-fold greater cleavage products) than with THF templates. Another example is shown in Fig. 8, B and C. This difference is unlikely to result from differential fraying of the ends of the duplex because SSB did not stimulate RuvC cleavage preferentially on either template (Fig. 8, D–F). It has been demonstrated previously that RecA binds UV-irradiated DNA with higher avidity than it does unirradiated DNA (40); we therefore considered that this difference might be the result of preferred binding of RecA to the templates carrying a CPD. This proved to be the case.
Small oligonucleotide substrates were prepared, where there was a 5′-single-stranded extension adjacent to a duplex region. The template lesions were present in the ssDNA at the junction with the dsDNA (Fig. 9A). This arrangement is identical to that at the stalled fork (Fig. 4) and allows for RecA nucleation on the ssDNA and its extension across the lesion into the double-stranded DNA. Gel mobility shift assays were performed using substrates that had either a CPD or THF lesion. RecA binding (as measured by substrate bound/total substrate) to both substrates gave large gel shifts, suggesting that filaments had formed. Binding to the CPD substrate was about 2-fold better than binding to the THF substrate. This differential binding may account for the results observed in Fig. 8.
DISCUSSION
Access of RecA to Stalled Replication Forks
In the replication system used here, replication forks stall because of a collision with leading-strand template damage. In the absence of any other fork-processing enzymes, the lagging-strand polymerase and the replication fork helicase continue slowly downstream still associated with the stalled leading-strand polymerase. Once DnaG synthesizes a primer on the leading-strand template downstream of the stall site that is sensed by the leading-strand polymerase, it dissociates from its β clamp and cycles forward to the new primer. Replication then continues downstream (13, 29). In order to observe other fork-processing reactions, such as RFR, we add ddNTPs to arrest DNA synthesis; however, under these conditions, the stalled polymerase is likely to remain stably bound (30), and we have demonstrated that unwinding by DnaB does continue under these conditions (14). SSB coats the exposed ssDNA.
Unlike RecG and RuvAB (14), RecA could not interact productively with these stalled forks to generate a RFR product that we could score either by RuvC cleavage or because a branch migration reaction unwound the nascent DNA. Furthermore, we noted little effect of RecA when it was added directly to replication reactions as well, and it did not interfere with RecG-stimulated RuvAB RFR. Indiani et al. (31) have reported that RecA, under conditions similar to those described here in terms of RecA and SSB concentrations, will slow replisome progression. Indiani et al. observe the most profound effects of RecA when it is present before DNA synthesis commences (31) in their rolling-circle replication system, which proceeds by origin-independent loading of the replisome. In our system, the replisome is loaded via DnaA-dependent events at oriC; thus, there could be significant differences in the opportunities available for RecA to bind to the DNA. These authors also reported that the addition of RecA to a moving replisome will slow it (31); however, the difference in the rates of replisome progression in the presence and absence of RecA, which could be observed directly early in the rolling circle reaction, was only about 20–25%. It is certainly possible that RecA slows replisome progression to a similar extent in our system, but because of the time frame of our experiments (e.g. the first data point in Fig. 1 is at 1 min), it is unlikely that we would notice much of a difference in the accumulation of SF.
Using deproteinized stalled forks as a substrate, it was clear that RecA could regress the fork in a reaction that presumably must initiate with RecA binding the single-stranded leading-strand template downstream of the damage site (Fig. 4). How large a gap is required for this to occur is unclear. We demonstrated that the presence of SSB prevented RecA from nucleating on this ssDNA in a productive fashion. We note, however, that although SSB clearly blocked RecA RFR on the deproteinized substrate, it may not be the only factor that dictates the success of RecA loading to a stalled fork where the replication proteins are present. The structure of this stalled fork could be different from the one assumed after deproteinization. Indeed, genetic assays have strongly suggested that RecA has access to forks stalled either by denaturation of a temperature-sensitive DnaB (24) or by UV photoproducts (25). It is not clear at the moment how to accommodate our results with the genetic assays. Our reaction conditions are very similar to the concentrations of template (about 1 nm for an average of 2 chromosomes/cell), RecA (9000 (41) to 15,000 (42) copies/cell, about 3–5 μm), and SSB (1000–2000 tetramers/cell (43), about 0.4–0.8 μm) found in log phase E. coli cells. RecA and SSB binding to ssDNA substrates is competitive, with the outcome being dependent on Mg2+ concentration and temperature (37). Under our conditions, RecA could not outcompete SSB for binding to the ssDNA. This seems counter to the findings of Robu et al. (26), who showed that RecA regression of a model substrate similar to the SF used here was stimulated by SSB. However, in those experiments, RecA was allowed to interact with the ssDNA gap first in the absence of SSB. It seems unlikely that such a reaction scheme would happen in vivo. RecA RFR in the presence of SSB would therefore appear to require assistance from other mediator proteins, most likely RecFOR (44), which can direct the loading of RecA specifically onto gapped DNA coated with SSB (45). Induction of the SOS response increases RecA concentration by 10-fold, whereas SSB concentration does not change (46). In an SOS-induced cell, then, one might expect RecA to win the competition with SSB. However, RecA-mediated RFR in the temperature-sensitive dnaB strain did not require SOS induction and was recF- and recO-independent (24), whereas UV-irradiation does induce the SOS response, but under these conditions, RecA RFR did require RecFOR (47). Further study will be required for a consistent explanation of these events to emerge.
Okazaki Fragments and RecA-promoted Branch Migration
We found that the generation of significant levels of RFR products by RecA required that the Okazaki fragments on the nascent lagging strand be sealed. There are two possible explanations for this observation. 1) Productive formation of an RFR product requires that the HJ formed by RecA branch migrate away from the stalled fork toward the origin (Fig. 4). It is possible that in the absence of Okazaki fragment sealing, there is a competing strand exchange reaction, where the 3′-end of the Okazaki fragment on the lagging-strand sister invades the leading-strand sister duplex. The HJ formed in this manner could only branch migrate toward the fork to the position of the leading-strand stall, but formation of the HJ might be sufficient to compete with the productive RFR reaction. Sealing the Okazaki fragments should prevent this competition. 2) Gaps in the nascent lagging strand might inhibit RecA-promoted branch migration. In modeling four strand exchange reactions (such as what happens here at the stalled fork), it has been established that RecA can branch migrate past a double-strand break (48) and deletions and insertions of from 4 to 38 bp, whereas those of 120 bp or greater did not support branch migration (49). Although the nature of Okazaki fragments synthesized in the oriC replication system has not been examined closely, gaps of varying lengths between Okazaki fragments synthesized in a rolling circle replication system have been observed (50).
Differential Binding of RecA to Template Damage
We found a small, but significant, difference in the extent of RecA RFR product formation when the template damage was a CPD compared with a THF. Interestingly, this difference manifested itself although the RecA concentration in the RFR reaction was in considerable excess over the concentration required to saturate binding to the oligonucleotide substrates. This difference may reflect the fact that the nascent leading-strand gaps downstream of the stalled leading strand are small and inhibit RecA nucleation. Another possibility is that the single-stranded leading-strand template DNA in the gap is likely to take the form of a loop (29), the conformation of which could also be inhibitory to RecA binding.
RecA has been shown previously to preferentially bind UV-irradiated DNA compared with unirradiated DNA, with DNA containing exclusively the 6-4 dipyrimidine photoproduct being bound better than DNA containing the CPD (40). The explanation proposed at the time was that this might be a mechanism for preferential targeting of SOS mutagenesis to 6-4 dipyrimidine sites. Abasic sites occur under normal cell growth conditions, whereas UV irradiation is required to generate a CPD. It is possible that the preferential DNA binding that we observe by RecA reflects a mechanism to make it less likely that under normal growth conditions, the SOS response is induced by endogenous DNA template damage.
This work was supported, in whole or in part, by National Institutes of Health Grant GM34557 (to K. J. M.).
- HJ
- Holliday junction
- RFR
- replication fork regression
- HR
- homologous recombination
- CPD
- cyclopyrimidine dimer
- THF
- tetrahydrofuran
- ddNTP
- 2′,3′-dideoxyribonucleoside 5′-triphosphate
- CP1 and CP2
- cleavage product 1 and 2, respectively
- NDD
- nascent DNA duplex
- SF
- stalled fork
- SSB
- E. coli single-stranded DNA-binding protein
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 2. Gabbai C. B., Marians K. J. (2010) Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair 9, 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeeles J. T., Poli J., Marians K. J., Pasero P. (2013) Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 5, a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGlynn P., Lloyd R. G. (2002) Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 3, 859–870 [DOI] [PubMed] [Google Scholar]
- 5. Atkinson J., McGlynn P. (2009) Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 37, 3475–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Michel B., Grompone G., Florès M. J., Bidnenko V. (2004) Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 101, 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R. (2007) Recombination proteins and rescue of arrested replication forks. DNA Repair 6, 967–980 [DOI] [PubMed] [Google Scholar]
- 8. Whitby M. C., Vincent S. D., Lloyd R. G. (1994) Branch migration of Holliday junctions: identification of RecG protein as a junction specific DNA helicase. EMBO J. 13, 5220–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsaneva I. R., Müller B., West S. C. (1992) ATP-dependent branch migration of Holliday junctions promoted by the RuvA and RuvB proteins of E. coli. Cell 69, 1171–1180 [DOI] [PubMed] [Google Scholar]
- 10. Cassuto E., West S. C., Mursalim J., Conlon S., Howard-Flanders P. (1980) Initiation of genetic recombination: homologous pairing between duplex DNA molecules promoted by recA protein. Proc. Natl. Acad. Sci. U.S.A. 77, 3962–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McEntee K., Weinstock G. M., Lehman I. R. (1979) Initiation of general recombination catalyzed in vitro by the recA protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 2615–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shibata T., Cunningham R. P., DasGupta C., Radding C. M. (1979) Homologous pairing in genetic recombination: complexes of recA protein and DNA. Proc. Natl. Acad. Sci. U.S.A. 76, 5100–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yeeles J. T. P., Marians K. J. (2011) The Escherichia coli replisome is inherently DNA damage tolerant. Science 334, 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta S., Yeeles J. T. P., Marians K. J. (August 19, 2014) Regression of replication forks stalled by leading-strand template damage. I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the Holliday junctions formed by RecG preferentially. J. Biol. Chem. 10.1074/jbc.M114.587881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuzminov A. (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63, 751–813, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kowalczykowski S. C. (2000) Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 25, 156–165 [DOI] [PubMed] [Google Scholar]
- 17. Rothstein R., Michel B., Gangloff S. (2000) Replication fork pausing and recombination or “gimme a break.” Genes Dev. 14, 1–10 [PubMed] [Google Scholar]
- 18. Lusetti S. L., Cox M. M. (2002) The bacterial reca protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 71, 71–100 [DOI] [PubMed] [Google Scholar]
- 19. Asai T., Sommer S., Bailone A., Kogoma T. (1993) Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. EMBO J. 12, 3287–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masai H., Asai T., Kubota Y., Arai K., Kogoma T. (1994) Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO J. 13, 5338–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kogoma T. (1996) Recombination by replication. Cell 85, 625–627 [DOI] [PubMed] [Google Scholar]
- 22. West S. C., Cassuto E., Howard-Flanders P. (1981) Heteroduplex formation by recA protein: polarity of strand exchanges. Proc. Natl. Acad. Sci. U.S.A. 78, 6149–6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox M. M., Lehman I. R. (1981) Directionality and polarity in recA protein-promoted branch migration. Proc. Natl. Acad. Sci. U.S.A. 78, 6018–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seigneur M., Ehrlich S. D., Michel B. (2000) RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol. Microbiol. 38, 565–574 [DOI] [PubMed] [Google Scholar]
- 25. Courcelle J., Donaldson J. R., Chow K. H., Courcelle C. T. (2003) DNA damage-induced replication fork regression and processing in Escherichia coli. Science 299, 1064–1067 [DOI] [PubMed] [Google Scholar]
- 26. Robu M. E., Inman R. B., Cox M. M. (2001) RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heller R. C., Marians K. J. (2005) The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell 17, 733–743 [DOI] [PubMed] [Google Scholar]
- 28. Shah R., Bennett R. J., West S. C. (1994) Genetic recombination in E. coli: RuvC protein cleaves Holliday junctions at resolution hotspots in vitro. Cell 79, 853–864 [DOI] [PubMed] [Google Scholar]
- 29. Yeeles J. T., Marians K. J. (2013) Dynamics of leading-strand lesion skipping by the replisome. Mol. Cell 52, 855–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McInerney P., O'Donnell M. (2007) Replisome fate upon encountering a leading strand block and clearance from DNA by recombination proteins. J. Biol. Chem. 282, 25903–25916 [DOI] [PubMed] [Google Scholar]
- 31. Indiani C., Patel M., Goodman M. F., O'Donnell M. E. (2013) RecA acts as a switch to regulate polymerase occupancy in a moving replication fork. Proc. Natl. Acad. Sci. U.S.A. 110, 5410–5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Connolly B., Parsons C. A., Benson F. E., Dunderdale H. J., Sharples G. J., Lloyd R. G., West S. C. (1991) Resolution of Holliday junctions in vitro requires the Escherichia coli ruvC gene product. Proc. Natl. Acad. Sci. U.S.A. 88, 6063–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu C. A., Zechner E. L., Marians K. J. (1992) Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate Okazaki fragment size. J. Biol. Chem. 267, 4030–4044 [PubMed] [Google Scholar]
- 34. West S. C., Cassuto E., Howard-Flanders P. (1982) Role of SSB protein in RecA promoted branch migration reactions. Mol. Gen. Genet. 186, 333–338 [DOI] [PubMed] [Google Scholar]
- 35. Cox M. M., Soltis D. A., Livneh Z., Lehman I. R. (1983) On the role of single-stranded DNA binding protein in recA protein-promoted DNA strand exchange. J. Biol. Chem. 258, 2577–2585 [PubMed] [Google Scholar]
- 36. Kowalczykowski S. C., Krupp R. A. (1987) Effects of Escherichia coli SSB protein on the single-stranded DNA-dependent ATPase activity of Escherichia coli RecA protein. Evidence that SSB protein facilitates the binding of RecA protein to regions of secondary structure within single-stranded DNA. J. Mol. Biol. 193, 97–113 [DOI] [PubMed] [Google Scholar]
- 37. Kowalczykowski S. C., Clow J., Somani R., Varghese A. (1987) Effects of the Escherichia coli SSB protein on the binding of Escherichia coli RecA protein to single-stranded DNA. Demonstration of competitive binding and the lack of a specific protein-protein interaction. J. Mol. Biol. 193, 81–95 [DOI] [PubMed] [Google Scholar]
- 38. Benson F. E., West S. C. (1994) Substrate specificity of the Escherichia coli RuvC protein. Resolution of three- and four-stranded recombination intermediates. J. Biol. Chem. 269, 5195–5201 [PubMed] [Google Scholar]
- 39. Ferrari M. E., Fang J., Lohman T. M. (1997) A mutation in E. coli SSB protein (W54S) alters intra-tetramer negative cooperativity and inter-tetramer positive cooperativity for single-stranded DNA binding. Biophys. Chem. 64, 235–251 [DOI] [PubMed] [Google Scholar]
- 40. Rosenberg M., Echols H. (1990) Differential recognition of ultraviolet lesions by RecA protein. Possible mechanism for preferential targeting of SOS mutagenesis to (6-4) dipyrimidine sites. J. Biol. Chem. 265, 20641–20645 [PubMed] [Google Scholar]
- 41. Sommer S., Boudsocq F., Devoret R., Bailone A. (1998) Specific RecA amino acid changes affect RecA-UmuD′C interaction. Mol. Microbiol. 28, 281–291 [DOI] [PubMed] [Google Scholar]
- 42. Stohl E. A., Brockman J. P., Burkle K. L., Morimatsu K., Kowalczykowski S. C., Seifert H. S. (2003) Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J. Biol. Chem. 278, 2278–2285 [DOI] [PubMed] [Google Scholar]
- 43. Bobst E. V., Bobst A. M., Perrino F. W., Meyer R. R., Rein D. C. (1985) Variability in the nucleic acid binding site size and the amount of single-stranded DNA-binding protein in Escherichia coli. FEBS Lett. 181, 133–137 [DOI] [PubMed] [Google Scholar]
- 44. Umezu K., Chi N. W., Kolodner R. D. (1993) Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc. Natl. Acad. Sci. U.S.A. 90, 3875–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morimatsu K., Kowalczykowski S. C. (2003) RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol. Cell 11, 1337–1347 [DOI] [PubMed] [Google Scholar]
- 46. Courcelle J., Khodursky A., Peter B., Brown P. O., Hanawalt P. C. (2001) Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158, 41–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chow K. H., Courcelle J. (2004) RecO acts with RecF and RecR to protect and maintain replication forks blocked by UV-induced DNA damage in Escherichia coli. J. Biol. Chem. 279, 3492–3496 [DOI] [PubMed] [Google Scholar]
- 48. West S. C., Howard-Flanders P. (1984) Duplex-duplex interactions catalyzed by RecA protein allow strand exchanges to pass double-strand breaks in DNA. Cell 37, 683–691 [DOI] [PubMed] [Google Scholar]
- 49. Hahn T. R., West S., Howard-Flanders P. (1988) RecA-mediated strand exchange reactions between duplex DNA molecules containing damaged bases, deletions, and insertions. J. Biol. Chem. 263, 7431–7436 [PubMed] [Google Scholar]
- 50. Kurth I., Georgescu R. E., O'Donnell M. E. (2013) A solution to release twisted DNA during chromosome replication by coupled DNA polymerases. Nature 496, 119–122 [DOI] [PMC free article] [PubMed] [Google Scholar]