

Background: Amyloid-β oligomers trigger Alzheimer disease pathophysiology via the interaction of cellular prion protein (PrPC) with metabotropic glutamate receptor 5 (mGluR5).
Results: PrPC region 91–153 interacts preferentially with the activated conformation of mGluR5.
Conclusion: Antibodies against PrPC region 91–153 and agonist/antagonist-driven mGluR5 conformations regulate the PrPC-mGluR5 interaction.
Significance: These findings have therapeutic implications for Alzheimer disease by identifying compounds that modulate the PrPC-mGluR5 interaction.
Keywords: Alzheimer Disease, Amyloid-β (AB), Metabotropic Glutamate Receptor (mGluR), Neurodegenerative Disease, Prion, Protein-Protein Interaction, Therapeutic Modulation
Abstract
Soluble Amyloid-β oligomers (Aβo) can trigger Alzheimer disease (AD) pathophysiology by binding to cell surface cellular prion protein (PrPC). PrPC interacts physically with metabotropic glutamate receptor 5 (mGluR5), and this interaction controls the transmission of neurotoxic signals to intracellular substrates. Because the interruption of the signal transduction from PrPC to mGluR5 has therapeutic potential for AD, we developed assays to explore the effect of endogenous ligands, agonists/antagonists, and antibodies on the interaction between PrPC and mGluR5 in cell lines and mouse brain. We show that the PrPC segment of amino acids 91–153 mediates the interaction with mGluR5. Agonists of mGluR5 increase the mGluR5-PrPC interaction, whereas mGluR5 antagonists suppress protein association. Synthetic Aβo promotes the protein interaction in mouse brain and transfected HEK-293 cell membrane preparations. The interaction of PrPC and mGluR5 is enhanced dramatically in the brains of familial AD transgenic model mice. In brain homogenates with Aβo, the interaction of PrPC and mGluR5 is reversed by mGluR5-directed antagonists or antibodies directed against the PrPC segment of amino acids 91–153. Silent allosteric modulators of mGluR5 do not alter Glu or basal mGluR5 activity, but they disrupt the Aβo-induced interaction of mGluR5 with PrPC. The assays described here have the potential to identify and develop new compounds that inhibit the interaction of PrPC and mGluR5, which plays a pivotal role in the pathogenesis of Alzheimer disease by transmitting the signal from extracellular Aβo into the cytosol.
Introduction
Soluble amyloid-β oligomers (Aβo)3 are potent synaptotoxins and key mediators of Alzheimer disease (AD) pathophysiology (1–7). There is a robust correlation between disease severity and the concentration of prefibrillar, soluble Aβo (8–10). In contrast, the load of insoluble fibrillar amyloid plaques correlates poorly with the degree of dementia (8, 9, 11–13). Recent progress in the field has improved our understanding of the mechanisms by which Aβo interacts with synapses and triggers synaptotoxicity. Cellular prion protein (PrPC) has been identified as high-affinity cell surface receptor for Aβo (14), which has been confirmed both in vivo and in vitro (15–17). Numerous AD-related deficits are dependent on the presence of PrPC, such as Aβo-triggered synaptic dysfunction, dendritic spine and synapse loss, serotonin axon degeneration, epileptiform discharges, spatial learning and memory impairment, and the reduced survival of APP/PS1 transgenic mice (1, 14, 18–22). Aβo-PrPC complexes are extractable from human AD brains, and human AD brain-derived Aβo inhibits synaptic function in a PrPC-dependent manner (15, 19, 23, 24). Furthermore, blockade of the interaction between Aβo and PrPC, which has been mapped to regions 23–27 and 95–110 in PrPC, prevents Aβo-induced inhibition of synaptic plasticity (14, 17). However, the role of PrPC as a mediator of Aβo-induced toxicity does not appear to apply to all Aβo conformers and all assay models. Both Kessels et al. (25) and Calella et al. (26) found Aβo-induced impairment of hippocampal LTP independent of the presence of PrPC (25, 26). Moreover, another study verified an Aβo-dependent decline of long term memory consolidation that was independent of PrPC (16). Variable outcomes in toxicity assays are most likely due to distinct compositions of different Aβo preparations. Several different isoforms of Aβo exist, and certain forms have been demonstrated to trigger specific AD-related toxic effects, some of which might be independent of PrPC (3, 27–29).
When Aβo/PrPC complexes form, they trigger AD pathophysiology by interacting with mGluR5 (30). Both PrPC and mGluR5 receptors are located in lipid raft-like domains, and these are hypothesized to be the key location of Aβo-triggered induction of synaptotoxicity (31–34). Consistent with this finding, Renner et al. (35) revealed a PrPC- and mGluR5-dependent binding of Aβo to synapses using live single particle tracking of labeled Aβo in hippocampal neurons. They claim that Aβo cause synaptic dysfunction by triggering an abnormal clustering and overstabilization of mGluR5 receptors within the plasma membrane (35). Moreover, mGluR5 receptors are implicated in excitotoxicity and in transducing signals from the cell surface receptor PrPC into the cytosol (36, 37). Participation of mGluR5 in AD-related synaptotoxicity is consistent with the observation that Aβo-induced suppression of LTP and enhancement of long term depression (LTD) can be imitated by mGluR5 agonists and suppressed by mGluR5 antagonists (1, 38–40). Furthermore, incubation of neurons with Aβo initiates secondary messenger cascades that mimic the activation of mGluR receptors (7). Therefore, it is not surprising that multiple Aβo-induced AD-related deficits are dependent on the presence of both PrPC and mGluR5. Some examples include Aβo-triggered reduction of LTP and enhancement of LTD, activation of intracellular Fyn kinase, Aβo-induced dendritic spine loss, and spatial learning and memory deficits in APP/PS1 transgenic mice (19, 30, 41, 42).
Assuming that the physical interaction of PrPC with mGluR5 is essential for the transmission of Aβo-induced neurotoxic signals to intracellular substrates, targeting the PrPC-mGluR5 interaction has potential clinical implications for AD. The development of therapeutic strategies would benefit from a more precise knowledge about the interaction between PrPC and mGluR5. The structures of both PrPC and mGluR5 have been characterized (43–45), potentially facilitating the study of their interaction and regulation by Aβo. In this study, we used a library of PrPC deletion mutants as well as antibody mapping experiments to identify the 91–153 region of PrPC as accounting for the interaction with mGluR5. Moreover, we provide evidence that the interaction of mGluR5 with PrPC can be manipulated by agonist/antagonist-induced conformational changes of mGluR5 or antibody blockade of PrPC. Our findings also reveal a significant enhanced interaction between PrPC and mGluR5 in the brains of mice expressing familial AD transgenes. This stimulatory effect of the APP transgene is mimicked by the artificial supply of Aβo and inhibited by both mGluR5-directed antagonists and PrPC-directed antibodies, which target the binding sites of Aβo and mGluR5 on PrPC.
EXPERIMENTAL PROCEDURES
Aβ42 Oligomer Preparation
Aβ42 oligomers were prepared as described previously (14). All concentrations are given in monomer equivalents, with 1 μm of total Aβ42 peptide corresponding to ∼10 nm oligomeric species (14). Aβo was prepared immediately before use in glutamate-free F12 medium to avoid direct stimulation of glutamate receptors.
Mouse Strains
All mouse strains have been described previously (18, 46, 47). Males and females were used in approximately equal numbers, and none were excluded.
Drugs and Antibodies
The following metabotropic glutamate receptor-directed compounds were used: S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadi-azol-5-yl]-piperidin-1-yl}-methanone (Selleckchem), DCB (3,3′-dichloro-benzaldazine, Tocris Bioscience), dihydroxyphenylglycine (Tocris Bioscience), 6-methoxy-N-(4-methoxyphenyl)-4-quinazolin-amine-hydrochloride (Tocris Bioscience), 3-((2-methyl-4-thiazolyl) ethynyl)pyridine (MTEP) hydrochloride (Tocris Bioscience), 6-methyl-2-(phenylazo)pyridin-3-ol (Tocris Bioscience), and 4-butoxy-N-(2,4-di-fluorophenyl)benzamide (Tocris Bioscience). The following antibodies were used: 6D11 (mouse monoclonal antibody, epitope between residues 97 and 100 of PrPC, Covance/Signet) and M20 (affinity-purified goat polyclonal antibody raised against the C-terminal part of mouse PrPC, Santa Cruz Biotechnology). The following antibodies were used for antibody mapping experiments: 6D11 (Covance, epitope between residues 97 and 100), 3F4 (Covance, epitope between residues 108 and 111), Pri308 (Cayman Chemical, epitope between residues 106 and 126), 6G3 (Santa Cruz Biotechnology, epitope between residues 130 and 150), Bar 233 (Cayman Chemical, epitope between residues 141 and 151), Bar221 (Cayman Chemical, epitope between residues 141 and 151), M20 (Santa Cruz Biotechnology, raised against the C-terminal part of mouse PrPC), 11C6 (Cayman Chemical, epitope between residues 142 and 160), and SAF70 (Cayman Chemical, epitope between residues 156 and 162).
Cell Culture and Preparation of Cell Lysates
HEK-293T cells were maintained in DMEM supplemented with 10% FCS, 1% l-glutamine (2 mm final concentration), 1% sodium pyruvate (1 mm final concentration) and 1% penicillin/streptomycin (100 units/ml). Cells were transfected using Lipofectamine 2000 transfection reagent (Invitrogen). To prepare detergent-solubilized cell lysates, cells were rinsed with ice-cold PBS and solubilized in radioimmune precipitation assay (RIPA) buffer containing 150 mm sodium chloride, 1.0% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris (pH 7.4), 1 mm EDTA, complete protease inhibitor mixture (Roche), and phosSTOP phosphatase inhibitor mixture (Roche). The insoluble fraction was removed by centrifugation at 20,000 × g, and the supernatant was used for protein assays.
Cell Surface Biotinylation
Cells were rinsed three times in ice-cold PBS to remove primary amine-containing culture medium and incubated in PBS containing 2 mm EZ-Link NHS-biotin (Thermo Scientific) for 30 min at 4 °C. Cells were rinsed three times in quenching buffer (100 mm glycine in PBS) to block any unreacted NHS-biotin. Proteins were extracted in RIPA lysis buffer, separated by SDS-PAGE, and analyzed by immunoblotting.
Preparation of RIPA Buffer-soluble Extracts from Brain Tissue
Mouse forebrains were homogenized in three volumes of ice-cold (w/v) 50 mm Tris-HCl, 150 mm NaCl (pH 7.4) (TBS), complete protease inhibitor mixture (Roche), and phosSTOP phosphatase inhibitor mixture (Roche) using a Teflon homogenizer. The homogenized brain extract was centrifuged at 100,000 × g for 20 min at 4 °C, and the pellet was resuspended in RIPA buffer. The resuspension was centrifuged at 100,000 × g for 20 min. The supernatant was used for protein assays.
Crude Membrane Preparations
HEK-293 cells or mouse forebrains were homogenized in homogenization buffer (10 mm Tris-HCl (pH 7.4), 1 mm EDTA, 200 mm sucrose, complete protease inhibitor mixture (Roche), and phosSTOP phosphatase inhibitor mixture (Roche)), and insoluble material was removed by centrifugation at 900 × g for 10 min at 4 °C. The supernatant was centrifuged at 110,000 × g for 75 min at 4 °C, and the membrane pellet was resuspended in solubilization buffer (10 mm Tris-HCl (pH 7.4), 1 mm EDTA, complete protease inhibitor mixture (Roche), and phosphatase inhibitor (Roche)) for 3 h to overnight at 4 °C. Proteins were extracted by 1.0% Nonidet P-40 for 1 h at 4 °C and used for protein assays.
Immunoprecipitation
One microgram of capture antibody was incubated overnight at 4 °C with 1 mg of detergent-solubilized lysate protein with continuous mixing. The antibodies used were anti-Myc (Sigma-Aldrich, catalog no. C3956) for anti-Myc immunoprecipitation and Saf32 (Cayman Chemical, catalog no. 189720) for anti-PrPC immunoprecipitation in all experiments except anti-PrPC immunoprecipitation experiments of PrPC deletion mutants, where a mixture of both Bar 233 (Cayman Chemical, catalog no. 10009036) and Saf32 (Cayman Chemical, catalog no. 189720) was used as capture antibodies. PureProteome protein A/G mix magnetic beads (Millipore, catalog no. LSKMAGAG10) or goat anti-rabbit IgG magnetic beads (New England Biolabs, catalog no. S1432S) were washed in wash buffer (PBS and 0.1% Tween 20 (pH 7.4)). The preformed antibody-antigen complex was added to the beads and incubated for 1 h (in the case of HEK-293 cell experiments) or 3 h (in the case of mouse brain experiments) at 4 °C with gentle rotation. For some experiments, antibodies were covalently coupled to protein A/G mix magnetic beads. Here beads were washed in wash buffer, incubated with double the amount of appropriate antibody for 1 h at 4 °C, and washed three times in wash buffer and once in cross-link buffer (20 mm sodium phosphate and 0.15 m NaCl (pH 7.4)). Antibodies were then immobilized on the beads by incubation with 2.5 mm Bis(sulfosuccinimidyl) suberate (BS3) cross-linker for 1 h at 4 °C. The reaction was quenched by 17 mm Tris-HCl (pH 7.4) and incubation for 1 h at 4 °C. Non-immobilized antibodies were removed by one wash in 0.2 m glycine-HCl (pH 2.5), followed by three washes in wash buffer. Beads were incubated with detergent-solubilized lysate overnight at 4 °C with gentle rotation and washed three times in wash buffer prior to elution of proteins in SDS-PAGE sample loading buffer. The immunoprecipitated complexes were then resolved by SDS-PAGE and immunoblotted.
Plate-based Binding Assay of PrPC-mGluR5
White 384-well MaxiSorp microplates (Nunc, catalog no. 460372) were coated with 20 μl/well of 150 μm purified recombinant PrPC (amino acids 23–230) overnight at 4 °C. Plates were washed and blocked with 110 μl/well of protein-free PBS-T20 blocking buffer (Pierce) for 3–5 h at room temperature. Immobilized PrPC was exposed to detergent lysates of HEK-293 cells expressing Myc-mGluR (1% N-nonanoyl-N-methylglucamine in PBS, complete protease inhibitor mixture (Roche), and phosphatase inhibitor (Roche)) in 3-fold serial dilutions and incubated overnight at 4 °C. Plates were washed and incubated with 20 μl/well of primary antibody solution (anti-Myc, 1:2000 dilution in PBS-T) for 2 h at room temperature. Plates were washed and incubated with 20 μl/well of secondary antibody solution (europium-conjugated, 1:8000 in dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) buffer) for 1–2 h at room temperature. Plates were washed, 20 μl/well of DELFIA enhancement solution was added, and imaging was performed using the Victor 3V microplate reader (PerkinElmer Life Sciences).
Immunoblots
Proteins were electrophoresed through precast 4–20% tris-glycine gels (Bio-Rad) and transferred to nitrocellulose membranes (Invitrogen) with an iBlotTM gel transfer device (Invitrogen). Membranes were blocked (blocking buffer for fluorescent Western blotting, Rockland, catalog no. MB-070-010) for 1 h at room temperature and incubated overnight in primary antibodies. The following antibodies were used: 6D11 (Covance, catalog no. 39810-500; 1:1000), 6E10 (Millipore MAB 1560, 1:1000), anti-actin (Sigma-Aldrich, catalog no. A2066, 1:10,000), anti-Myc (Sigma-Aldrich, catalog no. C3956, 1:1000), anti-mGluR5 (Millipore, catalog no. AB5675, 1:500), Bar 233 (Cayman Chemical, catalog no. 10009036, 1:200), Saf32 (Cayman Chemical, catalog no. 189720, 1:200), and IRDye Streptavidin 680 (Odyssey, 1:20,000). Secondary antibodies were applied for 1 h at room temperature (Odyssey donkey anti-mouse or donkey anti-rabbit IRDye 680 or 800), and proteins were visualized with a Licor Odyssey infrared imaging system. Quantification of band intensities was performed within a linear range of exposure.
RESULTS
Mapping the mGluR5-interacting Region in PrPC
The mGluR5 binding regions in PrPC were mapped using PrPC deletion mutants (Fig. 1) and antibody mapping experiments (Fig. 2). All PrPC deletion mutants expressed at similar levels in HEK-293 cells (Fig. 1, A, bottom panel, and C, bottom panel). Trafficking defects for the mutants were excluded by cell surface biotinylation of living cells with the membrane-impermeable chemical EZ-Link NHS-Biotin. A comparable streptavidin signal was observed in anti-PrPC immunoprecipitates of cells expressing deletion mutants and the full-length version of PrPC (Fig. 1A, top panel). This indicates that deletions do not prevent PrPC mutants from reaching the plasma membrane, which is a requirement to evaluate their interaction with mGluR5. Then, evaluation of the interaction between Myc-mGluR5 and different versions of PrPC was performed (Fig. 1C). We found that deletions spanning residues 91–153 reduced the interaction of PrPC with mGluR5. Most strikingly, we observed a reduction in the amount of PrPC-d91–111 pulled down after Myc-mGluR5 immunoprecipitation (Fig. 1D, 23 ± 11%, n = 4, blue bar) and the complementary reduction of the Myc-mGluR5 signal in PrPC-d91–111 immunoprecipitation (Fig. 1D, 16 ± 8.6%, n = 4, blue bar) compared with the full-length PrPC. Similarly, deletion of the β-sheet-rich region in PrPC decreased the PrPC signal in anti-Myc immunoprecipitates (Fig. 1D, 40 ± 16%, n = 4, red bar). Moreover, deletion of helix 1 in PrPC showed a reduction in the Myc-mGluR5 signal in PrPC immunoprecipitation (Fig. 1D, 40 ± 9.5%, n = 4, yellow bar). These results indicate that the region spanning residues 91–153 is involved in binding Myc-mGluR5. The absence of a reduced coimmunoprecipitation signal with the PrPC deletion mutants that lack elements outside of region 91–153 imply that regions other than 91–153 are not essential for the interaction with mGluR5.
FIGURE 1.

Myc-mGluR5 binds to residues 91–153 of PrPC. A, cell surfaces of HEK-293 cells transfected with plasmids directing the expression of either PrPC-Fl or each of the indicated PrPC deletion mutants were biotinylated. Detergent-solubilized lysates (input) were immunoblotted with anti-PrPC, and anti-PrPC immunoprecipitates (IP) were immunoblotted with Streptavidin. B, schematic of the PrPC structure and deletion (d) locations (gray, residues 23–51; green, octa-repeat (OR); blue, residues 91–111; red, β-sheet-rich region; yellow, helix 1 (H1); purple, helix 2 (H2); orange, helix 3 (H3). The Aβo binding sites in PrPC are highlighted in dark blue (residues 23–27 and 95–110), and the mGluR5 binding sites are highlighted in dark red (residues 91–153). SP, signal peptide; GPI, glycosylphosphatidylinositol. C, HEK-293 cells were transfected with either empty pcDNA3 vector or vector for Myc-mGluR5 or PrPC-Fl (full-length) or cotransfected for either Myc-mGluR5 and PrPC-Fl or PrPC deletion mutants, as indicated. Detergent-solubilized lysates (Input), anti-Myc immunoprecipitates, and anti-PrPC immunoprecipitates were immunoblotted with either anti-Myc or anti-PrPC, as indicated. D, the quantified PrPC deletion mutant signal in anti-Myc immunoprecipitates is normalized to the PrPC-Fl signal in anti-Myc immunoprecipitates. Data are mean ± S.E. from four experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC-d91–111 and Myc-mGluR5 with PrPC-dBeta is reduced significantly. *, p = 0.0359; **, p < 0.0059; one-sample Student's t test. E, the quantified Myc signal in anti-PrPC deletion mutant immunoprecipitates is normalized to the Myc signal in anti-PrPC-Fl immunoprecipitates. Data are mean ± S.E. from four experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC-d91–111 (**, p = 0.0023 by one-sample t test) and coimmunoprecipitation of Myc-mGluR5 with PrPC-dHelix1 is reduced significantly (**, p = 0.0081 by one-sample Student's t test).
FIGURE 2.

Antibodies directed against region 91–153 of PrPC block the Myc-mGluR5 binding to immobilized PrPC. A and D–F, relative binding of detergent-solubilized Myc-mGluR to immobilized recombinant PrPC. A, immobilized PrPC strongly interacts with Myc-mGluR5 lysates but not with Myc-mGluR8 lysates or control lysates. B, Myc-mGluR lysates used in A were immunoblotted with anti-Myc. C, schematic of the PrPC structure and antibody epitopes. Antibodies used in mapping experiments are 6D11 (epitope, 97–100), 3F4 (epitope, 108–111), Pri308 (epitope, 106–126), 6G3 (epitope, 130–150), Bar221 and Bar 233 (Bar221/3; epitope, 141–151), Saf70 (epitope, 156–162), 11C6 (epitope, 142–160), and M20 (epitope, C-terminal residues) (73–75). SP, signal peptide; OR, octa-repeat; GPI, glycosylphosphatidylinositol. D, antibodies recognizing region 91–111 of PrPC (6D11, 3F4, and Pri308) disrupt the interaction between Myc-mGluR5 and immobilized PrPC dose-dependently. E, antibodies directed against the β-sheet-rich region and helix 1 of PrPC (BAR233, 6G3, and BAR221) blocked the Myc-mGluR5 binding to PrPC dose-dependently. F, no disruption of the Myc-mGluR5 binding to immobilized PrPC was initiated by control antibodies not recognizing PrPC (GAPDH) or antibodies recognizing exclusively domains other than region 91–153 of PrPC (SAF61, M20, and 11C6).
To confirm these results, we took a different approach to map the regions of PrPC interacting with mGluR5. Recombinant full-length PrPC was used to coat MaxiSorp microplates, which were then incubated with detergent-soluble membrane fractions prepared from HEK-293 cells expressing Myc-mGluR (Fig. 2). A robust signal was detected with Myc-mGluR5 lysates (Fig. 2A, black dotted line). Even though Myc-mGluR8 expression was higher than Myc-mGluR5 expression (Fig. 2B), the closely related protein Myc-mGluR8 (Fig. 2A, red dotted line) and control cell lysates (Fig. 2A, green dotted line) produced no detectable signal in the plate-based binding assay of PrPC-mGluR, demonstrating the specificity of this assay toward Myc-mGluR5. Using this assay, we screened a panel of anti-prion protein antibodies for their ability to disrupt the interaction between Myc-mGluR5 with immobilized PrPC (Fig. 2C). Antibodies recognizing the 91–111 region of PrPC (6D11, 3F4, and Pri308) blocked the protein interaction in a dose-dependent manner (Fig. 2D). In addition, antibodies recognizing the β-sheet-rich region and helix 1 of PrPC (BAR233, 6G3, and BAR221) showed a similar interaction inhibition (Fig. 2E). In contrast, control antibodies not recognizing PrPC (GAPDH) and antibodies recognizing domains of PrPC outside of region 91–153 (SAF70, M20, 11C6, and others not shown) had no effect on the interaction (Fig. 2F). These data are consistent with deletion mapping results indicating that region 91–153 of PrPC mediates the interaction with mGluR5.
Regulation of the PrPC-mGluR5 Interaction
We analyzed whether the interaction between PrPC and Myc-mGluR5 can be regulated by agonist/antagonist-driven conformational changes of mGluR5 (Fig. 3). Our results indicate that negative allosteric modulators weaken the interaction between PrPC and Myc-mGluR5, and the strongest effect was seen with MTEP. This drug reduces the coimmunoprecipitation of PrPC with mGluR5 (Fig. 3C, 33 ± 5.2%, n = 12) and the complementary coimmunoprecipitation (Fig. 3D, 46 ± 6.9%, n = 10) compared with the full interaction signal of untreated cells. We observed that this MTEP-triggered negative regulation of the PrPC-mGluR5 coimmunoprecipitation is dose-dependent (Fig. 4). On the other hand, agonists and positive allosteric modulators increased the coimmunoprecipitation of PrPC and Myc-mGluR5, and the strongest effect was seen by treating cells and detergent solubilized lysates with the functional glutamate analog DHPG (Fig. 3, C and D). In the presence of DHPG, the PrPC-mGluR5 interaction increased (Fig. 3C, 260 ± 24%, n = 11) when mGluR5 was immunoprecipitated, and, in the same fashion, complementary coimmunoprecipitation was increased (Fig. 3D, 263 ± 20%, n = 11) compared with the amount of coimmunoprecipitation in untreated cells. DCB is a silent allosteric modulator of mGluR5, competing with MTEP but not inhibiting the receptor (48). Application of DCB alone did not alter the interaction between PrPC and Myc-mGluR5 (Fig. 3B). However, incubation of cells with DCB 10 min prior to application of MTEP prevented the blocking of the PrPC-Myc-mGluR5 interaction triggered by MTEP (Fig. 3B, sixth lane). These results indicate that treatment with DCB prevents the negative allosteric modulator MTEP from inducing conformational changes that could alter the interaction of PrPC and Myc-mGluR5.
FIGURE 3.

Agonist/antagonist-driven conformational states of mGluR5 regulate the interaction of HEK-293 cell-expressed PrPC and Myc-mGluR5. A, HEK-293 cells were transfected with either empty pcDNA3 vector or vector for PrPC or Myc-mGluR5 or cotransfected for PrPC and Myc-mGluR5. Cells were incubated for 10 min at 37 °C with 2.5 μm indicated drug. Detergent-solubilized lysates (Input) were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates (IP) and anti-PrPC immunoprecipitates were supplied with 2.5 μm indicated drug and immunoblotted with either anti-Myc or anti-PrPC. VU-0357121, 4-butoxy-N-(2,4-difluorophenyl)benzamide; SIB-1757, 6-methyl-2-(phenylazo)pyridin-3-ol; ADX-47273, S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadi-azol-5-yl]-piperidin-1-yl}-methanone; LY-456236, 6-methoxy-N-(4-methoxyphenyl)-4-quinazolinamine hydrochloride. B, cells were incubated for 10 min at 37 °C with indicated drug concentrations. One culture was preincubated for 10 min with 25 μm DCB prior to incubation for 10 min at 37 °C with 2.5 μm MTEP. Detergent-solubilized lysates were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates and anti-PrPC immunoprecipitates were supplied with the indicated drug concentrations and immunoblotted with either anti-Myc or anti-PrPC as indicated. C and D, positive allosteric modulators and agonists are shown in green, silent allosteric modulators are shown in yellow, and negative allosteric modulators are shown in red. ns, not significant. C, quantification of the PrPC signal in anti-Myc immunoprecipitates is normalized to the signal of untreated samples. Data are mean ± S.E. from four experiments, apart from DHPG, DCB and MTEP application from 11, 6 and 12 independent experiments, respectively. Coimmunoprecipitation of PrPC with Myc-mGluR5 is enhanced significantly by DHPG (***, p = 0.00049 by Wilcoxon signed-rank test) and reduced significantly by MTEP (***, p = 0.00024 by Wilcoxon signed-rank test). On the contrary, LY-456236, a selective mGluR1 receptor antagonist, did not significantly alter the PrPC signal in anti-Myc immunoprecipitates. D, quantification of the Myc signal in anti-PrPC immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from four experiments, apart from DHPG and MTEP application from 11 independent experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC is enhanced significantly by DHPG (***, p = 0.00098 by Wilcoxon signed-rank test) and reduced significantly by MTEP (***, p = 0.00049 by Wilcoxon signed-rank test). In contrast, LY-456236 did not significantly change the Myc signal in anti-PrPC immunoprecipitates.
FIGURE 4.

Antagonist-driven conformational states of mGluR5 regulate the interaction of HEK-293 cell-expressed PrPC and Myc-mGluR5 in a dose-dependent manner. A, HEK-293 cells were cotransfected for PrPC and Myc-mGluR5. Cells were incubated for 10 min at 37 °C with the indicated MTEP concentration, and detergent-solubilized lysates (Input) were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates (IP) and anti-PrPC immunoprecipitates were supplied with the indicated MTEP concentration and immunoblotted with either anti-Myc or anti-PrPC as indicated. B, quantification of the PrPC signal in anti-Myc immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from four experiments, apart from 2.5 μm MTEP application from 11 independent experiments. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; one-sample Student's t test. C, quantification of the Myc signal in anti-PrPC immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from four experiments, apart from 2.5 μm MTEP application from 10 experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC is reduced significantly by MTEP. *, p < 0.05; **, p < 0.01; ****, p < 0.0001; one-sample Student's t test).
Confirmation of Drug Specificity with Chimeric mGluRs
To further determine the drug specificity of alterations produced on the coimmunoprecipitation of PrPC and Myc-mGluR5, driven by agonist/antagonist induced conformational alterations of mGluR5, we investigated the effect of mGluR5-directed endogenous ligand and agonists/antagonists on the interaction between PrPC and different Myc-mGluR chimeras (Fig. 5). As a control, we cotransfected PrPC and Myc-mGluR8. The coimmunoprecipitation of these proteins was significantly lower compared with the coimmunoprecipitation signal of Myc-mGluR5 with PrPC (Fig. 5, C and D, red bars versus black bars, p = 0.0006 and p = 0.0005 by one-sample Student's t test, respectively). Moreover, both chimeric Myc-mGluR-N5/C8 and Myc-mGluR-N8/C5 proteins coimmunoprecipitated less effectively with PrPC compared with Myc-mGluR5 (Fig. 5, C and D, green and purple bars versus black bars). These chimeric proteins contain the extracellular domain of either Myc-mGluR5 or Myc-mGluR8 and the transmembrane-spanning domain of the other metabotropic receptor, respectively (Fig. 5B). As seen before, PrPC and Myc-mGluR5 coimmunoprecipitated more effectively in the presence of glutamate and DHPG but less effectively in the presence of MTEP (Fig. 5, E and F). In contrast, MTEP did not show any effect on the coimmunoprecipitation of PrPC and Myc-mGluR-N5/C8 (Fig. 5, G and H). Moreover, the PrPC-Myc-mGluR-N8/C5 coimmunoprecipitation signal was not affected by DHPG (Fig. 5, I and J). Therefore, the highly specific mGluR5-directed drugs MTEP and DHPG failed to alter the interaction between PrPC and Myc-mGluR when their implicated receptor binding element was missing. In contrast, glutamate effects are observable across all classes of mGluRs. These results provide further mechanistic support for the specificity of the mGluR5 conformational regulation of PrPC association.
FIGURE 5.

Agonist/antagonist-driven conformational states of mGluR5 regulate the interaction of PrPC with Myc-mGluR5 but not PrPC with Myc-mGluR8 and only partially of PrPC with chimeric Myc-mGluR proteins. A, schematics showing the design of Myc-tagged mGluR mutants and location of ligand binding. B, HEK-293 cells were cotransfected with vectors for PrPC and different Myc-tagged mGluRs as indicated. Cells were incubated for 10 min at 37 °C with 100 μm glutamate or 2.5 μm indicated drug, and detergent-solubilized lysates (Input) were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates (IP) and anti-PrPC immunoprecipitates were incubated with 100 μm glutamate or 2.5 μm indicated drug and immunoblotted with either anti-Myc or anti-PrPC as indicated. C, quantification of the PrPC signal in anti-Myc immunoprecipitates is normalized to the PrPC signal in anti-Myc-mGluR5 immunoprecipitates. Data are mean ± S.E. from four experiments. The PrPC signal in anti-Myc-mGluR8 immunoprecipitates (***, p = 0.0006 by one-sample Student's t test) and in Myc-mGluR-N5/C8 immunoprecipitates (*, p = 0.0329 by one-sample Student's t test) is reduced significantly. D, quantification of the Myc signal in anti-PrPC immunoprecipitates is normalized to the Myc-mGluR5 signal in anti-PrPC immunoprecipitates. Data are mean ± S.E. from four experiments. The interaction of Myc-mGluR8 and PrPC is reduced significantly (***, p = 0.0005 by one-sample Student's t test). E, G, and I, quantification of the PrPC signal in anti-Myc immunoprecipitates is normalized to the signal of untreated samples. Data are mean ± S.E. from two to ten experiments. ns, not significant. F, H, and J, quantification of the Myc signal in anti-PrPC immunoprecipitates is normalized to the signal of untreated samples. E, coimmunoprecipitation of PrPC with Myc-mGluR5 is enhanced significantly by glutamate (*, p = 0.0313 by Wilcoxon signed-rank test) and DHPG (**, p = 0.0020 by Wilcoxon signed-rank test) and reduced significantly by MTEP (**, p = 0.0020 by Wilcoxon signed-rank test). F, coimmunoprecipitation of Myc-mGluR5 with PrPC is enhanced significantly by glutamate (*, p = 0.0313 by Wilcoxon signed-rank test) and DHPG (**, p = 0.0020 by Wilcoxon signed-rank test) and reduced significantly by MTEP (**, p = 0.0020 by Wilcoxon signed-rank test). G, coimmunoprecipitation of PrPC with Myc-mGluR-N5/C8 is not altered significantly by conformational mGluR changes because of low sample size (n = 2). However, a trend is clearly observable. H, coimmunoprecipitation of Myc-mGluR-N5/C8 with PrPC is enhanced significantly by glutamate (*, p = 0.0332 by one-sample Student's t test) and DHPG (*, p = 0.0492 by one-sample Student's t test) but not altered by MTEP. I, coimmunoprecipitation of PrPC with Myc-mGluR-N8/C5 is reduced significantly by MTEP (*, p = 0.0498 by one-sample Student's t test). J, coimmunoprecipitation of Myc-mGluR-N8/C5 with PrPC is enhanced significantly by glutamate (*, p = 0.0421 by one-sample Student's t test).
Conformational Regulation of mGluR5 Requires a Membrane Environment
To determine whether the modulation of PrPC-mGluR5 complex strength by mGluR5 conformational changes (agonist/antagonist binding) is dependent on the stability of the plasma membrane, we analyzed how this modulation is affected by the administration of agonist/antagonist to different cellular and subcellular fractions (Fig. 6). PrPC and Myc-mGluR5 coimmunoprecipitate less effectively when MTEP is applied constantly at all steps of the immunoprecipitation process, first to the intact cells and later to the detergent solubilized lysates. Similarly, DHPG is more effective when the drug is applied at all steps of the immunoprecipitation process (cells and detergent-solubilized lysates) (Fig. 6A). This effect is even stronger when membrane preparations of untreated cells expressing PrPC and Myc-mGluR5 are subsequently incubated with MTEP and DHPG (Fig. 6B). Membrane fractions were prepared in the absence of SDS, which confirms that the coimmunoprecipitation of PrPC with Myc-mGluR5 does not occur in aggregated protein complexes and is not dependent on non-native protein interactions induced by SDS. Moreover, the regulation of this protein-protein interaction by mGluR5-directed drugs is still observable in the absence of denaturing detergent. However, compound-induced modulation of the PrPC-Myc-mGluR5 interaction is less effective when cells or detergent-solubilized lysates alone are incubated with MTEP or DHPG (Fig. 6, C and D), with the lowest modulation seen in Myc-mGluR5 coimmunoprecipitation with PrPC after treatment of detergent-solubilized lysates only (Fig. 6D). These results indicate that agonist/antagonist-induced modulations of the PrPC-Myc-mGluR5 interaction are strong only when mGluR5 receptors are treated in their native membrane-embedded conformation and drugs are present throughout coimmunoprecipitation.
FIGURE 6.

The modulatory effect of agonist/antagonist-driven changes on the interaction of PrPC with Myc-mGluR5 is strongest after treatment of HEK-293 cell membrane preparations. HEK-293 cells were cotransfected with vectors for PrPC and Myc-mGluR5. A, cells were incubated for 10 min at 37 °C with 2.5 μm indicated drug, and detergent-solubilized lysates (Input) were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates (IP) and anti-PrPC immunoprecipitates were supplied with 2.5 μm indicated drug and immunoblotted with either anti-Myc or anti-PrPC as indicated. B, membrane fractions were prepared in the absence of SDS, incubated for 3 h at 4 °C with 2.5 μm indicated drug, and then membrane proteins were extracted by Nonidet P-40. Membrane extractions (Input), anti-Myc immunoprecipitates, and anti-PrPC immunoprecipitates of membrane extractions were immunoblotted with either anti-Myc or anti-PrPC as indicated. C, cells were incubated for 10 min at 37 °C with 2.5 μm indicated drug, and detergent-solubilized lysates (Input), anti-Myc immunoprecipitates, and anti-PrPC immunoprecipitates were immunoblotted with either anti-Myc or anti-PrPC as indicated. D, detergent-solubilized cell lysates were supplied with 2.5 μm indicated compound, and detergent solubilized lysates (Input), anti-Myc immunoprecipitates, and anti-PrPC immunoprecipitates were immunoblotted with either anti-Myc or anti-PrPC as indicated.
Conformational Regulation of Endogenous Brain PrPC-mGluR5 Interaction
We further investigated whether or not agonist/antagonist-driven conformational states of mGluR5 are also able to regulate the brain PrPC-mGluR5 interaction (Fig. 7). We first determined that coimmunoprecipitation of brain PrPC with mGluR5 requires both proteins and is absent in either single Grm5−/− or Prnp−/− knockout mouse brain. Treatment of detergent-solubilized membrane fractions of WT brain, cleared by 100,000 × g centrifugation after the extraction of membrane proteins by Nonidet P-40, showed no agonist/antagonist-dependent regulation of the mGluR5 signal in anti-PrPC immunoprecipitates even though coimmunoprecipitation is strong (Fig. 7A). However, the mGluR5 signal in anti-PrPC immunoprecipitates was altered significantly when membrane fractions of WT brains were treated with mGluR5 agonists/antagonists followed by the extraction of proteins with Nonidet P-40 and removal of large particulate material at 20,000 × g (Fig. 7B). As seen in HEK membranes, MTEP-induced changes in mGluR5 conformation trigger a less effective mGluR5-PrPC interaction (Fig. 7C, red bar), whereas DHPG-induced changes in the mGluR5 conformation cause mGluR5 to coimmunoprecipitate more efficiently with PrPC (Fig. 7C, green bar). This effect is seen in the absence of SDS, which further verifies that a modulatory interaction between brain PrPC and mGluR5 is not dependent on non-native protein interactions. However, even after drug treatment of membrane fractions, if smaller proteolipid complexes are removed by 100,000 × g ultracentrifugation in the presence of Nonidet P-40, then ligand regulation of the protein-protein association is lost. These experiments demonstrate that drug-induced conformational changes of mGluR5 can regulate the brain PrPC-mGluR5 complex interaction in a certain array of proteins and membrane environment.
FIGURE 7.

Agonist/antagonist-driven conformational states of mGluR5 regulate the interaction of PrPC with mGluR5 in brain-derived membrane fractions. Each immunoprecipitation was performed from one Grm5−/−, Prnp−/−, or WT mouse brain hemisphere. For each experiment, 1.5 WT brain hemispheres were combined, and three membrane pellets were prepared to ensure an equal amount of protein in each membrane aliquot. Membrane fractions were prepared in the absence of SDS and incubated overnight at 4 °C with 2.5 μm indicated drug. Membrane proteins were extracted by Nonidet P-40, and membrane extractions (Input) and anti-PrPC immunoprecipitates of membrane extractions were immunoblotted with either anti-mGluR5 or anti-PrPC as indicated. A, membrane extractions cleared by high-speed ultracentrifugation spin did not show an agonist/antagonist-dependent regulation of the mGluR5 signal in anti-PrPC immunoprecipitates (IP). Co-IP, coimmunoprecipitation. B, membrane extractions cleared by low-speed microcentrifugation spin showed an agonist/antagonist-dependent regulation of the mGluR5 signal in anti-PrPC immunoprecipitates. C, the quantified mGluR5 dimer and monomer signal in anti-PrPC immunoprecipitates from membrane extractions cleared by low-speed microcentrifugation spin were combined because the ratio between the dimer and the monomer was not changed by treatment. The signal in anti-PrPC immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from four individual experiments, i.e. from six WT brains total, with one immunoprecipitation being performed from one hemisphere each. Coimmunoprecipitation of mGluR5 with PrPC is reduced significantly by MTEP (**, p = 0.0014 by one-sample Student's t test) and enhanced significantly by DHPG (**, p = 0.0087 by one-sample Student's t test).
Conformational Regulation of mGluR5 by Endogenous Ligands
The immunoprecipitation assays described here can also be used to examine the effects of the endogenous ligands of PrPC and mGluR5, Aβo, and glutamate, respectively, on the coimmunoprecipitation of PrPC and mGluR5 interaction between them (Fig. 8). Both glutamate and Aβo enhance the coimmunoprecipitation of PrPC in Myc immunoprecipitates in a similar manner as the mGluR5-directed agonist DHPG (Fig. 8B; gray bar, 278 ± 28%, n = 7; blue bar, 214 ± 34%, n = 8; green bar, 260 ± 24%, n = 11). Similar effects were seen in the complementary PrPC immunoprecipitation (Fig. 8C; gray bar, 242 ± 27%, n = 7; blue bar, 218 ± 31%, n = 8; green bar, 263 ± 20%, n = 11). However, preincubation of cells with DHPG prior to Aβo did not further increase the coimmunoprecipitation signal of PrPC and Myc-mGluR5 (Fig. 8, B and C, yellow bars), indicating occlusive action of the glutamate analog DHPG and the endogenous ligand Aβo. The coimmunoprecipitation of PrPC with Myc-mGluR5 in cells preincubated with MTEP prior to Aβo was not different to the coimmunoprecipitation of these proteins in untreated cells (Fig. 8, B and C, orange bars versus black bars). Therefore, Aβo lose their ability to promote the formation of the complex when mGluR5 is in an inhibited conformation. Taken together, our results indicate that the endogenous ligands glutamate and Aβo enhance the interaction between PrPC and Myc-mGluR5 and that this increased interaction can be reversed by MTEP-induced conformational changes of Myc-mGluR5.
FIGURE 8.

The endogenous ligands glutamate and Aβo enhance the interaction of PrPC with Myc-mGluR5 in a similar manner as the agonist DHPG. A, HEK-293 cells were cotransfected for PrPC and Myc-mGluR5 and incubated for 10 min at 37 °C with 100 μm glutamate, 1 μm Aβo, or 2.5 μm drug as indicated. Some cultures were preincubated for 10 min with 2.5 μm indicated drug prior to incubation for 10 min at 37 °C with 1 μm Aβo or 100 μm glutamate. Detergent-solubilized lysates (Input) were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates (IP) and anti-PrPC immunoprecipitates were treated with 100 μm glutamate, 1 μm Aβo, 2.5 μm drug, or a combination of ligand and drug, as indicated, and immunoblotted with either anti-Myc or anti-PrPC as indicated. B, quantification of the PrPC signal in anti-Myc immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from 4–12 experiments. Coimmunoprecipitation of PrPC with Myc-mGluR5 is enhanced significantly by glutamate (*, p = 0.0156 by Wilcoxon signed-rank test), Aβo (*, p = 0.0119 by Wilcoxon signed-rank test), and DHPG (***, p = 0.00049 by Wilcoxon signed-rank test) and reduced significantly by MTEP (***, p = 0.00024 by Wilcoxon signed-rank test). Coimmunoprecipitation of PrPC with Myc-mGluR5 in cells preincubated with MTEP prior to Aβo is not significantly different to the coimmunoprecipitation of PrPC with Myc-mGluR5 in untreated cells. ns, not significant. C, quantification of the Myc signal in anti-PrPC immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from 4–11 experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC is enhanced significantly by glutamate (*, p = 0.0156 by Wilcoxon signed-rank test), Aβo (*, p = 0.0312 by Wilcoxon signed-rank test), and DHPG (***, p = 0.00098 by Wilcoxon signed-rank test) and reduced significantly by MTEP (***, p = 0.00049 by Wilcoxon signed-rank test). Coimmunoprecipitation of Myc-mGluR5 with PrPC in cells preincubated with MTEP prior to Aβo is not significantly different to the coimmunoprecipitation of these proteins in untreated cells.
Aβo-dependent Regulation of the PrPC-mGluR5 Interaction Requires Intact Lipid Rafts
Aβo effects are proposed to occur in lipid raft-like domains to which PrPC and mGluR5 are known to localize (31–33). We show that disruption of lipid rafts by pretreatment of PrPC and Myc-mGluR5 coexpressing HEK293 cells with methyl-β-cyclodextrin (MβCD) prevented Aβo-induced alterations of the PrPC-Myc-mGluR5 interaction (Fig. 9).
FIGURE 9.
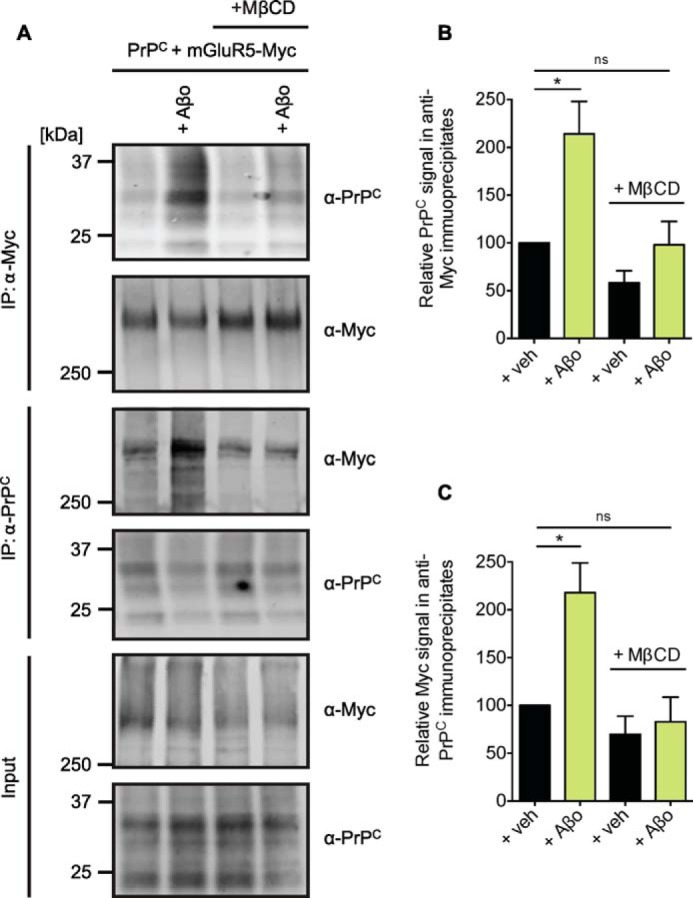
Aβo-induced enhancement of the coimmunoprecipitation of PrPC with mGluR5 requires intact lipid rafts. A, HEK-293 cells were cotransfected with PrPC and Myc-mGluR5 and incubated for 1 h at 37 °C with 5 mg/ml MβCD prior to 1 μm Aβo exposure for 10 min at 37 °C. Detergent-solubilized lysates (Input) were immunoblotted with either anti-Myc or anti-PrPC as indicated. Anti-Myc immunoprecipitates (IP) and anti-PrPC immunoprecipitates were treated with 1 μm Aβo and immunoblotted with either anti-Myc or anti-PrPC as indicated. B, quantification of the PrPC signal in anti-Myc immunoprecipitates after Aβo exposure is normalized to the signal of vehicle-treated samples. Data are mean ± S.E. from three experiments. Coimmunoprecipitation of PrPC with Myc-mGluR5 is enhanced significantly by Aβo (*, p = 0.0234 by Wilcoxon signed-rank test). Coimmunoprecipitation of PrPC with Myc-mGluR5 in cells preincubated with MβCD prior to Aβo is not significantly different to the coimmunoprecipitation of PrPC with Myc-mGluR5 in vehicle-treated cells. ns, not significant; Veh, vehicle. C, quantification of the Myc signal in anti-PrPC immunoprecipitates after treatment is normalized to the signal of vehicle-treated samples. Data are mean ± S.E. from three experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC is enhanced significantly by Aβo (*, p = 0.0313 by Wilcoxon signed-rank test). Coimmunoprecipitation of Myc-mGluR5 with PrPC in cells preincubated with MβCD prior to Aβo is not significantly different to the coimmunoprecipitation of these proteins in untreated cells.
Reversal of the Aβo-triggered Augmented PrPC-mGluR5 Interaction
Because the Aβo-triggered augmented PrPC-mGluR5 interaction is a potential step in the process of neurodegeneration, blocking this event might have therapeutic significance. To test whether PrPC-directed antibodies or mGluR5-directed drugs other than MTEP could prevent the Aβo-triggered augmentation of the PrPC-Myc-mGluR5 interaction, we analyzed their effect prior to Aβo administration on the coimmunoprecipitation of PrPC with Myc-mGluR5 in the absence of SDS (Fig. 10). Aβo increase the PrPC coimmunoprecipitation with Myc-mGluR5 in HEK-293 cells (Fig. 10B, 214 ± 34%, n = 8, black bar). We analyzed a series of known therapeutic molecules to evaluate their effect on the pathological increased interaction between PrPC and Myc-GluR5 promoted by Aβo. First we show that, in the absence of Aβo, only the application of MTEP significantly reduced the normal interaction between PrPC and Myc-mGluR5 (Fig. 10B, 33 ± 5.2%, n = 12, red bar). Also, the sole application of DCB, 6D11, and Bar221 triggered a slight but not significant decline of the steady-state interaction between PrPC and Myc-mGluR5 (Fig. 10B; yellow, purple, and blue bars, respectively). Note that 6D11 and Bar221 reduced association in the plate-based format (Fig. 2, D and E), suggesting that association may be more resistant to regulation when formed in the cell membrane. We then tested whether PrPC-directed antibodies or mGluR5-directed drugs reverse the Aβo-induced increase on the coimmunoprecipitation of PrPC with Myc-mGluR5. Our findings revealed that not only the mGluR5-directed antagonist MTEP but also the silent allosteric modulator DCB reversed the Aβo-triggered increase of the coimmunoprecipitation of PrPC and Myc-mGluR5 (Fig. 10C, red and yellow bars, respectively). Moreover, two antibodies binding within the 91–153 region of PrPC, 6D11 and Bar221, reversed the increase in the PrPC-mGluR5 interaction triggered by Aβo (Fig. 10C, purple and blue bars, respectively). In contrast, the antibody M20 binding outside of the 91–153 region of PrPC did not reverse the enhanced coimmunoprecipitation signal of PrPC and mGluR5 triggered by Aβo (Fig. 10C, green bar). These experiments demonstrate that mGluR5-directed drugs and PrPC-directed antibodies targeting the Aβo- and/or mGluR5-binding site on PrPC, but not antibodies targeting regions outside of this binding site, can reverse the Aβo-induced stimulation of the PrPC-mGluR5 interaction.
FIGURE 10.

Aβo-induced enhancement of the coimmunoprecipitation of PrPC with mGluR5 in membrane fractions can be reversed by mGluR5-directed antagonists and antibodies directed against region 91–153 of PrPC. A, HEK-293 cells were cotransfected for PrPC and Myc-mGluR5. Membrane fractions were prepared in the absence of SDS and incubated for 3 h at 4 °C with either 2.5 μm MTEP, 25 μm DCB, 0.1 μm antibody, 1 μm Aβo, or a combination of Aβo and therapeutic molecule, as indicated. Membrane proteins were extracted by Nonidet P-40, and membrane extractions (Input) and anti-Myc immunoprecipitates (IP, using goat anti-rabbit IgG magnetic beads) of membrane extractions were immunoblotted with either anti-Myc or anti-PrPC. B,C: Quantification of the PrPC signal in anti-Myc immunoprecipitates after treatment is normalized to the signal of untreated samples. B, data are mean ± S.E. from 5–12 experiments. Coimmunoprecipitation of PrPC with Myc-mGluR5 is reduced significantly by MTEP (***, p = 0.00098 by Wilcoxon signed-rank test) and enhanced significantly by Aβo (*, p = 0.0234 by Wilcoxon signed-rank test). C, data are mean ± S.E. from three to eight experiments. Coimmunoprecipitation of Myc-mGluR5 with PrPC is enhanced significantly by Aβo (*, p = 0.0234 by Wilcoxon signed-rank test). This augmentation can be reversed by simultaneous incubation with MTEP, DCB, 6D11, and Bar 221 to a level that is not significantly different to untreated samples. ns, not significant.
mGluR5 Conformational Regulation in an Alzheimer Model Mouse Brain
We further analyzed whether an increase of the coimmunoprecipitation signal of PrPC with mGluR5 is caused exclusively by an acute synthetic Aβo administration or whether this effect can also be observed in a transgenic AD mouse model brain because of endogenous Aβo in vivo (Fig. 11). We observed that the mGluR5 coimmunoprecipitation with PrPC was increased 2.5-fold in APP/PS1+ transgenic brain compared with WT brain (Fig. 11C, red bar; Fig. 10D, 309 ± 76%, gray bar; n = 9). This is similar to treatment of WT brain-derived membrane fractions with exogenous Aβo. Here Aβo enhanced the mGluR5 signal in PrPC immunoprecipitates 1.9-fold compared with untreated membrane fractions (Fig. 11D, 189 ± 27%, green bar). To further elucidate whether or not a drug- or antibody-induced modulatory effect can reverse this Aβo-induced increase in the PrPC-mGluR5 interaction, we prepared brain membrane fractions of WT and APP/PS1+ transgenic animals in the absence of SDS and incubated these with either Aβo, mGluR5-directed compounds, the PrPC-directed antibody 6D11, or a combination of Aβo and therapeutic molecules. We observed that the Aβo-dependent increase in the coimmunoprecipitation signal was reduced significantly by MTEP (Fig. 11D, red bar). A trend for the reversal of the Aβo-triggered increase of PrPC-mGluR5 coimmunoprecipitation in WT brain membrane fractions by 6D11 was also observable (Fig. 11D, purple bar). Moreover, we found that incubation of APP/PS1+ transgenic brain-derived membrane fractions with either the mGluR5-directed antagonist MTEP or the PrPC-directed antibody 6D11 fully reversed the enhanced PrPC-mGluR5 coimmunoprecipitation triggered by the presence of the APP/PS1+ transgenic background (Fig. 11D, blue and orange bars, respectively). Application of the silent allosteric modulator DCB produced a trend to recover the increased interaction of PrPC and mGluR5 in brain-derived membrane fractions of APP/PS1+ transgenic animals (Fig. 11D, yellow bar). These results imply a mechanism by which the APP/PS1+ background in AD transgenic mice or acute Aβo administration enhance the interaction between brain PrPC and mGluR5, which can be reversed by mGluR5-directed drugs or PrPC-directed antibodies targeting the binding site of mGluR5 and Aβo on PrPC.
FIGURE 11.

The coimmunoprecipitation of PrPC with mGluR5 is enhanced dramatically in APP/PS1+ mouse brain or WT brain incubated with Aβo, which can be reversed by mGluR5-directed antagonists and PrPC-directed antibodies. A, brain lysates from WT and APP/PS1+ mice were immunoblotted with either anti-mGluR5, anti-PrPC, or anti-APP, as indicated. Actin is the loading control. IP, immunoprecipitation. B, two WT brain homogenizations and two APP/PS1+ brain homogenizations were combined, and four membrane fractions were prepared in the absence of SDS for each genotype to ensure an equal amount of protein in either WT or APP/PS1+ brain membrane aliquots. Membrane fractions were treated overnight at 4 °C with either 1 μm Aβo, 2.5 μm MTEP, 25 μm DCB, 0.1 μm antibody, or a combination of Aβo and therapeutic molecule, as indicated. Membrane proteins were extracted by Nonidet P-40. Membrane extractions (Input) and anti-PrPC immunoprecipitates (using Saf32-cross-linked protein A/G-coupled beads) of membrane extractions were immunoblotted with either anti-mGluR5, anti-PrPC, or anti-APP, as indicated. Actin is the loading control. C, quantification of the combined mGluR5 dimer and monomer signal in anti-PrPC immunoprecipitates is normalized to actin. Data are mean ± S.E. from nine individual 4- to 13-month-old animals per genotype. The mGluR5 signal in anti-PrPC immunoprecipitates is increased significantly in APP/PS1+ brain (***, p = 0.0005 by Mann-Whitney test). D, the combined mGluR5 dimer and monomer signal in anti-PrPC immunoprecipitates after treatment is normalized to the signal of untreated samples. Data are mean ± S.E. from three individual experiments, one experiment performed from two WT and two APP/PS1+ brains each, as described above. The mGluR5 signal in anti-PrPC immunoprecipitates of Aβo-treated brain-derived membrane extractions is increased compared with untreated membrane extractions. This enhanced mGluR5-PrPC coimmunoprecipitation signal is reduced significantly by MTEP (**, p = 0.0073 by one-sample Student's t test). The mGluR5 signal in anti-PrPC immunoprecipitates is increased significantly in APP/PS1+ brain-derived membrane extractions compared with to WT brain-derived membrane extractions (**, p = 0.0039 by Wilcoxon signed-rank test). The mGluR5 signal in anti-PrPC immunoprecipitates derived from MTEP-treated APP/PS1+ membrane preparations is reduced significantly compared with untreated APP/PS1+ membrane preparations (**, p = 0.0024 by one-sample Student's t test). The coimmunoprecipitation of mGluR5 with PrPC is reduced significantly in 6D11-treated APP/PS1+ membrane preparations compared with untreated APP/PS1+ membrane preparations (**, p = 0.0053 by one-sample Student's t test).
DISCUSSION
This study provides important insights into the interaction between PrPC and mGluR5, which has therapeutic significance for the treatment of AD. We determined the site of interaction between mGluR5 and PrPC to be exclusively dependent on region 91–153 of PrPC. Our report further demonstrates that pharmacological manipulation of the interaction between PrPC and mGluR5 rescues Aβo-triggered AD-related phenotypes. These findings provide further evidence to support the role of both PrPC and mGluR5 in Aβo-induced pathophysiology.
Significance of PrPC and mGluR5 in AD-related Phenotypes
PrPC is a high-affinity cell surface receptor for Aβo and is involved in a number of AD-related phenotypes (14, 15, 18–23). Despite the consistent finding of Aβo binding to PrPC, some conflicting reports exist concerning the role of PrPC in Aβo-induced synaptotoxicity and memory consolidation (16, 25, 26). Kessels et al. (25) found an Aβo-induced impairment of hippocampal LTP independent of the genetic Prnp background. Also, Calella et al. (26) observed an Aβo-triggered decrease of synaptic plasticity unaffected by ablation or overexpression of PrPC. Moreover, Balducci et al. (16) found Aβo-dependent reduced consolidation of long term recognition memory independent of PrPC. These studies challenged the role of PrPC as a mediator of Aβo-induced toxicity. However, the composition of Aβo preparations between different studies varies greatly and is likely to account for inconsistent outcomes of functional Aβo-dependent experiments (24). This stresses the need for a thorough characterization of Aβo preparations prior to functional studies to prevent Aβo-induced nonspecific toxicity that is independent of cell surface receptors like PrPC.
Less is known about the events downstream of the Aβo-PrPC complex, with a crucial element being the transmission from Aβo-PrPC complexes onto intracellular targets. Electrophysiological studies regarding the synaptotoxic effects of Aβo provided the first strong evidence for a critical role of mGluR5 receptors in Aβo-triggered AD-related phenotypes. Several studies have demonstrated the recovery of Aβo-induced inhibition of LTP by mGluR5-directed antagonists (1, 38–40). Further support comes from a comparison of mGluR5 glutamate- and Aβo-triggered intracellular signaling. Glutamate binding to the extracellular region of mGluRs induces conformational changes, which triggers G-protein activation and intracellular responses (49, 50). Activation of group I mGluRs, comprising mGluR1 and mGluR5, activates phospholipase Cβ1 (PLCβ1) via Gαq/11 proteins (51). This triggers hydrolysis of phosphatidylinositol-4,5-bisphosphate membrane phospholipids to inositol-1,4,5-trisphosphate and diacylglycerol, which causes the release of intracellular Ca2+ and activation of PKC (52, 53). Interestingly, incubation of mature neurons with Aβo mimics the decline of the phosphatidylinositol-4,5-bisphosphate level and the increase of intracellular Ca2+ seen by activation of mGluR5 (7, 19, 30). Further evidence for these indications has been provided by identification of mGluR5 as a coreceptor for Aβo bound to PrPC (54).
Mapping the mGluR5-interacting Regions in PrPC
Aβo-PrPC binding to mGluR5 triggers some aspects of AD pathophysiology. Pharmacological strategies targeting the PrPC-mGluR5 interaction would largely benefit from a better understanding of the interaction between PrPC and mGluR5. Human PrPC is a 209-residue glycoprotein anchored into the membrane of lipid rafts by a glycosylphosphatidylinositol anchor (32, 55). It contains two potential glycosylation sites at residues Asn-181 and Asn-197, respectively. Region 23–111 of PrPC is intrinsically unstructured, preceded by a 22-residue long signal peptide. The intrinsically unstructured part of PrPC is subdivided into the so-called octa-repeat region (residues 60–91), a charged cluster (residues 91–111), and a hydrophobic, β-sheet containing region (residues 112–134). The C-terminal domain of PrPC is mainly α-helical, harboring three individual α-helices. Helix 2 and helix 3 are connected by a disulfide bond between residues Cys-179 and Cys-214, respectively (43).
Here we demonstrate that amino acids 91–153 of PrPC mediate mGluR5 binding. Our findings are on the basis of coimmunoprecipitation experiments of PrPC deletion mutants and mGluR5. These experiments revealed that PrPC region 91–111 is necessary for mGluR5 binding. Our results further implicate that the adjacent structural elements, the β-sheet rich region and helix 1, are also involved in mGluR5-binding. We hypothesize that the entire region 91–153 mediates the binding to mGluR5 or that deletion of the β-sheet rich region/helix 1 triggers conformational changes in region 91–111 that inhibit the interaction of PrPC-dBeta or PrPC-dHelix-1 with mGluR5. These results were further verified in an anti-prion protein antibody screen. Antibodies directed against region 91–111, β-sheet rich region, or helix 1 of PrPC largely reduced Myc-mGluR5 binding to immobilized PrPC. In contrast, deletion of structural elements outside of region 91–153 or antibodies recognizing domains of PrPC other than region 91–153 had no effect on PrPC-mGluR5 interaction.
Mapping the PrPC-interacting Regions in mGluR5
The mGluR structure is composed of an extracellular region, a seven transmembrane-spanning region, and a cytoplasmic region (44, 45, 56). To determine the region in mGluR5 accounting for interaction with PrPC, we used chimeric proteins composed of the extracellular region of either Myc-mGluR5 or Myc-mGluR8 and the transmembrane spanning region of the other receptor in coimmunoprecipitation experiments with PrPC. As a control, we cotransfected the closely related Myc-mGluR8 receptor and PrPC. The coimmunoprecipitation of these proteins was reduced significantly compared with the coimmunoprecipitation signal of Myc-mGluR5 with PrPC. Both chimeric Myc-mGluR-N5/C8 and Myc-mGluR-N8/C5 proteins revealed intermediate levels of binding to PrPC. We observed a similar trend before (30), which indicates that the PrPC-interacting regions are spread throughout the protein rather than localized in either the extracellular or the transmembrane-spanning mGluR region alone.
Pharmacological Manipulation of the PrPC-mGluR5 Interaction
mGluR5 is implicated in a number of neurological diseases, including fragile X syndrome, amyotrophic lateral sclerosis, multiple sclerosis, AD, Parkinson disease, Huntington disease, epilepsy, schizophrenia, and drug addiction, and its pharmacological tangibility has been studied extensively (51, 57–62). Moreover, anti-prion protein therapeutics have been developed as putative treatments for prion disease (reviewed in Ref. 63) and are available for screening their efficiency in regulating the PrPC-mGluR5 interaction. Because the PrPC-mGluR5 interaction is implicated in AD pathogenesis, we decided to develop assays to study the modulatory effect of therapeutic molecules on the interaction between mGluR5 and PrPC. Our results demonstrated that agonist/antagonist-induced conformational changes of mGluR5 and PrPC-directed antibodies alter the interaction between PrPC and mGluR5 in a dose-dependent manner both in HEK-293 cells and mouse brains.
Alternatives to Negative Allosteric Modulators
Our findings demonstrate a strong inhibitory effect of negative allosteric modulators, such as MTEP, on PrPC-mGluR5 interaction. However, mGluR5 function is important for healthy brain aging, and intervention should, therefore, be aimed at regulating the PrPC-mGluR5 interaction without modifying its physiological function in a negative way (53, 64). mGluR5-directed antagonists inhibit glutamate signaling, thereby negatively affecting normal cell signaling. A better pharmacological strategy for disease intervention is the use of so-called silent allosteric modulators. These do not affect glutamate signaling and, therefore, reduce possible side effects. However, they alter the conformation of metabotropic glutamate receptors and prevent the action of other allosteric modulators (48), for example. Our results show that application of the silent allosteric modulator DCB did not alter the PrPC-mGluR5 interaction in the absence of Aβo. However, preincubation of cells with DCB prior to application of the negative allosteric modulator MTEP blocks an inhibitory effect of MTEP on PrPC-Myc-mGluR5 interaction. Our findings are consistent with DCB occupancy preventing MTEP from binding to mGluR5 (48), which explains the lack of effect on the coimmunoprecipitation signal of PrPC with Myc-mGluR5 in untreated cells compared with DCB and MTEP double-treated cells.
Confirmation of Drug Specificity with Chimeric mGluRs
To provide further mechanistic support for the specificity of the assays we developed, we tested the effect of mGluR5-directed compounds on the interaction of PrPC with either Myc-mGluR5, Myc-mGluR-N5/C8, Myc-mGluR-N8/C5, or Myc-mGluR8 as negative control. The coimmunoprecipitation signal of PrPC with Myc-mGluR5 was regulated by glutamate, DHPG, and MTEP, as seen before. However, MTEP did not regulate the coimmunoprecipitation of PrPC with Myc-mGluR-N5/C8. Also, DHPG failed to modulate the coimmunoprecipitation of PrPC and Myc-mGluR-N8/C5. These findings provide evidence for the drug specificity in the assays we developed because the Myc-mGluR-N5/C8 mutant protein does not contain the transmembrane-spanning part of mGluR5 that is targeted by MTEP (61). DHPG, on the other hand, is highly specific for the extracellular binding pocket of metabotropic group 1 receptors, which includes mGluR5 but not mGluR8 (61, 65).
Conformational Regulation of mGluR5 Requires a Membrane Environment
We found that a modulatory effect on the interaction between PrPC and mGluR5 is only observable when mGluR5 receptors are manipulated in their membrane-embedded conformation. Extraction of receptors from the lipid bilayer hinders agonist/antagonist-triggered conformational changes of mGluR5, which prevents an alteration of its interaction with PrPC. However, our findings also show a less efficient modulation of the mGluR5-PrPC interaction after agonist/antagonist treatment of cells only compared with the treatment of membrane preparations or both cells and detergent-solubilized lysates. This effect is most likely due to a washout of compounds during the harvesting and lysis of cells as well as during the time-consuming immunoprecipitation.
Moreover, we failed to modulate the PrPC-mGluR5 interaction in brain-derived membrane fractions cleared by 100,000 × g centrifugation after the extraction of membrane proteins. In contrast, conformational regulation of the mGluR5-PrPC coimmunoprecipitation was observable when extracted proteins were cleared by 20,000 × g centrifugation removal. These findings suggest that small proteolipid complexes contain PrPC and mGluR5 in a pharmacologically vulnerable conformation. 100,000 × g centrifugation removes complexes needed to observe a compound-induced modulatory effect on PrPC-mGluR5 interaction. Taken together, a conformational change can only occur when mGluR5 is in its native lipid-associated conformation and environment. A conformational change can only trigger a modulation of the coimmunoprecipitation signal of Myc-mGluR5 and PrPC if the same compound concentration is supplied throughout all steps of coimmunoprecipitation to prevent a washout of compounds and if complexes are not removed by 100,000 × g centrifugation after detergent addition.
Similarities between mGluR5 Agonist-induced Conformational Changes and the Effect of Aβo
We further report that soluble Aβo consistently induced an enhancement of PrPC-mGluR5 coimmunoprecipitation both in HEK-293 cells and mouse brain. Preincubation of HEK-293 cells with the mGluR5-directed agonist DHPG prior to Aβo application did not further stimulate the PrPC-mGluR5 signal. This indicates an occlusive action of DHPG and Aβo. One possible explanation is an overlapping binding site of Aβo-PrPC complexes and DHPG on mGluR5, the latter being located in the extracellular binding pocket (61, 65). The effect could also be explained by DHPG-triggered conformational changes of mGluR5, which prevent Aβo-PrPC complexes from binding and inducing further conformational alterations. Our previous results showed that exclusive application of Aβo or DHPG trigger eEF2 phosphorylation in neurons (30). Application of both Aβo and DHPG did not further increase eEF2 phosphorylation, which is in accordance with the findings of HEK-293 cell experiments highlighted here. We further demonstrated that the Aβo-dependent enhancement of the PrPC-mGluR5 interaction is dependent on the existence of lipid raft-like domains. Pretreatment of PrPC- and Myc-mGluR5-expressing cell cultures with MβCD destroyed lipid rafts and prevented an Aβo-dependent modulation of the PrPC-Myc-mGluR5 interaction. This is in accordance with the fact that Aβo effects are proposed to occur in lipid raft-like domains, where PrPC and mGluR5 receptors are located (31–34).
mGluR5 Conformational Regulation in an Alzheimer Model Mouse Brain
Furthermore, we provide evidence for a strongly enhanced mGluR5 signal in anti-PrPC immunoprecipitates of APP/PS1+ transgenic mouse brain. These findings strongly support the potential value of therapeutically targeting the PrPC-mGluR5 interaction in AD pathogenesis. The effect of the APP/PS1+ transgene or artificial supply of Aβo was rescued by the mGluR5-directed antagonist MTEP. MTEP induces a strong conformational change of mGluR5, which reverses the Aβo-induced enhanced interaction of PrPC with mGluR5. This is of high biological relevance because of findings of previous studies that demonstrated the reversal of Aβo-induced effects in cell-based toxicity assays by the mGluR5-specific antagonist MTEP (19, 30). Moreover, MTEP treatment rescues AD-related learning and memory deficits in APP/PS1+ transgenic mice, which is in accordance with the MTEP-induced reversal of AD-related molecular phenotypes (30) described here. However, more feasible therapeutic agents for AD are silent allosteric modulators that do not affect endogenous glutamate signaling. In our experiments, the silent allosteric modulator DCB fully rescued Aβo-induced association in transfected cell lysates and partially rescued the APP/PS1+ transgenic-dependent enhancement of the PrPC-mGluR5 interaction. Moreover, application of antibodies directed against the putative PrPC-mGluR5 binding site (6D11 and Bar233) or the Aβo-PrPC binding site (6D11) prohibited the acute Aβo-induced or APP/PS1+ transgene-dependent augmentation of PrPC-mGluR5 coimmunoprecipitation. In contrast, M20, a polyclonal antibody targeting the C-terminal region of PrPC, did not significantly alter Aβo-triggered changes in PrPC-mGluR5 coimmunoprecipitation. These findings are in line with the mapping of mGluR5-interacting regions in PrPC to residues 91–153. Notably, the exclusive application of 6D11 and Bar 221 did not reveal a strong effect on the coimmunoprecipitation of PrPC and Myc-mGluR5. In contrast, 6D11 and Bar 221 showed a robust blockade of the Myc-mGluR5 binding to immobilized recombinant PrPC in antibody mapping experiments of the binding site of Myc-mGluR5 on PrPC. This indicates that the PrPC-directed antibodies 6D11 and Bar 221 cannot easily access PrPC interacting with mGluR5. In contrast, 6D11 and Bar 221 antibody binding to immobilized recombinant PrPC blocks further binding of Myc-mGluR5. Interestingly, coincubation of membrane fractions of PrPC- and Myc-mGluR5-expressing HEK-293 cells with the Aβo- and PrPC-directed antibodies 6D11 and Bar 221 altered the PrPC-mGluR5 interaction to a larger extent than the exclusive application of PrPC-directed antibodies. These findings indicate that Aβo trigger a conformational change of the PrPC-Myc-mGluR5 complex that renders PrPC more vulnerable to antibody treatment by enabling the binding of anti-prion protein antibodies.
The Putative Role of mGluR5 in AD
We hypothesize that mGluR5 plays a crucial role in Alzheimer disease pathogenesis by transmitting neurotoxic signals from extracellular Aβo-PrPC complexes into the cytosol. Beraldo et al. (36) report that binding of laminin to PrPC alters neuronal plasticity and memory by mGluR1/5-mediated transmission of signals into the cytosol. Similar events are likely to occur after binding of Aβo to PrPC, such as mGluR5-mediated transmission of signals onto intracellular substrates. Different substrates are feasible, one of which is Fyn kinase, whose activation provides a link to the NR2B subunit phosphorylation and redistribution of NMDA receptors observed after acute Aβo treatment (19, 30, 42). NMDA receptors are involved in LTP, and their significance for AD is stressed by the symptomatic benefits of pharmacological NMDA receptor antagonists like memantine (66, 67). One possibility to explain the Aβo-PrPC-induced signal transmission mediated by mGluR5 is the redistribution and overstabilization of mGluR5 receptors after Aβo-PrPC binding, as seen by Renner et al. (35). mGluR5 receptors are normally laterally mobile within the membrane (68). It is feasible that Aβo-PrPC complexes act like an extracellular scaffold stabilizing mGluR5, thereby preventing their lateral diffusion. A reduced diffusion efficiency of mGluR5 causes disruptive Ca2+ signaling, which alters NMDA receptor activity (69). Preventing Aβo-PrPC complexes from binding to mGluR5 could ameliorate these putative neurotoxic events.
Future Directions
The assays described here can be used to identify therapeutic molecules that inhibit the interaction between PrPC and mGluR5, whose signaling is implicated in AD pathogenesis. Whether prohibiting the binding of PrPC to mGluR5 will eventually reduce neuronal loss and memory deficits in AD still needs to be determined. Moreover, activation of mGluR5 receptors is known to stimulate several signaling pathways, some of which are involved in cell survival and proliferation, such as the ERK and AKT pathways (70, 71). Future research is necessary to determine the role of Aβo-PrPC-mGluR5 complexes in these pathways. mGluR5 receptors are also known to be part of large multimolecular complexes (72). Further studies could determine additional modulators of the Aβo-PrPC-mGluR5 interaction, and such factors could potentially have tangibility for AD therapeutic research. Despite extensive research in the field, a preventive or disease-modifying treatment for AD is still not available, generating one of the biggest threats to public health of this century. This stresses the need to find and characterize novel pharmacological targets for AD therapeutic intervention.
Acknowledgments
We thank Stefano Sodi for assistance with mouse husbandry.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AG034924 (to S. M. S.). This work was also supported by grants from the BrightFocus Foundation, Alzheimer's Association, and Falk Medical Research Trust (to S. M. S.). S. M. S. is a cofounder of Axerion Therapeutics, seeking to develop NgR- and PrP-based therapeutics.
- Aβo
- Amyloid-β oligomer(s)
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- DCB
- 3,3-dichloro-benzaldazine
- DHPG
- dihydroxyphenylglycine
- LTD
- long-term depression
- LTP
- long term potentiation
- MβCD
- methyl-β-cyclodextrin
- MTEP
- 3-((2-methyl-4-thiazolyl) ethynyl)pyridine
- PrPC
- cellular prion protein.
REFERENCES
- 1. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 [DOI] [PubMed] [Google Scholar]
- 3. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 4. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li S., Hong S., Shepardson N. E., Walsh D. M., Shankar G. M., Selkoe D. (2009) Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palop J. J., Mucke L. (2010) Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berman D. E., Dall'Armi C., Voronov S. V., McIntire L. B., Zhang H., Moore A. Z., Staniszewski A., Arancio O., Kim T. W., Di Paolo G. (2008) Oligomeric amyloid-β peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci. 11, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., Rogers J. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 10. Wang J., Dickson D. W., Trojanowski J. Q., Lee V. M. (1999) The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 158, 328–337 [DOI] [PubMed] [Google Scholar]
- 11. Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 [DOI] [PubMed] [Google Scholar]
- 12. Katzman R., Terry R., DeTeresa R., Brown T., Davies P., Fuld P., Renbing X., Peck A. (1988) Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 23, 138–144 [DOI] [PubMed] [Google Scholar]
- 13. Dickson D. W., Crystal H. A., Bevona C., Honer W., Vincent I., Davies P. (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 16, 285–298; discussion 298–304 [DOI] [PubMed] [Google Scholar]
- 14. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou W. Q., Xiao X., Yuan J., Puoti G., Fujioka H., Wang X., Richardson S., Zhou X., Zou R., Li S., Zhu X., McGeer P. L., McGeehan J., Kneale G., Rincon-Limas D. E., Fernandez-Funez P., Lee H. G., Smith M. A., Petersen R. B., Guo J. P. (2011) Amyloid-β42 interacts mainly with insoluble prion protein in the Alzheimer brain. J. Biol. Chem. 286, 15095–15105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balducci C., Beeg M., Stravalaci M., Bastone A., Sclip A., Biasini E., Tapella L., Colombo L., Manzoni C., Borsello T., Chiesa R., Gobbi M., Salmona M., Forloni G. (2010) Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen S., Yadav S. P., Surewicz W. K. (2010) Interaction between human prion protein and amyloid-β (Aβ) oligomers: role OF N-terminal residues. J. Biol. Chem. 285, 26377–26383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gimbel D. A., Nygaard H. B., Coffey E. E., Gunther E. C., Laurén J., Gimbel Z. A., Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Um J. W., Nygaard H. B., Heiss J. K., Kostylev M. A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E. C., Strittmatter S. M. (2012) Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bate C., Williams A. (2011) Amyloid-β-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J. Biol. Chem. 286, 37955–37963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kudo W., Lee H. P., Zou W. Q., Wang X., Perry G., Zhu X., Smith M. A., Petersen R. B., Lee H. G. (2012) Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chung E., Ji Y., Sun Y., Kascsak R. J., Kascsak R. B., Mehta P. D., Strittmatter S. M., Wisniewski T. (2010) Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barry A. E., Klyubin I., Mc Donald J. M., Mably A. J., Farrell M. A., Scott M., Walsh D. M., Rowan M. J. (2011) Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Freir D. B., Nicoll A. J., Klyubin I., Panico S., Mc Donald J. M., Risse E., Asante E. A., Farrow M. A., Sessions R. B., Saibil H. R., Clarke A. R., Rowan M. J., Walsh D. M., Collinge J. (2011) Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kessels H. W., Nguyen L. N., Nabavi S., Malinow R. (2010) The prion protein as a receptor for amyloid-β. Nature 466, E3–E4; discussion E4–E5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calella A. M., Farinelli M., Nuvolone M., Mirante O., Moos R., Falsig J., Mansuy I. M., Aguzzi A. (2010) Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gandy S., Simon A. J., Steele J. W., Lublin A. L., Lah J. J., Walker L. C., Levey A. I., Krafft G. A., Levy E., Checler F., Glabe C., Bilker W. B., Abel T., Schmeidler J., Ehrlich M. E. (2010) Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-β oligomers. Ann. Neurol. 68, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reed M. N., Hofmeister J. J., Jungbauer L., Welzel A. T., Yu C., Sherman M. A., Lesné S., LaDu M. J., Walsh D. M., Ashe K. H., Cleary J. P. (2011) Cognitive effects of cell-derived and synthetically derived Aβ oligomers. Neurobiol. Aging 32, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Cheng I. H., Scearce-Levie K., Legleiter J., Palop J. J., Gerstein H., Bien-Ly N., Puoliväli J., Lesné S., Ashe K. H., Muchowski P. J., Mucke L. (2007) Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 282, 23818–23828 [DOI] [PubMed] [Google Scholar]
- 30. Um J. W., Kaufman A. C., Kostylev M., Heiss J. K., Stagi M., Takahashi H., Kerrisk M. E., Vortmeyer A., Wisniewski T., Koleske A. J., Gunther E. C., Nygaard H. B., Strittmatter S. M. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 79, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Francesconi A., Kumari R., Zukin R. S. (2009) Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J. Neurosci. 29, 3590–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Agostini F., Dotti C. G., Pérez-Cañamás A., Ledesma M. D., Benetti F., Legname G. (2013) Prion protein accumulation in lipid rafts of mouse aging brain. PLoS ONE 8, e74244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zampagni M., Evangelisti E., Cascella R., Liguri G., Becatti M., Pensalfini A., Uberti D., Cenini G., Memo M., Bagnoli S., Nacmias B., Sorbi S., Cecchi C. (2010) Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J. Mol. Med. 88, 597–608 [DOI] [PubMed] [Google Scholar]
- 34. Rushworth J. V., Griffiths H. H., Watt N. T., Hooper N. M. (2013) Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 288, 8935–8951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Renner M., Lacor P. N., Velasco P. T., Xu J., Contractor A., Klein W. L., Triller A. (2010) Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beraldo F. H., Arantes C. P., Santos T. G., Machado C. F., Roffe M., Hajj G. N., Lee K. S., Magalhães A. C., Caetano F. A., Mancini G. L., Lopes M. H., Américo T. A., Magdesian M. H., Ferguson S. S., Linden R., Prado M. A., Martins V. R. (2011) Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 25, 265–279 [DOI] [PubMed] [Google Scholar]
- 37. Benarroch E. E. (2008) Metabotropic glutamate receptors: synaptic modulators and therapeutic targets for neurologic disease. Neurology 70, 964–968 [DOI] [PubMed] [Google Scholar]
- 38. Rammes G., Hasenjäger A., Sroka-Saidi K., Deussing J. M., Parsons C. G. (2011) Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of β-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology 60, 982–990 [DOI] [PubMed] [Google Scholar]
- 39. Wang Q., Walsh D. M., Rowan M. J., Selkoe D. J., Anwyl R. (2004) Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24, 3370–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruno V., Ksiazek I., Battaglia G., Lukic S., Leonhardt T., Sauer D., Gasparini F., Kuhn R., Nicoletti F., Flor P. J. (2000) Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology 39, 2223–2230 [DOI] [PubMed] [Google Scholar]
- 41. Hu N. W., Nicoll A. J., Zhang D., Mably A. J., O'Malley T., Purro S. A., Terry C., Collinge J., Walsh D. M., Rowan M. J. (2014) mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 5, 3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larson M., Sherman M. A., Amar F., Nuvolone M., Schneider J. A., Bennett D. A., Aguzzi A., Lesné S. E. (2012) The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological Tau changes in Alzheimer's disease. J. Neurosci. 32, 16857–16871a [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Antonyuk S. V., Trevitt C. R., Strange R. W., Jackson G. S., Sangar D., Batchelor M., Cooper S., Fraser C., Jones S., Georgiou T., Khalili-Shirazi A., Clarke A. R., Hasnain S. S., Collinge J. (2009) Crystal structure of human prion protein bound to a therapeutic antibody. Proc. Natl. Acad. Sci. U.S.A. 106, 2554–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu H., Wang C., Gregory K. J., Han G. W., Cho H. P., Xia Y., Niswender C. M., Katritch V., Meiler J., Cherezov V., Conn P. J., Stevens R. C. (2014) Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muto T., Tsuchiya D., Morikawa K., Jingami H. (2007) Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 3759–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jankowsky J. L., Xu G., Fromholt D., Gonzales V., Borchelt D. R. (2003) Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 62, 1220–1227 [DOI] [PubMed] [Google Scholar]
- 47. Lu Y. M., Jia Z., Janus C., Henderson J. T., Gerlai R., Wojtowicz J. M., Roder J. C. (1997) Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J. Neurosci. 17, 5196–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Brien J. A., Lemaire W., Chen T. B., Chang R. S., Jacobson M. A., Ha S. N., Lindsley C. W., Schaffhauser H. J., Sur C., Pettibone D. J., Conn P. J., Williams D. L., Jr. (2003) A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol. Pharmacol. 64, 731–740 [DOI] [PubMed] [Google Scholar]
- 49. Rondard P., Goudet C., Kniazeff J., Pin J. P., Prézeau L. (2011) The complexity of their activation mechanism opens new possibilities for the modulation of mGlu and GABAB class C G protein-coupled receptors. Neuropharmacology 60, 82–92 [DOI] [PubMed] [Google Scholar]
- 50. Schwartz T. W., Frimurer T. M., Holst B., Rosenkilde M. M., Elling C. E. (2006) Molecular mechanism of 7TM receptor activation: a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519 [DOI] [PubMed] [Google Scholar]
- 51. Ribeiro F. M., Paquet M., Cregan S. P., Ferguson S. S. (2010) Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol. Disord. Drug Targets 9, 574–595 [DOI] [PubMed] [Google Scholar]
- 52. Pin J. P., Duvoisin R. (1995) The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34, 1–26 [DOI] [PubMed] [Google Scholar]
- 53. Lüscher C., Huber K. M. (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Um J. W., Strittmatter S. M. (2013) Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 7, 37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yusa S., Oliveira-Martins J. B., Sugita-Konishi Y., Kikuchi Y. (2012) Cellular prion protein: from physiology to pathology. Viruses 4, 3109–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pin J. P., Galvez T., Prézeau L. (2003) Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol. Ther. 98, 325–354 [DOI] [PubMed] [Google Scholar]
- 57. Gregory K. J., Dong E. N., Meiler J., Conn P. J. (2011) Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 60, 66–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sheffler D. J., Gregory K. J., Rook J. M., Conn P. J. (2011) Allosteric modulation of metabotropic glutamate receptors. Adv. Pharmacol. 62, 37–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gasparini F., Spooren W. (2007) Allosteric modulators for mGlu receptors. Curr. Neuropharmacol. 5, 187–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gregory K. J., Noetzel M. J., Rook J. M., Vinson P. N., Stauffer S. R., Rodriguez A. L., Emmitte K. A., Zhou Y., Chun A. C., Felts A. S., Chauder B. A., Lindsley C. W., Niswender C. M., Conn P. J. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol. Pharmacol. 82, 860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Molck C., Harpsoe K., Gloriam D. E., Mathiesen J. M., Nielsen S. M., Brauner-Osborne H. (2014) mGluR5: exploration of orthosteric and allosteric ligand binding pockets and their applications to drug discovery. Neurochem. Res. 10.1007/s11064-014-1248-8 [DOI] [PubMed] [Google Scholar]
- 62. Bruno V., Battaglia G., Copani A., D'Onofrio M., Di Iorio P., De Blasi A., Melchiorri D., Flor P. J., Nicoletti F. (2001) Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow Metab. 21, 1013–1033 [DOI] [PubMed] [Google Scholar]
- 63. Trevitt C. R., Collinge J. (2006) A systematic review of prion therapeutics in experimental models. Brain 129, 2241–2265 [DOI] [PubMed] [Google Scholar]
- 64. Xu J., Zhu Y., Contractor A., Heinemann S. F. (2009) mGluR5 has a critical role in inhibitory learning. J. Neurosci. 29, 3676–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wiśniewski K., Car H. (2002) (S)-3,5-DHPG: a review. CNS Drug Rev. 8, 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reisberg B., Doody R., Stöffler A., Schmitt F., Ferris S., Möbius H. J., and Memantine Study Group. (2003) Memantine in moderate-to-severe Alzheimer's disease. N. Engl. J. Med. 348, 1333–1341 [DOI] [PubMed] [Google Scholar]
- 67. Lipton S. A. (2005) The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr. Alzheimer Res. 2, 155–165 [DOI] [PubMed] [Google Scholar]
- 68. Sergé A., Fourgeaud L., Hémar A., Choquet D. (2002) Receptor activation and homer differentially control the lateral mobility of metabotropic glutamate receptor 5 in the neuronal membrane. J. Neurosci. 22, 3910–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rosenmund C., Feltz A., Westbrook G. L. (1995) Calcium-dependent inactivation of synaptic NMDA receptors in hippocampal neurons. J. Neurophysiol. 73, 427–430 [DOI] [PubMed] [Google Scholar]
- 70. Mao L., Yang L., Tang Q., Samdani S., Zhang G., Wang J. Q. (2005) The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J. Neurosci. 25, 2741–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hou L., Klann E. (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tu J. C., Xiao B., Naisbitt S., Yuan J. P., Petralia R. S., Brakeman P., Doan A., Aakalu V. K., Lanahan A. A., Sheng M., Worley P. F. (1999) Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592 [DOI] [PubMed] [Google Scholar]
- 73. Leclerc E., Liemann S., Wildegger G., Vetter S. W., Nilsson F. (2000) Selection and characterization of single chain Fv fragments against murine recombinant prion protein from a synthetic human antibody phage display library. Hum. Antibodies 9, 207–214 [PubMed] [Google Scholar]
- 74. Féraudet C., Morel N., Simon S., Volland H., Frobert Y., Créminon C., Vilette D., Lehmann S., Grassi J. (2005) Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J. Biol. Chem. 280, 11247–11258 [DOI] [PubMed] [Google Scholar]
- 75. Spinner D. S., Kascsak R. B., Lafauci G., Meeker H. C., Ye X., Flory M. J., Kim J. I., Schuller-Levis G. B., Levis W. R., Wisniewski T., Carp R. I., Kascsak R. J. (2007) CpG oligodeoxynucleotide-enhanced humoral immune response and production of antibodies to prion protein PrPSc in mice immunized with 139A scrapie-associated fibrils. J. Leukocyte Biol. 81, 1374–1385 [DOI] [PubMed] [Google Scholar]