

Background: MLCK in cell migration remains controversial.
Results: MLCK deletion causes enhanced cell protrusion along with a reduction of membrane tension and is rescued by kinase-dead MLCK or five-DFRXXL motif.
Conclusion: MLCK regulates cell migration not by myosin regulatory light chain phosphorylation but possibly through a membrane tension-based mechanism.
Significance: Our results shed light on a novel regulatory mechanism of protrusion during cell migration.
Keywords: Actin, Cell Migration, Cell Motility, Cytoskeleton, Myosin, Plasma Membrane, Cortical Cytoskeleton, Myosin Light Chain Kinase, Membrane Protrusion, Membrane Tension
Abstract
Myosin light chain kinase (MLCK) has long been implicated in the myosin phosphorylation and force generation required for cell migration. Here, we surprisingly found that the deletion of MLCK resulted in fast cell migration, enhanced protrusion formation, and no alteration of myosin light chain phosphorylation. The mutant cells showed reduced membrane tether force and fewer membrane F-actin filaments. This phenotype was rescued by either kinase-dead MLCK or five-DFRXXL motif, a MLCK fragment with potent F-actin-binding activity. Pull-down and co-immunoprecipitation assays showed that the absence of MLCK led to attenuated formation of transmembrane complexes, including myosin II, integrins and fibronectin. We suggest that MLCK is not required for myosin phosphorylation in a migrating cell. A critical role of MLCK in cell migration involves regulating the cell membrane tension and protrusion necessary for migration, thereby stabilizing the membrane skeleton through F-actin-binding activity. This finding sheds light on a novel regulatory mechanism of protrusion during cell migration.
Introduction
Cell migration that comprises multiple orchestrated steps is essential for developmental, physiological, and pathological processes in all multicellular organisms. The first step of a migration cycle is the formation of a membrane protrusion at the front edge of a motile cell (1, 2). The protrusions comprise lamellipodia and filopodia that are driven by the polymerization of actin filaments (3) and/or the addition of new membrane at the end of the endocytotic cycles (4). The second step is adhesion that stabilizes the protrusions by linking the actin cytoskeleton to the extracellular matrix (ECM)3 proteins, in which traction forces on the substratum are generated (5–8). The final steps are moving the cell body forward and detaching it from the substrate by contractile forces (1, 2, 9). During these processes, myosin II-mediated actomyosin contraction is believed to be required for force generation (1, 10, 11). Myosin II may also play a role in membrane protrusion at the leading edge (12, 13). Considering that myosin II activity is primarily regulated by the signals converging at regulatory light chain (RLC) phosphorylation (14, 15) and that MLCK serves as a dedicated kinase for RLC phosphorylation, MLCK is believed to be essential for the force generation necessary for cell migration (16, 17). On the other side, the cells alter their shapes, membrane structures and motility as well as other properties of the membrane during cell migration. The migration process might also be considered a dynamic process of membrane movements in which membrane tension presumably acts as a key regulatory factor. Cell migration can be affected by membrane tension modified by external stimuli (18–21). A critical concern is whether such an alteration of membrane tension could be regulated by an intrinsic mechanism. If so, what is/are the effector molecule(s)? Defining this intrinsic mechanism might significantly increase our understanding of the processes of cell migration.
MLCK activates myosin Mg-ATPase activity by phosphorylating Thr-18/Ser-19 on the RLC of myosin II, thereby initiating cross-bridge movements of both smooth muscle and non-muscle (22, 23). The MLCK locus encodes three transcripts from alternative promoters and produces two MLCK isoforms (short MLCK and long MLCK) and a C-terminal Ig module (telokin) (24, 25). Long MLCK (l-MLCK) is identical to short MLCK (S-MLCK) apart from a unique N-terminal extension containing two extra DFRXXL motifs and six Ig modules (26). The DFRXXL motif has potent F-actin binding activity, which aids interaction with F-actin filaments during cross-bridge movement (27, 28). MLCK functions in cytoskeleton organization, cytokinesis, and aggregation in a kinase-independent manner (29–31). A significant role of MLCK in cell migration has also been highlighted, e.g. MLCK regulates myosin II-mediated periodic lamellipodial contraction at the cell periphery (32). Because most proposed hypotheses are based on evidence from the application of nonspecific MLCK inhibitors (16, 33–35), the role of MLCK in cell migration remains controversial. For example, MLCK is activated at the leading edge, but myosin II is nearly non-existent in this area (36). In this report, we examined MLCK-deficient mouse smooth muscle cells (SMCs) cultured in vitro and surprisingly found that the absence of MLCK led to rapid cell spreading/protrusion formation, accelerated migration velocity in SMCs, and reduced membrane tension. Our results suggest that MLCK is not required for myosin light chain phosphorylation in cell migration but negatively controls migration by maintaining membrane tension.
EXPERIMENTAL PROCEDURES
Primary Culture of Intestinal SMCs
All experiments were conducted in accordance with the guidelines set by the Animal Care and Use Committee of Model Animal Research Center of Nanjing University (Nanjing, China). We generated smooth muscle-specific MLCK-knock-out mice as previously described (37). Eight- to 12-week-old mice were killed by cervical dislocation. Segments of the jejunum were excised and washed at least three times in ice-cold Hanks' solution (137.93 mm NaCl, 5.33 mm KCl, 4.17 mm NaHCO3, 0.441 mm KH2PO4, 0.338 mm Na2HPO4, 5.56 mm d-glucose, 500 units/ml penicillin, and 500 μg/ml streptomycin). The mesentery and related tissues were removed under a dissecting microscope. Muscularis propria were carefully teased away from the remaining intestine segments with micro-tweezers. Then, the muscle layers were minced into fragments (less than 1 cubic millimeter) in Dulbecco's modified Eagle's medium containing 20% (v/v) fetal bovine serum (FBS), l-glutamine, non-essential amino acids (NEAA) (Invitrogen), 100 units/ml penicillin and 100 μg/ml streptomycin. The suspension was transferred to culture dishes coated with 0.1% gelatin (Sigma-Aldrich) and then incubated at 37 °C in a humidified atmosphere of 5% CO2. The intestinal SMCs started to migrate from the explants after 4 to 6 days. Finally, the cell monolayer was passaged after 10 to 12 days and cultured in DMEM containing 10% (v/v) FBS, l-glutamine, NEAA, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Adenovirus Infection and Nucleofection for Rescue Experiments
Primary cultured intestinal SMCs were infected with full-length chicken l-MLCK-expressing adenovirus (Adv-MLCK) or N-terminal 2Ig (containing the first two Ig-like modules (1–251 aa) of chicken l-MLCK)-expressing adenovirus (Adv-2Ig) (MOI = 4) and then cultured up to 48 h before experiments. Adv-MLCK and Adv-2Ig were prepared by releasing l-MLCK and 2Ig fragments from pEGFP-MLCK210 (described in Yang et al. and Zhang et al.) (29, 30) and introducing them into the pShuttle-IRES-hrGFP-1 vector followed by virus preparation according to the AdEasyTM Adenoviral Vector System kit (Stratagene). For the nucleofection rescue experiments, we subcloned kinase-dead MLCK (with a deletion of the ATP-binding domain (1460–1482 aa, GSGKFGQVFRLVEKKTGKVWAGK) in chicken l-MLCK) (KD-MLCK), and a five-DFRXXL-motif (5DFRXXL)-containing fragment (29) into pShuttle-IRES-hrGFP-1 using a ClonExpressTM II One Step Cloning Kit (Vazyme). The SMCs were nucleofected with these recombinant plasmids by a NucleofectorTM 2b (LONZA) according to a protocol provided by the vendor and then cultured up to 48 h before experiments.
Immunofluorescence and Laser Scanning Confocal Microscopy
Immunofluorescence was performed on adherent cells grown on 0.1% gelatin-coated glass coverslips for 48 h, fixed in 4% paraformaldehyde or in a cold methanol/acetone mixture (3:7), permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS), and incubated with 5% normal goat serum (NGS) in PBS for 1 h to avoid nonspecific binding. After fixation and the blocking procedure, cell lines were incubated with the following antibodies as needed: anti-phospho-histone H3 (Ser-10) (PH3; Cell Signaling Technologies, dilution 1:100), anti-phospho-myosin light chain 2 (Ser-19) (p-RLC; Cell Signaling Technologies, dilution 1:200), anti-phospho-myosin light chain 2 (Thr-18/Ser-19) (2p-RLC; Cell Signaling Technologies, dilution 1:200), anti-smooth muscle α-actin (SMA; Thermo Fisher Scientific, dilution 1:250), anti-smooth muscle myosin heavy chain (SMMHC; Sigma-Aldrich, dilution 1:300), anti-MLCK (clone K36) (Sigma-Aldrich, dilution 1:100), anti-focal adhesion kinase (FAK; Bioworld, dilution 1:250), anti-myosin IIA (Cell Signaling Technologies, dilution 1:150), and anti-myosin IIB (Cell Signaling Technologies, dilution 1:200). For detection, appropriate secondary antibodies (diluted 1:250 in PBS containing 5% NGS) conjugated to Alexa Fluor 488, 555, or 633 (Molecular Probes, Invitrogen) were used. Nuclei were counterstained with TO-PRO-3 (Invitrogen). Cell fluorescence was then evaluated with a Leica TCS SP2 confocal laser scanning microscope (Leica) or a FluoView FV1000 confocal laser scanning microscope (Olympus). Signals from different fluorescent probes were taken in sequential double fluorescence mode, which allows for the elimination of cross-talk between channels; the co-localization was detected in an overlay model. Image acquisition and processing were conducted using the Multicolor Analysis Leica program or OLYMPUS FLUOVIEW Ver.2.0c.
Western Blot Analysis
Regulatory myosin light chain phosphorylation was measured by urea/glycerol-polyacrylamide gel electrophoresis (PAGE) and immunoblotting as previously described (38, 39). Cells were seeded on 60-mm Petri dishes and harvested by completely removing the medium followed by directly lysing with 1 ml of ice-cold 10% trichloroacetic acid containing 10 mm dithiothreitol in acetone. The pellets were washed three times with diethyl ether and resuspended with 8 m urea, 23 mm glycine, 234 mm sucrose, 10.4 mm dithiothreitol, 0.01% bromphenol blue, and 20 mm Tris-HCl (pH 8.6). Protein concentration was measured with a BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific). Equal amounts of protein were loaded into urea/glycerol-PAGE gels, followed by protein transfer to a 0.22-μm PVDF membrane (Millipore). The membrane was then probed with anti-RLC monoclonal antibody (Sigma-Aldrich, dilution 1:4000) and an appropriate secondary antibody (Pierce, Thermo Fisher Scientific) sequentially. The membrane was incubated in Super Signal West Pico substrate (Pierce, Thermo Fisher Scientific) before exposure to film. Non-phosphorylated, mono-phosphorylated, and di-phosphorylated RLC were quantified by densitometric analysis using the ImageJ software package (NIH).
Time-lapse Video Microscopy
Intestinal SMCs were cultured in a chamber containing 5% CO2 at 37 °C. Living-cell images were captured by a differential interference contrast (DIC) motorized inverted research microscope (IX 81; Olympus) equipped with a digital camera (QImaging) driven by Image-Pro Plus 6.2 software. Time-series images were acquired by 10× or 20× dry objective lens.
Wound Healing Assay
SMCs were seeded at an initial density of 1 × 105 cells/cm2 on 0.1% gelatin-coated 24-well plates. When the cells formed a confluent monolayer, a wound was made by scraping a 200-μl pipette tip across the monolayer; the cells were then rinsed with PBS and fed fresh culture medium containing 10% FBS. Beginning at 6 h after wounding, the gap areas were photographed every 3 h. Migrated distances of SMCs front were measured using Image-Pro Plus 6.2 software.
Measurement of Lamellipodial Extension Rate
We measured the lamellipodial extension rate of SMCs on gelatin-coated coverslips in an assay similar to that described earlier (19). Successive positions of the SMC leading edge were imaged every minute under phase-contrast microscopy. To measure the lamellipodial extension rate, the position of the active leading edge was recorded for calculating protruded distance. The distance moved was defined as the distance between the final and initial position of the lamellipodial edge. Extended distances of SMCs were measured using Image-Pro Plus 6.2 software.
Cell Spreading Assay
Glass coverslips were placed in 6-well plates and coated with 0.1% gelatin. Intestinal SMCs were dissociated and harvested by incubating with TrypLETM Express (Invitrogen) and then resuspended in culture medium containing 10% FBS. Cells (2 × 104) were seeded in each well and then incubated at 37 °C in a humidified atmosphere of 5% CO2. At designated time points (15, 30, and 60 min), cells were fixed and stained with SMA antibody and TO-PRO-3. Membrane protrusion and spreading were observed by immunofluorescence confocal microscopy based on the staining. Each coverslip was photographed at several randomly selected fields. The spreading area of the intestinal SMCs was measured using Image-Pro Plus 6.2 software.
Laser Tweezer Measurement of Cell Membrane Tension
We measured the tether force with optical tweezers as described previously (40). The key components consist of a 2000 milliwatt Nd:YAG laser source (1064 nm) connected to a 100× oil-immersion inverted microscope (Zeiss), highly sensitive cold CCD camera (Cascade), and a three-dimensional piezoelectric ceramic stage. When the distance between the center of the trapped microbead and the optical trap is within a small range (∼150 nm), the force exerted on the trapped object is proportional to the distance, that is, F = k × Δx, in which F is the tether force, k is trapping stiffness, and Δx is the distance between the center of the microbead and the optical trap. In our experiment, 5-μm polystyrene microbeads coated with 1 mg/ml mouse IgG (Sigma-Aldrich) (or 2% poly-l-lysine in PBS) were attached to the plasma membrane by holding them on the membrane surface (19), and tethers were formed by pulling on the microbeads with the laser tweezers. Images were taken to measure the distances between the center of the trapped microbead and that of the optical trap, and then we calculated the tether force.
Transmission Electron Microscopy
Primary cultured intestinal SMCs were washed with PBS followed by fixation with 2.5% glutaraldehyde in PBS. A detailed protocol was followed as previously described by Bozzola with minimal modification (41). Ultrathin sections were post-stained and examined using a Hitachi transmission electron microscope.
Fibronectin Pull-down Assay and Co-immunoprecipitation Assay
Confluent cells were washed twice with PBS and resuspended in a lysis buffer composed of 50 mm Tris-HCl pH 8.0, 400 mm NaCl, 0.5% Triton X-100, 0.5 mm EDTA, 0.5 mm EGTA, 1 mm PMSF, and Protease Inhibitor Mixture (Roche Applied Science). After 30 min on ice, the lysate was clarified by centrifugation at 16,000 × g for 15 min at 4 °C. The supernatant was diluted 1:3 in 20 mm Tris-HCl pH 7.5, 0.5% Triton X-100, 0.5 mm EDTA, 1 mm PMSF with Protease Inhibitor Mixture. For the pull-down of the FN related protein complexes, acid-washed glass beads (Sigma-Aldrich) were coated by incubation with 50 μg/ml FN (Calbiochem, Merck) in PBS for 1 h at room temperature and washed five times with PBS. Then, the FN-coated beads were blocked with 5 mg/ml bovine serum albumin in PBS for 30 min at room temperature. An equal amount of the diluted supernatant was incubated with the FN-coated beads with rotation for 3 h at 4 °C. RGD or RGES peptide (at a concentration of 250 μg/ml) was added as indicated. The beads with protein complexes were collected by centrifugation and washed three times with 20 mm Tris-HCl pH 7.5, 150 mm NaCl, 0.5% Triton X-100. For co-immunoprecipitation, equal amounts of the diluted supernatant were precleared with protein G slurry (GE Healthcare) and then incubated with specific antibodies (MLCK (K36) mAb, Sigma-Aldrich; MLCK (N17) pAb, Santa Cruz; Fibronectin pAb, Abcam; Integrin α5 pAb, Millipore) for 2 h at 4 °C. Fifty microliters of protein G slurry was added to each immune reaction, and these were rotated overnight at 4 °C. The immunoprecipitates were washed three times as above. Laemmli sample buffer was added to the pellets of glass beads or protein G slurry and boiled. Then, the proteins were separated by SDS-PAGE followed by immunoblotting using specific antibodies as indicated (Pan-actin mAb, Thermo Fisher Scientific; Vinculin mAb, Sigma-Aldrich).
Statistical Analysis
All of the measurements were performed with ImageJ (NIH) or Image-Pro Plus 6.2 (Media Cybernetics), and statistical analysis was performed with GraphPad Prism 5 software (GraphPad Software). Data are expressed as the mean ± S.E. from at least three experiments and were analyzed by Student's t test with significance defined as *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
RESULTS
Characterization of MLCK-deficient SMCs
Because of the high knock-out efficiency of smooth muscle-specific Cre for MLCK deletion and the unique advantages of smooth muscle cell in migration or signal transduction analysis, here we used primary jejunum SMCs as a model. To ablate MLCK expression in the smooth muscle cells, we generated Mlckflox/flox:SM22CreERT2 (ki) (MLCKSMKO) mice and then induced MLCK deletion specifically in smooth muscle by tamoxifen as previously described (37). The primary MLCK-deficient (Mlckflox/flox:SM22CreERT2, also MLCK−/− or KO) and control (Mlck+/flox:SM22CreERT2, also MLCK+/− or CTR) cells were prepared from the smooth muscle tissue of jejunum. Under our culture conditions, ∼95% of the cells were SMCs as evidenced by staining with the anti-smooth muscle α-actin (SMA) antibody and the anti-smooth muscle myosin heavy chain (SMMHC) antibody (Fig. 1A). In control cells, most MLCK signals co-localized with stress fibers, and some were located at the membrane periphery. A diffuse MLCK signal could also be observed throughout the cells (Fig. 1B). In knock-out cultures, nearly no MLCK signal was found in SMA-positive cells, but a clear signal was found in SMA-negative cells (Fig. 1C). Western blots consistently showed a significant reduction of both S-MLCK and l-MLCK proteins in knock-out cultures compared with control cultures (Fig. 1D). The band of residual MLCK may have been primarily due to the residual non-smooth muscle cells in the culture. Note that the expression of l-MLCK in cultured control cells was elevated during adaption to culture conditions, consistent with other reports (42).
FIGURE 1.
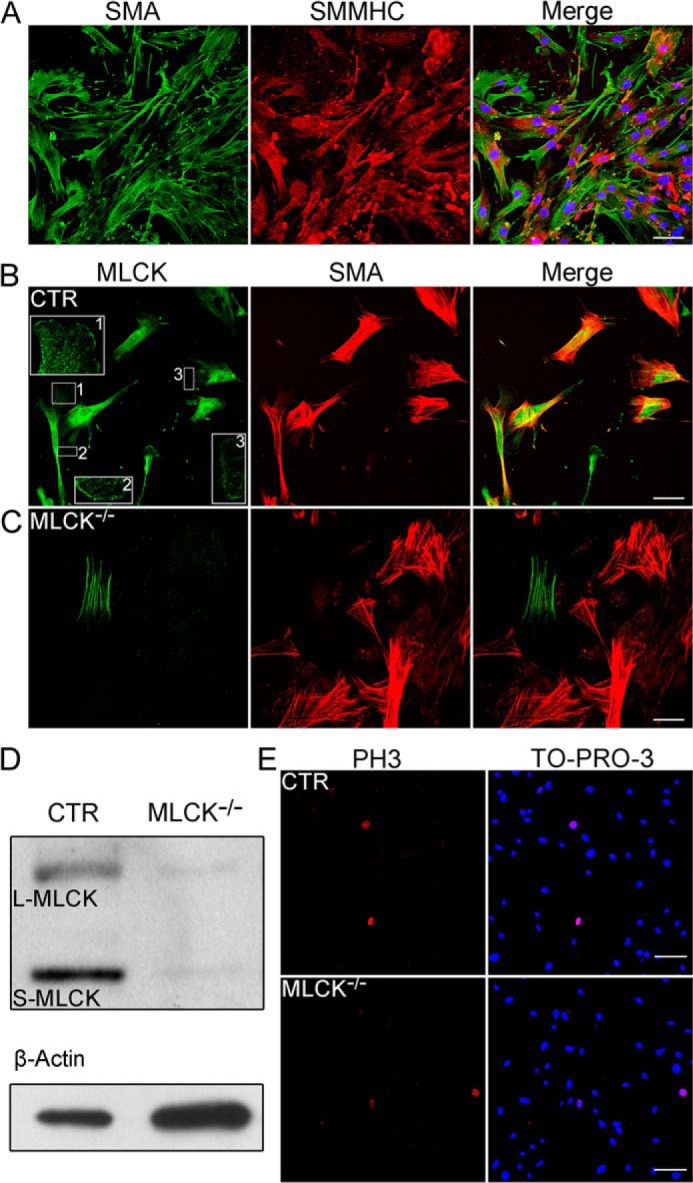
Ablation of MLCK expression in primary cultured SMCs. A, immunofluorescence analysis of smooth muscle-specific markers expressed in primary cultured intestinal SMCs. Specific antibodies to smooth muscle α-actin (SMA; green) and smooth muscle myosin heavy chain (SMMHC; red) were used as primary antibodies. Nuclei were marked with TO-PRO-3 (blue). B and C, immunofluorescence analysis of MLCK in the cultured SMCs with the K36 monoclonal antibody. MLCK (green) was expressed in control but not in MLCK-deficient SMCs. Boxed areas are magnified for detailed features. D, Western blot of MLCK in control and MLCK-deficient SMCs cultured for 1 week. β-Actin was used as a loading control. E, phospho-histone H3 (Ser-10) (PH3; red) immunofluorescence staining of control and MLCK-deficient SMCs cultured in vitro. Nuclei were marked with TO-PRO-3 (blue) stain. Scale bars are 50 μm (A, B, C) and 100 μm (E).
To examine the proliferation of MLCK-deficient cells, we measured the percentage of phospho-histone H3 (Ser10)-positive cells in SMCs by an immunofluorescence assay. The mutant SMCs had 3.2% proliferating cells, which was comparable to control SMCs (3.4%; Fig. 1E). This result suggested that SMC proliferation was not affected by MLCK deletion.
Absence of MLCK Promotes SMC Migration
The prevalent view is that MLCK promotes cell retraction through actomyosin contraction and that the inhibition of MLCK will reduce migration velocity (16, 34, 43). To examine the migration behaviors after MLCK deletion, we first performed explant assays in culture, which resembled the tissue environment for cell migration (44). Whereas control SMCs started to migrate out from the explants at days 6–8 of culture, MLCK-deficient SMCs started their outgrowth as early as at the day 4. The left panel of Fig. 2A represents the typical morphologies of control and knock-out explants cultured for 6 days. The percentages of the explants with outgrown cells were calculated as shown in the right panel of Fig. 2A. After culture for 5 days, ∼60% of the MLCK-deficient explants exhibit outgrown cells (KO: 62 ± 11% versus CTR: 25 ± 10%, p < 0.05; Fig. 2A, right panel). To examine the migration in a monolayer, wound-healing assays were performed. The results showed that MLCK-deficient SMCs healed the gap faster than control cells (Fig. 2B, left panel). Quantification showed a larger area of wound closure in the MLCK-deficient SMCs (KO: 693,360 ± 18,402 μm2 versus CTR: 493,199 ± 18,570 μm2, p < 0.001; Fig. 2B, right panel). We also examined the migration behavior of individual cells (Fig. 2D, left panel). Both control and knock-out cells migrated in random directions, but the accumulated migrated distances of knock-out cells were longer than those of controls at different time points (at 8.5 h, KO: 292.4 ± 50.5 μm versus CTR: 134.6 ± 12.7 μm, p < 0.01; Fig. 2D, middle panel). Furthermore, MLCK-deficient SMCs showed a higher average migrating velocity than control cells (KO: 34.4 ± 5.9 μm/hr versus CTR: 15.8 ± 1.5 μm/hr, p < 0.01; Fig. 2D, right panel). Taken together, these results indicate enhanced cell migration in MLCK-deficient SMCs.
FIGURE 2.

MLCK-deficient SMCs display enhanced migration. Explants of jejunal smooth muscle tissues were cultured on gelatin-coated dishes, and the outgrowth cells were photographed at different time points. A, typical morphologies of control and MLCK-deficient SMCs cultured for 6 days are presented. Positive explants are defined as the explants with ≥1 cell outside after culture. The percentages of the positive explants of control (n = 24–38) and MLCK-deficient (n = 26–40) cells cultured for 5, 7, and 9 days were calculated. B, wound healing assay of control and MLCK-deficient SMCs in vitro. Phase-contrast photographs were taken at 0, 12, and 24 h after the wound was made. Cell migration distances at 12 h were measured and statistically analyzed (n = 102 and 160 respective to CTR and KO). C, consecutive photographs for protruding SMCs were taken within the period of 0–30 min. The extended distance of the leading edge was measured (n = 25 and 30 for CTR and KO, respectively) and statistically analyzed. D, 13 control and 8 MLCK-deficient SMCs were randomly selected for cell migration trace analysis as shown in the left panel. The accumulated migrated distance (middle) and average migrating velocity (right) were measured and statistically analyzed. E, the adhesion properties of control and MLCK-deficient SMCs cultured for 7 days were examined by fixing the cells in situ and co-staining them with anti-SMA (green) and anti-FAK antibodies (blue). Scale bars are 200 μm (A, B) and 20 μm (C, D, E).
Protrusion Is Enhanced in MLCK-deficient SMCs
As cell migration is primarily initiated by membrane protrusion formation, we assessed the protrusion of MLCK-deficient SMCs in culture. In contrast to control cells, the mutant SMCs displayed larger protrusions formed at the leading edge (Fig. 2C, left panel) and a faster protruding rate. Quantification revealed that the average lamellipodial extension distance per 30 min in mutant SMCs was 21.9 ± 1.9 μm, significantly greater than control cells (15.0 ± 2.2 μm, p < 0.05; Fig. 2C, right panel). To determine the focal adhesions of these formed protrusions, we visualized focal adhesion kinase (FAK) in the SMCs using an anti-FAK antibody. The protrusions of both mutant and control cells had strong staining of FAK proteins (Fig. 2E). This observation suggested that the over-produced protrusion was capable of forming focal adhesions. As cell spreading shares a similar property of protrusion and is usually used as an important parameter for protrusion formation, we also characterized the cell spreading properties of MLCK-deficient SMCs (Fig. 3, A–D) by an immunofluorescence assay using an anti-SMA antibody. The control cells spread relatively slow with growing filopodia structures, while knock-out cells spread fast with fewer filopodia. Based on the video results (supplemental Movies S1 and S2), these structures were unlikely produced by retraction fiber formation. MLCK-deficient SMCs showed a similar spreading area compared with control SMCs 15 min after cell inoculation but larger after that time. At 60 min after the initiation of spreading, the sizes of MLCK-deficient SMCs increased 1.9-fold compared with those of the control (KO: 5646 ± 526 μm2 versus CTR: 3000 ± 260 μm2, p < 0.0001; Fig. 3E). The increase of spreading in mutant SMCs appeared not to be due to cell volume alterations because the cell volume was not affected after MLCK deletion, as evidenced by comparable cell sizes before 15 min of spreading (Fig. 3E). We thus concluded that the deletion of MLCK resulted in rapid membrane protrusion. To further verify this conclusion, we introduced a full-length MLCK-expressing adenovirus (Adv-MLCK) into the mutant cells. After infection with Adv-MLCK, the 60 min-spreading area of MLCK-deficient SMCs significantly decreased from 6987 ± 652 μm2 to 3261 ± 523 μm2 (p < 0.0001), which was close to that of control SMCs with Adv-GFP infection (3197 ± 238 μm2; Fig. 3F). In control SMCs, the overexpression of MLCK by infection with Adv-MLCK caused a further decrease in the spreading area (CTR+Adv-GFP: 3197 ± 238 μm2 versus CTR+Adv-MLCK: 1634 ± 126 μm2, p < 0.0001; Fig. 3F). When infected cells with 2Ig domain-expressing adenovirus (Adv-2Ig) as a MLCK control, both mutant and control SMCs showed no significant reduction of cell size (KO+Adv-GFP: 6987 ± 652 μm2 versus KO+Adv-2Ig: 5947 ± 1037 μm2, p > 0.05; CTR+Adv-GFP: 3197 ± 238 μm2 versus CTR+Adv-2Ig: 4618 ± 617 μm2, p = 0.0107; Fig. 3F). This rescue effect supported the conclusion that the altered membrane protrusion in the mutant cells was attributable to the role of MLCK.
FIGURE 3.
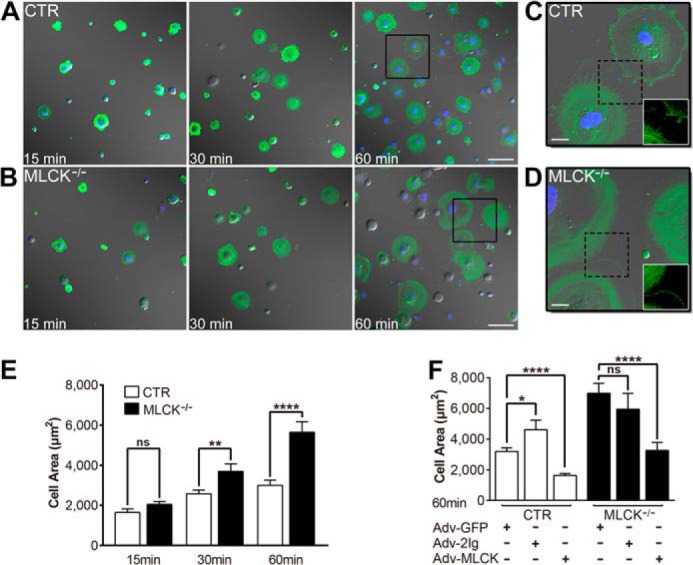
MLCK-deficient SMCs show accelerated protruding and spreading in vitro. A and B, the spreading assay for the SMCs was performed by seeding the suspended SMCs on gelatin-coated coverslips followed by imaging at 15, 30, and 60 min. The SMCs were visualized with an anti-SMA antibody (green) and TO-PRO-3 (blue). DIC images merged with fluorescence signals are shown. C and D, magnified images derived from the boxed areas in panels A and B, respectively. Fluorescent images of dashed boxed areas are shown in white boxes. E, the area of spreading cells was measured (n = 46–89) and statistically analyzed. F, rescue assay for MLCK-deficient SMCs. Control and MLCK-deficient SMCs were infected with control adenovirus (Adv-GFP) and MLCK-expressible variants (Adv-MLCK, Adv-2Ig; MOI = 4) for 48 h, and then the 60-min spreading area was examined. n = 32–88. Scale bars are 100 μm (A, B) and 20 μm (C, D).
MLCK Controls Cell Membrane Tension
As protrusion formation underlies the membrane events of a migrating cell, we examined the membrane of MLCK-deficient SMCs through live imaging analysis. The spreading edges of adhering MLCK-deficient SMCs appeared smoother and larger than control cells (Fig. 3, C and D; CTR: supplemental Movie S1 and KO: supplemental Movie S2). Interestingly, when the MLCK-deficient SMCs were suspended for 20 min, many cells formed blebs around the membrane periphery (Fig. 4A). Quantification showed that 56 ± 9% of mutant SMCs had blebs, which was significantly higher than control SMCs (19 ± 2%, p < 0.05; Fig. 4B). Increased bleb formation usually reflects an impaired association of the plasma membrane with the skeleton underneath (45). Our collective observations thus imply an increase in membrane movement in MLCK-deficient SMCs. As membrane tension is an important factor for the modulation of membrane dynamics (18, 46), we then directly measured the tether force of SMC membrane with laser tweezers (40). This method did not work accurately for bleb-forming cells; therefore, we only chose the cells without obvious bleb formation for tether force measurement. The results showed that the average tether force of MLCK-deficient SMCs was significantly lower than that of control cells with either mouse IgG- or poly-l-lysine-coated microbeads (Fig. 4C, mouse IgG-coated microbeads, KO: 96 ± 8 pN versus CTR: 145 ± 19 pN, p < 0.01; poly-l-lysine-coated microbeads, KO: 118 ± 6 pN versus CTR: 152 ± 12 pN, p < 0.01). Thus, we concluded that MLCK deletion led to decreased membrane tension.
FIGURE 4.

Membrane tether force is reduced in MLCK-deficient SMCs. The SMCs cultured in vitro were isolated by TrypLETM Express digestion and then subjected to tether force measurement with laser tweezers. A, DIC images show many membrane blebs around MLCK-deficient SMCs (below) after suspending over 20 min, but very few were observed around controls (above). Arrows point to typical membrane blebs. The scale bar is 10 μm. B, percentages of the blebbed cells were calculated (CTR: n = 302; KO: n = 193). C, average membrane tether forces measured by laser tweezers with mouse IgG- and poly-l-lysine-coated microbeads.
MLCK Regulates Membrane F-actin Skeleton
Because the membrane tension of nonspherical animal cells is largely determined by membrane-cytoskeleton adhesion (18, 45, 47), we speculated that the membrane skeleton was impaired after MLCK deletion. The bleb formation in suspended MLCK-deficient cells also supports this speculation because the incomplete formation of the cytoskeleton underneath the membrane causes membranous blebs (48). We then examined the F-actin skeleton of spreading SMCs. Compared with control SMCs, MLCK-deficient SMCs had much weaker F-actin signals in the membrane periphery, but the signals at other areas were comparable (Fig. 5, A–C). We analyzed the fluorescence intensities of bound phalloidin in peripheral areas (defined as 10% of diameter from cell edge) relative to whole cells with ImageJ (within the Intensity Correlation Analysis plugin) (49). The peripheral F-actin content in knock-out SMCs decreased from 40 ± 1% (CTR, n = 99) to 28 ± 1% (KO, n = 94, p < 0.0001) (Fig. 5D). During the later stages of spreading, control SMCs showed clearly visible filopodia around the membrane, but MLCK-deficient SMCs did not. The average number of filopodia structures per cell (longer than 5 μm) was quantified, showing that the mutant cells exhibited significantly fewer numbers of filopodia structures per cell than control cells (CTR: 4.8 ± 0.5 versus KO: 0.7 ± 0.2, p < 0.0001, both n = 100).
FIGURE 5.

MLCK affects F-actin cytoskeleton reorganization during cell spreading. A and B, immunofluorescence analysis of F-actin (green) and alpha-tubulin (red) expressed in control and MLCK-deficient SMCs 60 min after spreading. C, angled and side views of three-dimensional reconstruction from merged immunofluorescence image series of F-actin (green) and α-tubulin (red) at 60 min. D, the ratios of bound phalloidin (representing F-actin) fluorescence intensities of the peripheral area (top right, defined as 10% of diameter from cell edge) to that in the whole cell (top left) were measured by ImageJ with the Intensity Correlation Analysis plugin. MLCK-deficient cells (28 ± 1%, n = 94) exhibited less peripheral F-actin content than control cells (40 ± 1%, n = 99). Scale bars are 10 μm (A, B, C).
RLC Phosphorylation Is Not Affected in MLCK-deficient Cells
MLCK is a dedicated kinase for myosin light chain phosphorylation and is required for RLC phosphorylation during smooth muscle contraction in vivo (23, 50). The RLC phosphorylation level in MLCK-deficient smooth muscle tissue is remarkably reduced either with or without stimulation (37, 39). However, when SMCs were cultured for prolonged periods, phosphorylated RLC was clearly detected; the ratio of phosphorylated RLC over total RLC was similar to control cells (Fig. 6A. Mono-phosphorylated RLC, CTR: 32 ± 3%, KO, 26 ± 5%, p > 0.05; Di-phosphorylated RLC, CTR: 35 ± 4%, KO, 42 ± 8%, p > 0.05). We also measured RLC phosphorylation with immunofluorescence assays, and similar results were obtained (Fig. 6, B–D). There was no significant difference in the mono- or di-phosphorylation level or distribution between control and MLCK-deficient SMCs (Fig. 6B). Furthermore, the patterns of myosin IIA and IIB with phosphorylated RLC were comparable in the leading edge of migrating control and MLCK-deficient SMCs (Fig. 6, C and D). In both cell types, myosin IIB was concentrated in the central part and almost absent in the cell periphery, while myosin IIA was present in the whole cell. Thus, our results indicate that RLC phosphorylation was not significantly altered in MLCK-deficient SMCs in primary culture and that the phenotype of mutant SMCs might not be caused by a lack of MLCK kinase activity.
FIGURE 6.

MLCK regulates cell spreading through 5DFRXXL in a kinase-independent manner. A–D, RLC phosphorylation is not affected by MLCK deletion in SMCs. A, RLC phosphorylation was measured by urea/glycerol-PAGE and immunoblotting with an anti-RLC antibody. The bands of non-, mono-, and di-phosphorylated RLCs are shown (above). Densitometric analysis of the three bands was conducted by ImageJ (bottom, CTR: n = 6; KO: n = 5). B, immunofluorescence analysis with co-staining by anti-mono-phospho-RLC (green) and anti-di-phospho-RLC antibodies (red). Merged images are overlaid with TO-PRO-3-stained nuclei (blue). C and D, representative immunofluorescence images showing the distribution of phospho-RLC (green) and myosin IIA/B (red) in the leading edge of migrating control and MLCK-deficient SMCs. The arrow indicates the direction of cell protrusion extension. E, control and MLCK-deficient SMCs were nucleofected with a control (GFP) and MLCK-expression plasmids (l-MLCK, KD-MLCK, 5DFRXXL), and then the 60-min spreading area was examined after 48 h. n = 54–266. Scale bars are 20 μm (B) and 5 μm (C, D).
MLCK Regulates Cell Protrusion through 5DFRXXL in a Kinase-independent Manner
No alteration of RLC phosphorylation in motile MLCK-deficient SMCs implies that MLCK kinase activity is not required for migration. To test this hypothesis, we introduced kinase-dead MLCK into MLCK-deficient cells. Surprisingly, the enhanced spreading by MLCK deletion was significantly restored (Fig. 6E). Upon nucleofection with a kinase-dead MLCK-expressing plasmid, the spreading area (60 min after inoculation) of the mutant cells decreased from 4242 ± 140 μm2 to 3099 ± 149 μm2 (p < 0.0001), which was comparable to control cells (KO+KD-MLCK: 3099 ± 149 μm2 versus CTR+GFP: 3216 ± 87 μm2, p > 0.05; Fig. 6E). This result indicates that the loss of MLCK kinase activity did not contribute to the phenotype of KO cells.
Considering the alteration of F-actin organization in KO cells and that the five-DFRXXL motif (5DFRXXL) of MLCK is a potent F-actin-binding site, we tested the role of this region in the spreading phenotype. After nucleofection with a 5DFRXXL-expressing plasmid, the enhanced spreading of MLCK-deficient SMCs was inhibited completely (KO+GFP: 4242 ± 140 μm2 versus KO+5DFRXXL: 2212 ± 123 μm2, p < 0.0001; Fig. 6E). The introduction of this plasmid into control cells also led to a significant reduction of the spreading area (Fig. 6E). Thus, 5DFRXXL was able to rescue the phenotypic spreading of MLCK-deficient SMCs.
MLCK Is Involved in Fibronectin-Integrin-Cytoskeleton Linkages
Physiologically, the membrane skeleton is critical for the tension formation of the membrane (47, 51, 52). At the leading edge of a migrating cell, the interaction of the membrane cytoskeleton with the ECM determines the protrusion formation. Because integrins forge links between the fibronectin (FN)-containing ECM and actomyosin cytoskeleton (53–56), we hypothesized that MLCK may participates in such a membrane complex in which it acts as a scaffold protein of the actomyosin cytoskeleton and hence regulates membrane tension, which acts against cell protrusion. We thus performed an in vitro pull-down assay with FN-coated glass beads. FN-coated beads clearly pulled down S-MLCK from SMC lysate (Fig. 7A). This effect was significantly inhibited by the RGD peptide, a specific inhibitor of FN-integrin interactions, but not by the RGES control peptide (FN: 100% versus FN with RGD: 39 ± 12%, p < 0.01; FN: 100% versus FN with RGES: 95 ± 16%, p = 0.76; FN with RGD: 39 ± 12% versus FN with RGES: 95 ± 16%, p < 0.05; n = 5; Fig. 7B). l-MLCK and integrin α5 were detected in the pulled down complexes with a similar pattern. This observation indicated a potential interaction between MLCK and FN ECM, possibly through the linkage of integrins. To examine the association of MLCK with transmembrane complexes of FN/integrins as well as other associated proteins, we performed co-immunoprecipitation (co-IP) assays. The SMC lysate was immunoprecipitated with monoclonal and polyclonal antibodies against MLCK (MLCK mAb and pAb), FN pAb or integrin α5 pAb, respectively. The immunoprecipitates were assayed by Western blotting. MLCK, integrin α5 and FN were clearly detected in all of the immunoprecipitates (Fig. 7C). In addition, an important mediator of FN-integrin-cytoskeleton linkages (vinculin) and fundamental components of cortical cytoskeleton (myosin IIA and actin) were found in these immunoprecipitates (Fig. 7, C and D). However, weaker signals of myosin IIA and vinculin were observed after the deletion of MLCK (Fig. 7D). These results indicate that MLCK may be involved in the formation of the FN-integrin-cytoskeleton complex in vivo.
FIGURE 7.

MLCK participates in the formation of fibronectin-integrin-cytoskeleton transmembrane protein complexes. A, with a fibronectin pull-down assay, MLCK isoforms and integrin α5 were detected in the protein complexes binding FN. In the “input” signal panel, total protein of SMC lysate used in the pull-down assay was loaded. In other signal panels, fibronectin pulled-down proteins were probed. RGD peptide was used to block the FN-integrin interaction, and RGES peptide was used as a negative control. B, quantitation of FN pulled-down S-MLCK (n = 5). C and D, co-immunoprecipitation was performed, and the immunoprecipitates were then subjected to immunoblotting (IB) using antibodies against MLCK, integrin α5, FN, pan-actin, myosin IIA, and vinculin. Whole-SMC lysate (Input) was used as a positive control. Protein G-Sepharose without antibody (No Ab) or with rabbit IgG (IgG) was used as a negative control.
DISCUSSION
Similar to the force produced by smooth muscle contraction, the force necessary for cell body contractility during migration is developed by the cross-bridge movement of actomyosin, which is initiated by RLC phosphorylation (57, 58). As an essential kinase for RLC phosphorylation, MLCK is thought to accelerate cell migration by regulating multiple processes, including retraction (59), actin retrograde flow (32), and membrane ruffling (16). However, these conclusions are mainly based on pharmacological evidence of inhibitors of MLCK with nonspecific effects. The use of MLCK-deficient cells allows us to directly examine the role of MLCK in migration without being influenced by these nonspecific effects. We surprisingly found that the deletion of MLCK in cultured SMCs did not affect RLC phosphorylation and did not inhibit cell migration, as expected, but promoted cell migration velocity. All of our evidence suggests that MLCK is not required for the RLC phosphorylation necessary for force generation in motile cells. This conclusion points to two possibilities for RLC phosphorylation: RLC phosphorylation by MLCK could be compensated for by other myosin light chain kinases, such as ILK, MRCK, ROCK, and ZIP kinase (60–63); or calcium-independent myosin light chain kinases, not MLCK, phosphorylate RLC during migration. We would favor the latter possible speculation because the intracellular ionized calcium concentration of a cell without stimuli is relatively low (∼100 nm) and cannot easily activate MLCK (64). Undoubtedly, the identification of the key kinase for RLC phosphorylation during cell migration will lead to an improved understanding of force development in this process.
During the migration of the stimulated cells, the membrane tension of cells is reduced, and the reduced tension is associated with enhanced lamellipodial extension rate (19). There is evidence that this membrane tension is important for driving cell migration both in vitro and in vivo (46, 47, 65). Chemically reducing membrane tension may accelerate cell spreading and lamellipodial extension (19), whereas increasing membrane tension by hypotonic treatment may cause lamellipodial and filopodial retraction (66). However, how the migrating cells intrinsically regulate membrane tension necessary for migration behaviors is still unclear. We found that the deletion of MLCK resulted in enhanced cell spreading along with reduced membrane tether force and increased membrane flexibility. This strongly suggests that MLCK may be an essential intrinsic kinase that regulates membrane tension during cell migration. Because its catalytic activity is not required, such regulation of membrane tension may not be controlled by calcium signaling. According to our rescue experiments with 5DFRXXL, membrane tension is primarily determined by the constitutive F-actin binding activity of MLCK. Other lines of evidence also support this proposal. For example, the absence of MLCK caused less F-actin underneath the cell membrane; the modification of F-actin stabilization with a low concentration of F-actin stabilizer led to a partial restoration of the spreading phenotype. Therefore, the intrinsic regulation of membrane tension by MLCK may be accomplished by binding with F-actin filaments. Abundant (∼4 μm) and ubiquitous MLCK in cells provides sufficient protein necessary for F-actin binding in motile cells (67).
During migration processes, fibronectin-containing ECM essentially influences the multiple biochemical and mechanical processes of cell migration simultaneously (68). The cells translocate when they engage the ECM with integrin receptors that link to the intracellular cytoskeleton. The transmembrane complex serves as a mechanical sensor of tether force, and the integrin signaling may switch the toggle between relaxed and tense states (69). Our pull-down and co-IP assays showed that MLCK was involved in the formation of this complex. The tense membrane conformation mediated by MLCK appears to be coupled with FN ECM through integrin linkage during cell migration. This proposal is supported by the similar cell protrusion phenotype of fibronectin-depleted or integrin α5β1-disrupted cells (70). The phenotype of the integrin-null cells showing amoeboid-like flexible protrusions after disengaging FN ECM-integrins also supports this proposal (71). Interestingly, myosin IIA-deficient cells also display increased cell migration and exaggerated membrane ruffling (72), and introduction of myosin IIA-expression restrains the spreading of COS-7 cells (73). Thus, we propose that MLCK may serve as an F-actin-binding protein stabilizing the F-actin/myosin II network of the membranecytoskeleton, thereby maintaining the plasma membrane at a high-tension state before protruding. To allow cell protrusion, the membrane complex releases the membrane tension. The formed protrusion with low tension may provide the entocyte with enough space to move in. In addition, the power produced by the different membrane pressures between the front and rear area may push the cytosolic substances to flow into the protrusive area. In in vivo conditions, the pressure from tissue environments should be included in this driving force.
Supplementary Material
This work was supported by the National Basic Research Program of China (973 Program, 2014CB964701) and the National Natural Science Foundation of China (31371356, 31200824, and 31330034).

This article contains supplemental Movies S1 and S2.
- ECM
- extracellular matrix
- MLCK
- myosin light chain kinase
- RLC
- regulatory light chain
- SMC
- smooth muscle cell
- FAK
- focal adhesion kinase
- SMMHC
- smooth muscle myosin heavy chain
- DIC
- differential interference contrast
- SMA
- smooth muscle α-actin.
REFERENCES
- 1. Lauffenburger D. A., Horwitz A. F. (1996) Cell migration: a physically integrated molecular process. Cell 84, 359–369 [DOI] [PubMed] [Google Scholar]
- 2. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 3. Pollard T. D., Blanchoin L., Mullins R. D. (2000) Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29, 545–576 [DOI] [PubMed] [Google Scholar]
- 4. Traynor D., Kay R. R. (2007) Possible roles of the endocytic cycle in cell motility. J. Cell Sci. 120, 2318–2327 [DOI] [PubMed] [Google Scholar]
- 5. Nobes C. D., Hall A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 6. Hu K., Ji L., Applegate K. T., Danuser G., Waterman-Storer C. M. (2007) Differential transmission of actin motion within focal adhesions. Science 315, 111–115 [DOI] [PubMed] [Google Scholar]
- 7. Parsons J. T., Horwitz A. R., Schwartz M. A. (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schiller H. B., Fässler R. (2013) Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 14, 509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cramer L. P. (2013) Mechanism of cell rear retraction in migrating cells. Curr. Opin. Cell Biol. 25, 591–599 [DOI] [PubMed] [Google Scholar]
- 10. Pollard T. D., Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 11. Mitchison T. J., Cramer L. P. (1996) Actin-based cell motility and cell locomotion. Cell 84, 371–379 [DOI] [PubMed] [Google Scholar]
- 12. Burnette D. T., Manley S., Sengupta P., Sougrat R., Davidson M. W., Kachar B., Lippincott-Schwartz J. (2011) A role for actin arcs in the leading-edge advance of migrating cells. Nat. Cell Biol. 13, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Betapudi V., Licate L. S., Egelhoff T. T. (2006) Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 66, 4725–4733 [DOI] [PubMed] [Google Scholar]
- 14. Sellers J. R., Pato M. D., Adelstein R. S. (1981) Reversible phosphorylation of smooth muscle myosin, heavy meromyosin, and platelet myosin. J. Biol. Chem. 256, 13137–13142 [PubMed] [Google Scholar]
- 15. Sellers J. R. (1991) Regulation of cytoplasmic and smooth muscle myosin. Curr. Opin. Cell Biol. 3, 98–104 [DOI] [PubMed] [Google Scholar]
- 16. Totsukawa G., Wu Y., Sasaki Y., Hartshorne D. J., Yamakita Y., Yamashiro S., Matsumura F. (2004) Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 164, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larsen M., Tremblay M. L., Yamada K. M. (2003) Phosphatases in cell-matrix adhesion and migration. Nat. Rev. Mol. Cell Biol. 4, 700–711 [DOI] [PubMed] [Google Scholar]
- 18. Sheetz M. P., Dai J. W. (1996) Modulation of membrane dynamics and cell motility by membrane tension. Trends Cell Biol. 6, 85–89 [DOI] [PubMed] [Google Scholar]
- 19. Raucher D., Sheetz M. P. (2000) Cell spreading and lamellipodial extension rate is regulated by membrane tension. J. Cell Biol. 148, 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tinevez J. Y., Schulze U., Salbreux G., Roensch J., Joanny J. F., Paluch E. (2009) Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. U.S.A. 106, 18581–18586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Batchelder E. L., Hollopeter G., Campillo C., Mezanges X., Jorgensen E. M., Nassoy P., Sens P., Plastino J. (2011) Membrane tension regulates motility by controlling lamellipodium organization. Proc. Natl. Acad. Sci. U.S.A. 108, 11429–11434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Somlyo A. P., Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 23. Kamm K. E., Stull J. T. (1985) The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu. Rev. Pharmacol. Toxicol. 25, 593–620 [DOI] [PubMed] [Google Scholar]
- 24. Kamm K. E., Stull J. T. (2001) Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 276, 4527–4530 [DOI] [PubMed] [Google Scholar]
- 25. Smith A. F., Bigsby R. M., Word R. A., Herring B. P. (1998) A 310-bp minimal promoter mediates smooth muscle cell-specific expression of telokin. Am. J. Physiol. 274, C1188–C1195; discussion C1187 [DOI] [PubMed] [Google Scholar]
- 26. Smith L., Parizi-Robinson M., Zhu M. S., Zhi G., Fukui R., Kamm K. E., Stull J. T. (2002) Properties of long myosin light chain kinase binding to F-actin in vitro and in vivo. J. Biol. Chem. 277, 35597–35604 [DOI] [PubMed] [Google Scholar]
- 27. Smith L., Su X., Lin P., Zhi G., Stull J. T. (1999) Identification of a novel actin binding motif in smooth muscle myosin light chain kinase. J. Biol. Chem. 274, 29433–29438 [DOI] [PubMed] [Google Scholar]
- 28. Lin P., Luby-Phelps K., Stull J. T. (1999) Properties of filament-bound myosin light chain kinase. J. Biol. Chem. 274, 5987–5994 [DOI] [PubMed] [Google Scholar]
- 29. Yang C. X., Chen H. Q., Chen C., Yu W. P., Zhang W. C., Peng Y. J., He W. Q., Wei D. M., Gao X., Zhu M. S. (2006) Microfilament-binding properties of N-terminal extension of the isoform of smooth muscle long myosin light chain kinase. Cell Res. 16, 367–376 [DOI] [PubMed] [Google Scholar]
- 30. Zhang W. C., Peng Y. J., He W. Q., Lv N., Chen C., Zhi G., Chen H. Q., Zhu M. S. (2008) Identification and functional characterization of an aggregation domain in long myosin light chain kinase. FEBS J. 275, 2489–2500 [DOI] [PubMed] [Google Scholar]
- 31. Kudryashov D. S., Chibalina M. V., Birukov K. G., Lukas T. J., Sellers J. R., Van Eldik L. J., Watterson D. M., Shirinsky V. P. (1999) Unique sequence of a high molecular weight myosin light chain kinase is involved in interaction with actin cytoskeleton. FEBS Lett. 463, 67–71 [DOI] [PubMed] [Google Scholar]
- 32. Giannone G., Dubin-Thaler B. J., Döbereiner H. G., Kieffer N., Bresnick A. R., Sheetz M. P. (2004) Periodic lamellipodial contractions correlate with rearward actin waves. Cell 116, 431–443 [DOI] [PubMed] [Google Scholar]
- 33. Katoh K., Kano Y., Amano M., Kaibuchi K., Fujiwara K. (2001) Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am. J. Physiol. Cell Physiol. 280, C1669–C1679 [DOI] [PubMed] [Google Scholar]
- 34. Niggli V., Schmid M., Nievergelt A. (2006) Differential roles of Rho-kinase and myosin light chain kinase in regulating shape, adhesion, and migration of HT1080 fibrosarcoma cells. Biochem. Biophys. Res. Commun. 343, 602–608 [DOI] [PubMed] [Google Scholar]
- 35. Connell L. E., Helfman D. M. (2006) Myosin light chain kinase plays a role in the regulation of epithelial cell survival. J. Cell Sci. 119, 2269–2281 [DOI] [PubMed] [Google Scholar]
- 36. Chew T. L., Wolf W. A., Gallagher P. J., Matsumura F., Chisholm R. L. (2002) A fluorescent resonant energy transfer-based biosensor reveals transient and regional myosin light chain kinase activation in lamella and cleavage furrows. J. Cell Biol. 156, 543–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He W. Q., Peng Y. J., Zhang W. C., Lv N., Tang J., Chen C., Zhang C. H., Gao S., Chen H. Q., Zhi G., Feil R., Kamm K. E., Stull J. T., Gao X., Zhu M. S. (2008) Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 135, 610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Word R. A., Casey M. L., Kamm K. E., Stull J. T. (1991) Effects of Cgmp on [Ca2+]I, Myosin Light Chain Phosphorylation, and Contraction in Human Myometrium. Am. J. Physiol. 260, C861–C867 [DOI] [PubMed] [Google Scholar]
- 39. Zhang W. C., Peng Y. J., Zhang G. S., He W. Q., Qiao Y. N., Dong Y. Y., Gao Y. Q., Chen C., Zhang C. H., Li W., Shen H. H., Ning W., Kamm K. E., Stull J. T., Gao X., Zhu M. S. (2010) Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J. Biol. Chem. 285, 5522–5531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y. J., Wen C., Xie H. M., Ye A. P., Yin Y. J. (2009) Mechanical property analysis of stored red blood cell using optical tweezers. Colloids and Surfaces B: Biointerfaces 70, 169–173 [DOI] [PubMed] [Google Scholar]
- 41. Bozzola J. J. (2007) Conventional specimen preparation techniques for transmission electron microscopy of cultured cells. Methods Mol. Biol. 369, 1–18 [DOI] [PubMed] [Google Scholar]
- 42. Blue E. K., Goeckeler Z. M., Jin Y., Hou L., Dixon S. A., Herring B. P., Wysolmerski R. B., Gallagher P. J. (2002) 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am. J. Physiol. Cell Physiol. 282, C451–C460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holzapfel G., Wehland J., Weber K. (1983) Calcium control of actin-myosin based contraction in triton models of mouse 3T3 fibroblasts is mediated by the myosin light chain kinase (MLCK)-calmodulin complex. Exp. Cell Res. 148, 117–126 [DOI] [PubMed] [Google Scholar]
- 44. Kenagy R. D., Hart C. E., Stetler-Stevenson W. G., Clowes A. W. (1997) Primate smooth muscle cell migration from aortic explants is mediated by endogenous platelet-derived growth factor and basic fibroblast growth factor acting through matrix metalloproteinases 2 and 9. Circulation 96, 3555–3560 [DOI] [PubMed] [Google Scholar]
- 45. Sheetz M. P. (2001) Cell control by membrane-cytoskeleton adhesion. Nat. Rev. Mol. Cell Biol. 2, 392–396 [DOI] [PubMed] [Google Scholar]
- 46. Diz-Muñoz A., Fletcher D. A., Weiner O. D. (2013) Use the force: membrane tension as an organizer of cell shape and motility. Trends Cell Biol. 23, 47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lieber A. D., Yehudai-Resheff S., Barnhart E. L., Theriot J. A., Keren K. (2013) Membrane Tension in Rapidly Moving Cells Is Determined by Cytoskeletal Forces. Curr. Biol. 23, 1409–1417 [DOI] [PubMed] [Google Scholar]
- 48. Dai J. W., Sheetz M. P. (1999) Membrane tether formation from blebbing cells. Biophys. J. 77, 3363–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glynn M. W., McAllister A. K. (2006) Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat. Protoc. 1, 1287–1296 [DOI] [PubMed] [Google Scholar]
- 50. Somlyo A. P., Somlyo A. V. (1994) Signal Transduction and Regulation in Smooth Muscle. Nature 372, 231–236 [DOI] [PubMed] [Google Scholar]
- 51. Raucher D., Sheetz M. P. (1999) Characteristics of a membrane reservoir buffering membrane tension. Biophys. J. 77, 1992–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doherty G. J., McMahon H. T. (2008) Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu. Rev. Biophys. 37, 65–95 [DOI] [PubMed] [Google Scholar]
- 53. Liu S., Calderwood D. A., Ginsberg M. H. (2000) Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 113, 3563–3571 [DOI] [PubMed] [Google Scholar]
- 54. Geiger B., Bershadsky A., Pankov R., Yamada K. M. (2001) Transmembrane extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2, 793–805 [DOI] [PubMed] [Google Scholar]
- 55. Brakebusch C., Fässler R. (2003) The integrin-actin connection, an eternal love affair. EMBO J. 22, 2324–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vicente-Manzanares M., Choi C. K., Horwitz A. R. (2009) Integrins in cell migration - the actin connection. J. Cell Sci. 122, 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vicente-Manzanares M., Zareno J., Whitmore L., Choi C. K., Horwitz A. F. (2007) Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 176, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Okeyo K. O., Adachi T., Sunaga J., Hojo M. (2009) Actomyosin contractility spatiotemporally regulates actin network dynamics in migrating cells. J. Biomech. 42, 2540–2548 [DOI] [PubMed] [Google Scholar]
- 59. Verkhovsky A. B., Svitkina T. M., Borisy G. G. (1999) Self-polarization and directional motility of cytoplasm. Curr. Biol. 9, 11–20 [DOI] [PubMed] [Google Scholar]
- 60. Deng J. T., Van Lierop J. E., Sutherland C., Walsh M. P. (2001) Ca2+-independent smooth muscle contraction - A novel function for integrin-linked kinase. J. Biol. Chem. 276, 16365–16373 [DOI] [PubMed] [Google Scholar]
- 61. Leung T., Chen X. Q., Tan I., Manser E., Lim L. (1998) Myotonic dystrophy kinase-related Cdc42-binding kinase acts as a Cdc42 effector in promoting cytoskeletal reorganization. Mol. Cell Biol. 18, 130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 63. Komatsu S., Ikebe M. (2004) ZIP kinase is responsible for the phosphorylation of myosin II and necessary for cell motility in mammalian fibroblasts. J. Cell Biol. 165, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mizuno Y., Isotani E., Huang J., Ding H. L., Stull J. T., Kamm K. E. (2008) Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am. J. Physiol. 295, C358–C364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Diz-Muñoz A., Krieg M., Bergert M., Ibarlucea-Benitez I., Muller D. J., Paluch E., Heisenberg C. P. (2010) Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 8, e1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dai J. W., Sheetz M. P., Wan X. D., Morris C. E. (1998) Membrane tension in swelling and shrinking molluscan neurons. J. Neurosci. 18, 6681–6692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tansey M. G., Luby-Phelps K., Kamm K. E., Stull J. T. (1994) Ca(2+)-dependent phosphorylation of myosin light chain kinase decreases the Ca2+ sensitivity of light chain phosphorylation within smooth muscle cells. J. Biol. Chem. 269, 9912–9920 [PubMed] [Google Scholar]
- 68. Rozario T., DeSimone D. W. (2010) The extracellular matrix in development and morphogenesis: a dynamic view. Dev. Biol. 341, 126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Friedland J. C., Lee M. H., Boettiger D. (2009) Mechanically activated integrin switch controls α5β1 function. Science 323, 642–644 [DOI] [PubMed] [Google Scholar]
- 70. Davidson L. A., Marsden M., Keller R., Desimone D. W. (2006) Integrin α5β1 and fibronectin regulate polarized cell protrusions required for Xenopus convergence and extension. Curr. Biol. 16, 833–844 [DOI] [PubMed] [Google Scholar]
- 71. Renkawitz J., Schumann K., Weber M., Lämmermann T., Pflicke H., Piel M., Polleux J., Spatz J. P., Sixt M. (2009) Adaptive force transmission in amoeboid cell migration. Nat. Cell Biol. 11, 1438–1443 [DOI] [PubMed] [Google Scholar]
- 72. Even-Ram S., Doyle A. D., Conti M. A., Matsumoto K., Adelstein R. S., Yamada K. M. (2007) Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat. Cell Biol. 9, 299–309 [DOI] [PubMed] [Google Scholar]
- 73. Betapudi V. (2010) Myosin II motor proteins with different functions determine the fate of lamellipodia extension during cell spreading. PLoS ONE 5, e8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.