

Background: Misfolded SOD1 is associated with sporadic and familial ALS.
Results: The epitope of C4F6, an antibody specific for misfolded SOD1, has been defined. Loops IV and VII in SOD1 modulate exposure of this epitope.
Conclusion: Exposure of the C4F6 epitope correlates with heightened SOD1-mediated toxicity.
Significance: Concealing the C4F6 epitope by stabilizing SOD1 loops IV and VII has therapeutic potential for ALS.
Keywords: Amyotrophic Lateral Sclerosis (ALS) (Lou Gehrig Disease), Microglia, Neurodegenerative Disease, Protein Cross-linking, Protein Misfolding, Superoxide Dismutase (SOD)
Abstract
Mutations and aberrant post-translational modifications within Cu,Zn-superoxide dismutase (SOD1) cause this otherwise protective enzyme to misfold, leading to amyotrophic lateral sclerosis (ALS). The C4F6 antibody selectively binds misfolded SOD1 in spinal cord tissues from postmortem human ALS cases, as well as from an ALS-SOD1 mouse model, suggesting that the C4F6 epitope reports on a pathogenic conformation that is common to misfolded SOD1 variants. To date, the residues and structural elements that comprise this epitope have not been elucidated. Using a chemical cross-linking and mass spectrometry approach, we identified the C4F6 epitope within several ALS-linked SOD1 variants, as well as an oxidized form of WT SOD1, supporting the notion that a similar misfolded conformation is shared among pathological SOD1 proteins. Exposure of the C4F6 epitope was modulated by the SOD1 electrostatic (loop VII) and zinc binding (loop IV) loops and correlated with SOD1-induced toxicity in a primary microglia activation assay. Site-directed mutagenesis revealed Asp92 and Asp96 as key residues within the C4F6 epitope required for the SOD1-C4F6 binding interaction. We propose that stabilizing the functional loops within SOD1 and/or obscuring the C4F6 epitope are viable therapeutic strategies for treating SOD1-mediated ALS.
Introduction
ALS2 is a quickly progressing, fatal neurodegenerative disorder that primarily targets motor neurons. The complex nature of ALS represents a hurdle in developing effective therapies for this incurable disease. A growing list of genes have been linked to ALS, where mutations in SOD1, TDP-43, FUS, and C9orf72 account for >50% of inherited or familial ALS (FALS) (1). However, much less is known about the cause(s) of sporadic ALS (SALS) that account for the majority (90%) of ALS cases (1). FALS and SALS are clinically indistinguishable, suggesting similar mechanisms are at play for both forms of this disease.
SOD1 (Cu,Zn-superoxide dismutase) represents a factor that is common to FALS and SALS. Mutations in SOD1 likely cause FALS through a gain of toxic mechanism induced by a misfolded conformation of the protein (2). Importantly, aberrant post-translational modifications cause WT SOD1 to adapt a similar misfolded conformation (3–11). These observations support an emerging, albeit controversial, hypothesis that WT SOD1 plays a pathogenic role in a subset of SALS, analogous to the role of mutant SOD1 in FALS (2). Over the past several years, conformation specific antibodies have been generated that are selective for misfolded SOD1 variants over the native, WT SOD1 protein (12–17), suggesting that the epitopes for these antibodies represent pathogenic motifs within misfolded SOD1. C4F6 is one such conformation specific monoclonal antibody and is reactive for several ALS-linked SOD1 variants (17–19) including an oxidized form of WT SOD1 (SOD1ox) that serves as a model protein for SALS (4). Importantly, C4F6 detected misfolded SOD1 species within human postmortem FALS and SALS spinal cord tissues (4, 18) and C4F6 reactivity correlated with disease progression in the spinal cords of SOD1 G93A transgenic mice (18). That C4F6 blocked the inhibitory effect of misfolded SOD1 on fast axonal transport in squid axoplasm supports the notion that the C4F6 epitope with SOD1 confers toxicity (4). Collectively, these observations indicate that the C4F6 antibody is a reliable reporter of pathogenic SOD1 species in ALS.
Despite the evidence that C4F6 is selective for pathogenic SOD1 species, very little is known about the amino acids and structural elements that comprise this epitope. Therefore, we developed a chemical cross-linking, site-directed mutagenesis and mass spectrometry approach to define the potentially toxic C4F6 epitope within misfolded SOD1 proteins. Our analyses reveal that the zinc binding (loop IV) and electrostatic (loop VII) loops within SOD1 mask the C4F6 epitope and support a model where ALS-linked mutations destabilize loop IV and VII (20, 21), thereby exposing the C4F6 epitope. In support of this model, WT SOD1 lacking loops IV and VII (SOD1ΔIV/ΔVII) exhibits high reactivity with C4F6 while maintaining a relatively stable tertiary fold (22, 23). Exposure of the C4F6 epitope within SOD1ΔIV/ΔVII directly correlates with SOD1-mediated microglia activation, indicative of enhanced SOD1 toxicity (7, 24). These findings put forth loops IV and VII, as well as the C4F6 epitope itself as therapeutic targets for SOD1-mediated ALS.
EXPERIMENTAL PROCEDURES
SOD1 Protein Expression and Purification
The pET3d vectors containing human SOD1 and SOD1ΔIV/ΔVII (amino acid substitutions: C6A, C111S, and C146A) were generous gifts from Dr. Jill Zitzewitz (University of Massachusetts Medical School). This vector was used to construct WT SOD1ΔIV/ΔVII used in this study (amino acid substitutions A6C, S111C, and C146A). The SOD1ΔIV construct was generated by Genscript and inserted into the pET3d vector using standard cloning techniques. All mutations in this study were generated using standard site-directed mutagenesis methods. pET3d-SOD1 was expressed in BL21(DE3) PlysS cells, and protein expression was induced with 1 mm isopropyl β-d-thiogalactopyranoside (Sigma) in the presence of 200 μm copper (II) chloride (Sigma) and 200 μm zinc chloride (Sigma) when the culture reached an optical density (A600) between 0.6 and 0.8. The bacterial culture was induced for 3 h at 37 °C (wild-type SOD1) or 30 °C (FALS-linked SOD1 and SOD1ΔIV/ΔVII variants). Cells were harvested, and SOD1 proteins were fully purified as previously described (25). Apo SOD1 and SOD1ox were generated from purified wild-type SOD1 as previously described (4, 26). Protein concentrations for all subsequent analyses were quantified with the Pierce BCA Protein Assay kit (Thermo) and/or A280 using the appropriate extinction coefficients for monomeric SOD1.
For the mutagenesis screen in Fig. 7, SOD1ΔIV/ΔVII proteins were “partially purified” by ammonium sulfate precipitation of the postsonication supernatant (25). Ammonium sulfate precipitation was accomplished by adding 0.16 g of ammonium sulfate per 500 μl of bacterial lysate, agitating the mixture on a rotating platform for 15 min at 4 °C and then pelleting the precipitate by centrifugation at 13,000 rpm for 15 min. The supernatant was collected and used as the source of SOD1 protein. The samples were subjected to both native and denaturing Western analyses (see “Immunoblots” below).
FIGURE 7.

Amino acids Asp92 and Asp96 in SOD1 are required for C4F6 binding. Mutagenesis pinpoints key amino acids in misfolded SOD1 responsible for C4F6 reactivity. A, Western analyses of SOD1ΔIV/ΔVII with the indicated mutations (residue number corresponds to full-length SOD1). Pan-SOD1 (green) and C4F6 (red) antibodies were employed to assess partially purified SOD1 expression and reactivity with C4F6, respectively (control = WT SOD1ΔIV/ΔVII). Native Western analysis (top panels) shows a loss of C4F6 reactivity for the following proteins: D90A/D92A/V94A, D96A/S98A/D101A, V97A/I99A/D101A, and F45A/V47A. Denaturing Western analysis (SDS) of SOD1ΔIV/ΔVII variants reveal a loss of C4F6 reactivity for the triple mutations V97A/I99A/D101A, D90A/D92A/V94A, and D96A/S98A/D101A. B and D, the same analyses described in A demonstrate the D92A and D96A mutations induce a loss of C4F6 reactivity for SOD1ΔIV/ΔVII (B) and full-length ALS-linked SOD1 variants (D) under both native and denaturing conditions. Control indicates WT SOD1ΔIV/ΔVII in B, whereas in D, Control refers to the respective variant without the additional mutation. C, in contrast to D92A and D96A, the V97A mutation significantly perturbs the secondary structure of SOD1ΔIV/ΔVII as indicated by CD spectroscopy. MRE, mean residue ellipticity; Control, WT SOD1ΔIV/ΔVII. E, ALS-linked proteins containing the D96A mutation retain the ability to activate microglia as indicated by TNFα Western analysis (bottom panel, performed as described in Fig. 5). Quantification of TNFα is displayed above. Statistically significant comparisons between ALS-linked SOD1 variants and their D96A counterparts are indicated (*, p < 0.05; n = 2). F, the location of amino acids Asp92 and Asp96 mapped onto SOD1ΔIV/ΔVII (Protein Data Bank code 4BCZ).
Circular Dichroism
SOD1 variants were analyzed at 10 μm for full-length proteins and 20 μm for loop deletion constructs in phosphate-buffered saline (pH 7.4). CD spectra were acquired at 195–260 nm using a 0.1-cm cuvette at 25 °C. CD spectra of SOD1 variants (WT SOD1, SOD1ΔIV, and SOD1ΔIV/ΔVII) were acquired with a Jasco J-810 spectrophotometer (five scans). CD spectra of SOD1 variants (D92A-SOD1ΔIV/ΔVII, D96A-SOD1ΔIV/ΔVII, and V97A-SOD1ΔIV/ΔVII) were acquired with an AVIV model 400 spectrophotometer (10 scans). Mean residue ellipticity was calculated as follows: (θ * 100,000)/(A * [SOD1] * L), where θ represents raw data in millidegrees with buffer blank subtraction, A represents the number of amino acids in the SOD1 variant, and L represents the path length in cm.
Metal Analysis
SOD1 variants were prepared for quantitative metal analysis by dialysis with LC-MS grade water (Pierce) overnight at 4 °C. SOD1 samples at concentrations ranging from 40 to 110 μm were then subjected to an elemental analysis in technical duplicate for copper and zinc using inductively coupled plasma optical emission spectroscopy (Center for Applied Isotope Studies, University of Georgia). Postdialysis LC-MS grade water was analyzed as a buffer control and subtracted from the SOD1 samples prior to analyses.
Incubation of SOD1 Proteins with Excess Copper and Zinc
SOD1 proteins (10.75 μm) were incubated in the presence of 4-fold molar excess copper (II) chloride and zinc sulfate for 24 h at 4 °C prior to native and denaturing Western analyses (see “Immunoblots” below for details).
Immunoblots
For denaturing Western analyses, samples were diluted in 6× sample buffer (Boston Bioproducts; BP-111R). Samples were separated by PAGE with 15% Tris-acrylamide gels (unless otherwise noted) in 1% SDS, 25 mm Tris, 192 mm glycine and transferred to PVDF membrane (Millipore) in 25 mm Tris, 192 mm glycine. Membranes were blocked for 1 h in Odyssey blocking buffer (LI-COR). Primary antibodies were incubated at 4 °C overnight at the following dilutions: (pan-SOD1 (27), 1:500; C4F6, 0.3 μg/ml; anti-TNFα, 1:200 (Santa Cruz; sc-1351); and anti-Fab fragment, 1:1000 (Sigma; B0529)). The blots were incubated for 1 h with fluorophore-conjugated secondary antibodies (LI-COR) prior to visualization by the Odyssey infrared imaging system (LI-COR). The Odyssey infrared imaging software (LI-COR) was used for densitometry calculations. For native Western analysis, samples were diluted 1:1 with native sample buffer (Bio-Rad; 161-0738), electrophoresed with a 7.5% Tris-acrylamide gel, and transferred to PVDF in 25 mm Tris, 192 mm glycine at 4 °C. Blots were processed as described above for denaturing Western analyses.
C4F6 Expression, Purification, and Fab Generation
A hybridoma cell line expressing the C4F6 monoclonal antibody was a generous gift from Dr. Jean-Pierre Julien (Laval University) (17). C4F6 was purified with a protein A-agarose column (Pierce) according to the manufacturer's instructions. Purified C4F6 was buffer exchanged into 0.85% NaCl with PD10 G25 desalting columns (GE Healthcare). Fab fragments of C4F6 were generated from purified C4F6 antibody using immobilized papain (Thermo Scientific) in the presence of cysteine according to the manufacturer's instructions.
Octet Binding Studies
Using the Octet QK automated system (ForteBio) in a 96-well format at ambient temperature, C4F6 was immobilized onto anti-mouse IgG Fc Capture Biosensor tips (ForteBio). The extent to which SOD1 bound to C4F6 was determined using curves generated by Bio-layer Interferometry, which is based on light interference on the biosensor tip in the presence of SOD1 compared with an internal reference. To generate binding curves, the C4F6-coated tips were immersed into wells containing 12, 15, or 25 μm of the respective SOD1 variant in PBS (pH 7.4) until the binding reached equilibrium (∼120 s) followed by immersion into PBS-containing wells for dissociation as described (28). Dissociation constants (Kd) were determined by fitting the generated curves using the ForteBio software with a 1:1 binding model. At least two independent experiments were averaged for each SOD1 variant.
Sequencing the C4F6 Antibody by Mass Spectrometry and RNA/DNA Analysis
For mass spectrometry sequencing of the C4F6 protein, tryptic fragments of C4F6 were prepared as previously described (29) and subjected to MALDI-TOF and LC-MS/MS analyses. For MALDI-TOF/TOF, the digested peptide samples prepared above were further purified using C18 Zip Tips (Millipore) according to the manufacturer's instructions. Samples were coapplied to the MALDI sample target with α-cyano-4-hydroxycinnamic acid matrix (5 mg/ml). Samples were analyzed with an Axima-TOF2 MALDI mass spectrometer (Shimadzu Scientific Instruments) following external calibration. The data were acquired in the positive ion reflectron mode using both collision-induced dissociation and postsource decay fragmentation modes. All spectra were processed and subjected to a database search using Mascot (Matrix Science). For MS searches, the Peptide Mass Fingerprint program was used with a peptide mass tolerance of 100 ppm. For collision-induced dissociation and postsource decay spectra, the MS/MS Ion Search program was used with a Precursor tolerance of 100 ppm and a fragment tolerance of 1.5 Da.
For LC-MS/MS analysis, tryptic peptides were reconstituted in 25 μl of 0.1% (v/v) TFA, and a 3 μl aliquot was directly loaded onto a 100-μm inner diameter custom packed trap column packed with 2 cm of 200 Å, 5μ C18AQ particles (Michrom Bioresources). Peptides were then separated on a 75 μm inner diameter analytical column packed with 25 cm of 100 Å, 5μ C18AQ (Michrom) to a gravity-pulled tip. Peptides were eluted using a Proxeon Easy nanoLC (Thermo Scientific) with a linear gradient from 100% solvent A (0.1% (v/v) formic acid in 5% (v/v) acetonitrile) to 35% solvent B (0.1% formic acid in acetonitrile) over 35 min at a flow rate of 300 nl/min. The data were acquired in positive ion electrospray mode using a Thermo Scientific LTQ Orbitrap Velos Pro mass spectrometer operating in the data-dependent mode over the range m/z 350–2000. MS scans were acquired in the Orbitrap at a resolving power of 60,000 followed by 10 MS/MS spectra acquired in the LTQ ion trap. Raw data files were processed with Extract_MSN (Thermo Scientific) and were searched against the mammalian index of the NCBInr database using Mascot (ver. 2.3). Parent mass tolerances were set to 10 ppm, and fragment mass tolerances were set to 0.5 Da, and variable modifications of acetyl (protein N-terminal), pyroglutamic for N-terminal glutamine, carbamidomethylation of cysteine, and oxidation of methionine were considered.
Further sequencing of C4F6 was accomplished by extracting total RNA from C4F6 hybridoma cells using RNeasy kit (Qiagen) and PCR amplifying C4F6 Fab RNA for both heavy and light chains. PCR primers were designed by comparing the partial C4F6 Fab sequence obtained through mass spectrometry analyses (described above) with known antibody sequences for the same isotype, IgG2a, as C4F6 (International Immunogenetics Information System). These primers were used in a OneStep RT-PCR Kit (Qiagen) reaction to generate corresponding cDNA, which was subjected to gel purification followed by DNA sequencing (Genewiz).
Cross-linking of SOD1 Proteins with C4F6 Fab
For the DSP (Thermo Scientific) and d8-DSP (ProteoChem) reaction, the respective SOD1 protein (20 μm) and the Fab fragment of C4F6 (5 μm) were combined in PBS and cross-linked according to the manufacturer's instructions. The samples were then mixed 1:5 with 6× nonreducing SDS loading buffer (Boston Bioproducts), heated at 90 °C for 5 min, and subjected to SDS-PAGE with a 10% Tris-acrylamide gel followed by Coomassie Blue staining (50 μl of reaction/well) or Western analysis (as described above; 15 μl of reaction/well). Tryptic (Promega) in-gel digest was performed essentially as described (29). Extracted peptides were concentrated to 30 μl and combined 1:1 (DSP:d8-DSP) prior to mass spectrometry analysis (as described below). For EDC (Thermo Scientific), SOD1 and the Fab fragment of C4F6 were combined to final concentrations of 100 and 9 μm, respectively. EDC cross-linking was performed according to the manufacturer's instructions. The reaction was quenched by mixing samples 1:5 (v/v) with 6× reducing loading buffer and heating at 90 °C for 5 min. Samples were analyzed as described above for DSP cross-linking.
Identification of DSP and EDC SOD1-C4F6 Fab Cross-linked Peptides by Mass Spectrometry
Tryptic digests were analyzed by LC-MS/MS as described above for sequencing the C4F6 antibody with several noted modifications. During the 120-min gradient elution, data-dependent acquisition acquired full MS scans from m/z 350–2000 in the Orbitrap Velos Pro (resolution 60,000) followed by higher energy collisional dissociation scans on the 10 most intense parent ions acquired in the Orbitrap. Parent ions with charge states of 1+, 2+, or 3+ were excluded from the analysis. For database searching, a concatenated peptide database was generated by xComb (30) containing all possible EDC- or DSP-linked tryptic peptides (allowing up to two missed cleavages) of the intermolecular interactions of SOD1 and C4F6. The raw data were searched in Proteome Discoverer 1.3 against the concatenated database, with parent tolerance of 15 ppm and fragment ion tolerance of 0.05 Da. Additionally, spectra were verified manually with the use of GPMAW 8.20 software (Lighthouse Data, Odense, Denmark). To minimize false positives, only cross-linked peptides with >50% sequence coverage were included in the list of identified cross-links.
Microglial Activation Assays
Primary Microglia were isolated from mouse embryonic cerebral cortices and maintained as previously described (31, 32). Microglia were plated at 100,000 cells/ml in a non-tissue culture-treated 96-well plate (Costar). Microglia were allowed to adhere to the plate for 48 h prior to treatment with SOD1 proteins. Purified recombinant SOD1 variants were added directly to culture medium of microglia at a concentration of 2.5 μm for 48 h prior to immunofluorescence and immunoblotting analyses. Where indicated, the anti-CD14 antibody (Cell Sciences; CPC401A) was added to wells at 1:200 dilution 24 h prior to SOD1 treatment. The animal protocol was approved by the institutional animal care and use committee at the University of Massachusetts Medical Center in compliance with the Animal Welfare Act, U.S. Public Health Service Policy, and the National Institute of Health guidelines. All statistics were determined with an unpaired, two-tailed t test with Welch's correction.
Immunofluorescence
Cells were fixed for 5 min with 4% paraformaldehyde followed by a 30-min block in PBS, 1% BSA, 0.5% Triton (PBSAT). Primary (anti-CD11 b/c, 1:200 (Pierce; PA1–46162); and anti-α-tubulin, 1:100 (Sigma; T9026)) and secondary antibodies (fluorophore-conjugated, 1:2000 (Jackson ImmunoResearch Laboratories)) were added for 1 h at ambient temperature in PBSAT followed by nuclear staining with DAPI (1:300,000; Invitrogen; D1306) and mounting with FluorSave reagent (Fisher; D00060).
RESULTS
C4F6 Is Selective for Misfolded Forms of SOD1 over Native Wild-type SOD1
The C4F6-SOD1 binding interaction was quantified in nondenaturing conditions using the Octet system, which utilizes Bio-Layer Interferometry technology to accurately determine binding affinities (28). No binding interaction was detected between C4F6 and WT SOD1; thus, the apparent dissociation constant (Kd) is below the limit of detection (Table 1). C4F6 exhibited the tightest binding affinity for SOD1 G93A (Kd = 0.40 ± 0.29 μm), the immunogen for this antibody (17). C4F6 also recognized SOD1 A4V and G85R with Kd values of 1.46 ± 0.41 and 3.00 ± 3.83 μm, respectively. Oxidation of native WT SOD1 at Cys 111 (SOD1ox) was also sufficient to produce a tight binding interaction with C4F6 (Kd = 1.13 ± 0.60 μm). These data suggest that C4F6 recognizes an aberrant conformation shared among multiple misfolded SOD1 variants.
TABLE 1.
Apparent dissociation constants (Kd) of the C4F6/SOD1 interaction as determined by the OCTET system
Kd values are shown ± standard deviation. n denotes the number of independent experiments. NBD, no binding detected.
| SOD1 variant | Kd | n |
|---|---|---|
| μm | ||
| WT SOD1 | NBD | 5 |
| WT SOD1ΔIV | 0.72 ± 0.24 | 2 |
| WT SOD1ΔIV/ΔVII | 0.46 ± 0.24 | 2 |
| SOD1ox | 1.13 ± 0.60 | 4 |
| SOD1oxΔIV | 1.29 ± 0.35 | 2 |
| SOD1oxΔIV/ΔVII | 0.32 ± 0.03 | 2 |
| SOD1 G93A | 0.40 ± 0.29 | 5 |
| SOD1 G93AΔIV | 0.28 ± 0.05 | 2 |
| SOD1 G85R | 3.00 ± 3.83 | 5 |
| SOD1 A4V | 1.46 ± 0.41 | 3 |
Hot Spots for C4F6 Cross-linking Are Located within the Electrostatic and Zinc Binding Loops of Misfolded SOD1 Variants
We employed a chemical cross-linking strategy with native SOD1 proteins and C4F6 to identify the C4F6 epitope within misfolded SOD1. Both DSP (cross-links lysines and has a 12 Å spacer) and EDC (cross-links lysines with glutamic or aspartic acid residues and has a 0 Å spacer) were used independently to cross-link SOD1 proteins to the Fab fragment of C4F6. In the presence of DSP (Fig. 1A) and EDC (Fig. 1B), C4F6 cross-linked to all tested ALS-associated SOD1 variants and SOD1ox, but not to WT SOD1. Although the sites of ALS-linked point mutations (e.g. A4V, G85R, and G93A) and oxidation (e.g. C111) are in different regions of the tertiary structure (Fig. 2), C4F6 binding indicates that these SOD1 variants are misfolding in a similar manner.
FIGURE 1.
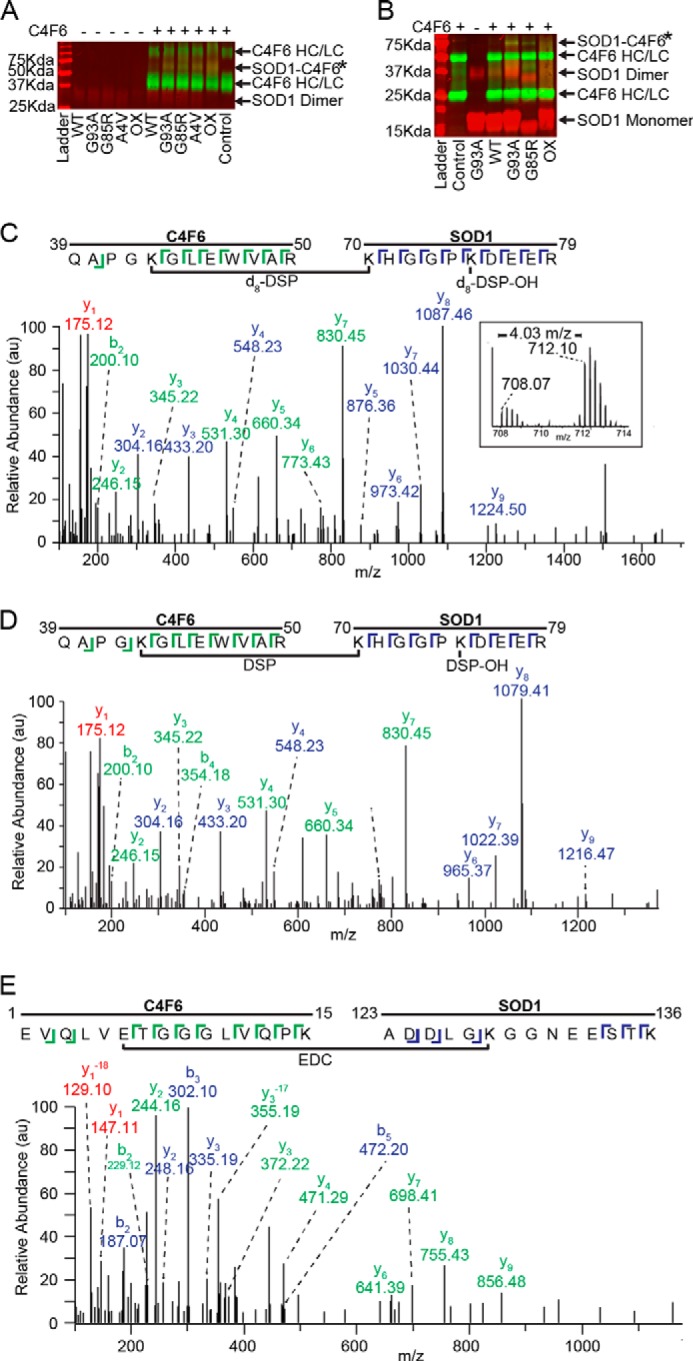
C4F6 specifically cross-links to misfolded SOD1 in the presence of DSP or EDC. A, a nonreducing Western analysis with pan-SOD1 (red) and anti-Fab (green) antibodies demonstrates the specific cross-linking with DSP between misfolded SOD1 variants and C4F6 Fab (SOD1-C4F6; yellow band at 66 kDa denoted by *). HC, heavy chain; LC, light chain. B, a band corresponding to the SOD1-C4F6 complex cross-linked with EDC (denoted by *) was detected at ∼70 kDa for SOD1 (G93A, G85R, and ox), but not WT SOD1. A similar SDS-PAGE and Western analysis was performed as in A, except a 12% gel was employed under reducing conditions. C and D, representative cross-linked peptides from a pooled (d8-DSP + DSP) sample where a 4.03 m/z shift was observed between the monoisotopic peaks (C, inset;16.13 Da mass shift/+4 charge state = 4.03 m/z) 708.07 m/z (DSP) and 712.10 m/z (d8-DSP). Both parent ions, 708.07 m/z (C, d8-DSP) and 712.10 m/z (D, DSP), were subjected to MS/MS analysis and identified as residues 70–79 of SOD1 and residues 39–50 of C4F6. E, exemplar MS/MS data (59% sequence coverage) for an EDC cross-linked peptide comprised of C4F6 (residues 1–15) and SOD1 G93A (residues 123–136). C–E, C4F6 (green) and SOD1 (blue) ions are depicted with the corresponding sequence displayed above. Peaks with identical ions in both C4F6 and SOD1 peptides are shown in red, where −18 corresponds to a loss of a water molecule for y1.
FIGURE 2.

C4F6 cross-links to loops IV and VII within misfolded SOD1. A, the structure of WT SOD1 (Protein Data Bank code 2C9V; copper, orange; zinc, yellow) with the sites of FALS-linked mutations (A4V, G85R, and G93A) and oxidation (Cys111). B, amino acids (red) that cross-link SOD1 G93A to C4F6 with DSP or EDC are highlighted onto the structure of SOD1 G93A (Protein Data Bank code 2WKO). The edge strands of β5 and β6 exposed upon removal of loops IV and VII are shown in purple (right panel). All potential side chains (Lys, Asp, and Glu) capable of cross-linking but that were not cross-linked to C4F6 are shown in gray. C, the amino acid sequences for SOD1 variants from this study are shown with the secondary structural elements illustrated below. Amino acids are highlighted as in B; loops IV (zinc binding) and VII (electrostatic) harbor a majority of cross-linked residues. SOD1ΔIV/ΔVII is denoted by ΔIV/ΔVII (loop IV, blue; loop VII, green).
To identify the amino acids involved in SOD1-C4F6 binding, the band corresponding to the cross-linked species (SOD1-C4F6*) was excised from a Coomassie-stained SDS-PAGE gel and subjected to tryptic digestion followed by mass spectrometry. The amino acid sequence for the C4F6 Fab fragment was determined using both mass spectrometry and RNA sequencing (see “Experimental Procedures”). To aid in DSP cross-link identification, samples were prepared with deuterium-labeled DSP (d8-DSP) in parallel, resulting in cross-linked peptides with an increased mass of 8.06 Da. Pooling both DSP and d8-DSP samples allowed for further validation of cross-linked peptides because of a charge state-dependent shift in the observed m/z of the parent ions. A representative cross-linked peptide from a pooled (d8-DSP + DSP) SOD1 G93A/C4F6 sample is illustrated in Fig. 1C (inset), where a 4.03 m/z shift was observed between the monoisotopic peaks 708.07 m/z (DSP) and 712.10 m/z (d8-DSP). This mass shift corresponds to the theoretical mass shift of a cross-linked peptide containing two cross-linkers, one hydrolyzed dead-end cross-linker and one directly joining the peptides, with DSP compared with d8-DSP (16.13 Da mass shift/+4 charge state = 4.03 m/z). Both parent ions, 712.10 m/z (d8-DSP) and 708.07 m/z (DSP) were subjected to MS/MS analysis and identified as residues 70–79 of SOD1 and residues 39–50 of C4F6 (Fig. 1, C and D). A representative cross-linked SOD1 G93A/C4F6 peptide from an EDC-treated sample is illustrated in Fig. 1E.
In total, 10 amino acids in SOD1 G93A were found to cross-link to C4F6, the majority of which fall in the zinc binding (loop IV, residues 49–81) and electrostatic (loop VII, residues 124–139) loops of SOD1 (Fig. 2 and Table 2). Both DSP and EDC cross-link these loop regions within SOD1 G93A to C4F6 (Fig. 2, B and C, and Table 2), demonstrating that these cross-linkers detect “hot spots” of C4F6 binding to SOD1 G93A. Interestingly, C4F6-binding hot spots within loops IV and VII are also detected for other misfolded SOD1 proteins, including apo-SOD1 and SOD1 (G85R, A4V, ox) variants (Fig. 2C and Table 2).
TABLE 2.
Amino acids that cross-link in the C4F6-SOD1 complex
DSP indicates combined data from deuterated and nondeuterated DSP analyses. HC, heavy chain; LC, light chain.
| SOD1 variant | SOD1 amino acid | C4F6 amino acid | Coverage | Efficacy | Cross-linker |
|---|---|---|---|---|---|
| % | |||||
| SOD1 G93A 94 ± 12%a | Lys70 | Lys43 HC | >59.1 | 3/4 | DSP |
| Lys70 | Lys54 HC | 66.7 | 1/4 | DSP | |
| Lys70 | Asp65 LC | >67.6 | 2/2 | EDC | |
| Lys70 | Lys112 LC | >60.0 | 4/4 | DSP | |
| Lys75 | Lys43 HC | 68.2 | 1/4 | DSP | |
| Lys75 | Lys54 HC | >55.6 | 2/4 | DSP | |
| Lys75 | Asp68 HC | 82.4 | 1/2 | EDC | |
| Asp76/Glu77 | Lys54 HC | 62.5 | 1/2 | EDC | |
| Glu77/78 | Lys54 HC | 61.5 | 1/2 | EDC | |
| Glu121 | Lys54 HC | 58.3 | 1/2 | EDC | |
| Glu121 | Lys67 HC | 50.0 | 1/2 | EDC | |
| Lys122 | Lys54 HC | 60.0 | 1/4 | DSP | |
| Asp124/125 | Lys54 HC | >52.2 | 2/2 | EDC | |
| Lys128 | Glu6 HC | 58.6 | 1/2 | EDC | |
| Glu132 | Lys54 HC | 52 | 1/2 | EDC | |
| Lys136 | Glu6 HC | 63.3 | 1/2 | EDC | |
| Lys136 | Asp65 LC | >53.8 | 2/2 | EDC | |
| Lys136 | Lys215 HC | 70.0 | 1/4 | DSP | |
| SOD1 G85R 97 ± 4%a | Lys70 | Asp64 HC | 60.9 | 1/1 | EDC |
| Lys70 | Asp65 LC | 66.7 | 1/1 | EDC | |
| Lys70 | Lys54 HC | 55.6 | 1/1 | DSP | |
| Asp76 | Lys54 HC | 55.6 | 1/1 | EDC | |
| Asp76/Glu77/Glu78 | Lys54 HC | 53.9 | 1/1 | EDC | |
| Glu121 | Lys54 HC | 54.2 | 1/1 | EDC | |
| Glu132 | Lys54 HC | 60.0 | 1/1 | EDC | |
| Lys136 | Asp65 LC | 50.0 | 1/1 | EDC | |
| SOD1 A4V 94%a | Lys70 | Lys54 HC | 55.6 | 1/2 | DSP |
| Lys75 | Lys54 HC | >51.9 | 2/2 | DSP | |
| Lys122 | Lys54 HC | 53.6 | 1/2 | DSP | |
| Lys136 | Lys43 HC | 66.7 | 1/2 | DSP | |
| Lys136 | Lys54 HC | >50.0 | 2/2 | DSP | |
| SOD1 OX 94%a | Lys70 | Lys43 HC | 63.6 | 1/2 | DSP |
| Lys70 | Lys54 HC | >70.4 | 2/2 | DSP | |
| Lys70 | Lys67 HC | 51.9 | 1/2 | DSP | |
| Lys70 | Lys112 LC | 86.7 | 1/2 | DSP | |
| Lys75 | Lys54 HC | 63.0 | 1/2 | DSP | |
| Lys136 | Lys215 HC | 70.0 | 1/2 | DSP | |
| Apo WT SOD1 90%a | Lys75 | Lys54 HC | >55.6 | 2/2 | DSP |
| Lys70 | Lys43 HC | 59.1 | 1/2 | DSP | |
| SOD1 ΔIV, VII 65%a | Lys122b | Lys54 HC | 65.6 | 1/2 | DSP |
| Lys122b | Lys43 HC | 63.0 | 1/2 | DSP | |
| Lys122b | Lys215 HC | 65.0 | 1/2 | DSP |
a SOD1 sequence coverage of trypsin digestion (± standard deviation, where applicable). Sequence coverage for the light and heavy chain of the C4F6 Fab fragment was found to be 90 ± 11 and 67 ± 7%, respectively. It is noted that the sequence coverage for the heavy chain is low; however, the sequence coverage of the variable region was consistently 100% (amino acids 1–120).
b Number corresponds to amino acid position in full-length SOD1.
Misfolding of Loops IV and VII within ALS-linked SOD1 Variants Exposes the C4F6 Epitope
Because loops IV and VII within SOD1 harbor the majority of amino acids involved in C4F6 binding (Fig. 2), we hypothesized that the C4F6 epitope is located within these loops. It follows that removal of loops IV and VII within misfolded SOD1 should eliminate the C4F6-SOD1 interaction. Loop deletion constructs were engineered to test this hypothesis. SOD1ΔIV represents WT SOD1 with loop IV replaced by a Gly-Ala-Gly linker. The SOD1ΔIV/ΔVII construct lacks loops IV and VII, both of which were replaced by Gly-Ala-Gly linkers (Fig. 2C, ΔIV/ΔVII). SOD1ΔIV and SOD1ΔIV/ΔVII are both apo- (Fig. 3) and monomeric proteins (23). SOD1ΔIV/ΔVII is a well folded protein as determined by x-ray crystallography (Fig. 4A) (22), NMR, protein stability (22, 23), and CD (Fig. 4B) analyses.
FIGURE 3.
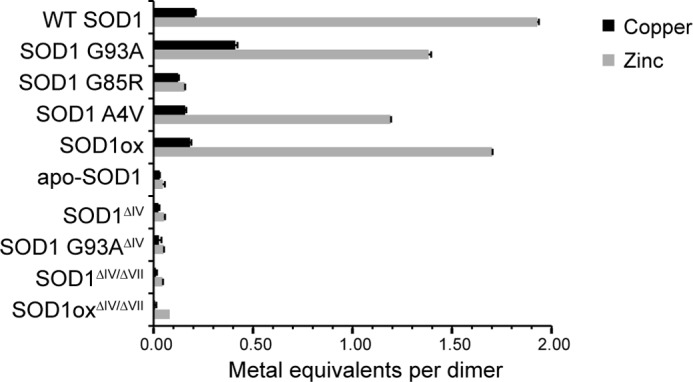
Metal analyses reveal deficient metal coordination in ALS-linked and loop deletion SOD1 variants. The metal content of as-isolated SOD1 variants determined by inductively coupled plasma optical emission spectroscopy. Full-length SOD1 (G93A, G85R, A4V, and ox) contain lower levels of zinc compared with WT SOD1. SOD1 (apo, WT ΔIV, G93A ΔIV, WT ΔIV/ΔVII, and ox ΔIV/ΔVII) are deficient in both copper and zinc. (apo refers to demetallated (see “Experimental Procedures”), ΔIV indicates deletion of loop IV, and ΔIV/ΔVII indicates deletion of loops IV and VII).
FIGURE 4.
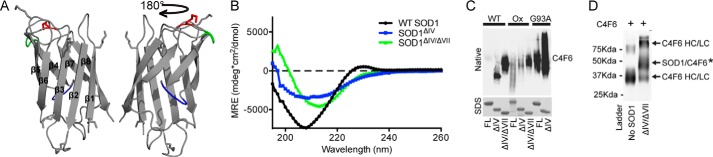
Deletion of loops IV and VII exposes the C4F6 epitope. A, the structure of SOD1ΔIV/ΔVII (Protein Data Bank code 4BCZ; loop IV, blue; loop VII, green; Lys122, red). B, CD spectra of WT SOD1 revealed a minimum ellipticity at 208 nm, indicative of a mixture of both β-sheets and α-helices within the secondary structure as previously reported (26, 53). A similar trend is seen for SOD1ΔIV. Because of the omission of α-helices within loops IV and VII, the minimum ellipticity is shifted toward a longer wavelength for SOD1ΔIV/ΔVII relative to WT SOD1 and SOD1ΔIV. C, the indicated recombinant SOD1 protein (FL, full-length; ΔIV, deletion of loop IV; ΔIV/ΔVII, deletion of loops IV and VII) was subjected to a native PAGE Western analysis with C4F6 (top panel), revealing that C4F6 reactivity is enhanced when the functional loops are removed. A denaturing SDS-PAGE (stained with Coomassie) serves as a loading control. D, WT SOD1ΔIV/ΔVII cross-links to C4F6 Fab through Lys122 in the presence of DSP (as described for Fig. 2).
Surprisingly, removal of loops IV and VII within SOD1 strengthened the C4F6-SOD1 interaction. Octet (Table 1) and native Western (Fig. 4C) analyses demonstrated that removal of these loops promoted a binding interaction between WT SOD1 and C4F6. WT SOD1ΔIV and WT SOD1ΔIV/ΔVII bound C4F6 with Kd values of 0.72 ± 0.24 and 0.46 ± 0.24 μm, respectively. Moreover, the affinity of SOD1ox for C4F6 increased 3-fold, from 1.13 ± 0.60 to 0.32 ± 0.03 μm, upon deletion of loops IV and VII. Therefore, C4F6 affinity for SOD1 proteins lacking loops IV and VII is increased relative to the respective full-length protein (Table 1). This trend is supported by native Western analyses, which show a robust increase in C4F6 reactivity for SOD1 G93A when loop IV is removed (G93A SOD1ΔIV/ΔVII could not be sufficiently expressed) and for both WT SOD1 and SOD1ox upon removal of loops IV and VII (Fig. 4C). WT SOD1ΔIV/ΔVII also cross-linked with C4F6 in the presence of DSP (Fig. 4D). Despite there being seven potential lysines available for cross-linking with DSP (Figs. 2C and 4A), cross-links were only identified at Lys122 (residue number corresponds to full-length SOD1). Lys122 is the only residue that cross-linked C4F6 in the context of full-length SOD1 G93A but was not removed by deletion of loops IV and VII (Figs. 2C and 4A). These results indicate that (i) the remaining six Lys residues in WT SOD1ΔIV/ΔVII are either not proximal to the C4F6 epitope or are not sufficiently positioned for cross-linking by DSP; (ii) the C4F6 epitope is not within, but rather is proximal to, loops IV and VII; (iii) deletion of loops IV and VII within WT SOD1 creates and/or unmasks the C4F6 epitope; and (iv) deletion of loops IV and VII in the context of misfolded ALS-linked variants further exposes the C4F6 epitope.
Exposure of the C4F6 Epitope within SOD1 Correlates with Microglial Activation
If the C4F6 epitope does indeed confer toxicity, then SOD1ΔIV/ΔVII proteins are expected to exhibit heightened toxicity. Chronic activation of microglia is evident in both ALS patients and mouse models (33, 34) and is tightly linked to motor neuron death (24, 35). Recombinant SOD1 G93A has been shown to activate microglia in vitro and, in turn, confer toxicity to cultured motor neurons (24). We utilized this sensitive assay to determine whether exposure of the C4F6 epitope exerts a disease-relevant effect onto microglia. Cultured primary murine microglia (>95% pure, as determined by CD11b/c-positive cells) were exposed to SOD1 for 48 h, and their activation was assessed by both cellular morphology and TNFα secretion. Treatment with all misfolded SOD1 variants employed in this study, but not WT SOD1, resulted in a similar morphological change from ramified/resting microglia to amoeboid, indicative of activation (36) (Fig. 5A). Consistent with these observations, elevated levels of secreted TNFα were detected in the media of microglia exposed to misfolded SOD1 proteins, but not WT SOD1, with the highest levels induced by WT SOD1ΔIV/ΔVII (Fig. 5, B and C). Consistent with previous studies using FALS-linked SOD1 proteins (24), the addition of an anti-CD14 antibody that competes with misfolded SOD1 for the CD14-TLR4 receptor in microglia partially suppressed SOD1-induced microglia activation for all SOD1 variants tested here (Fig. 5). We note that suppression of SOD1 G93A and WT SOD1ΔIV by the CD14 antibody did not achieve statistical significance; however, that an attenuation in TNFα signal was observed for all misfolded SOD1 proteins studied here indicates these proteins activate microglia through a common mechanism. Based on these data, we propose a model whereby the enhanced flexibility of loops IV and VII within ALS-linked variants (20, 21, 37) partially exposes a toxic domain that is recognized by C4F6, and this domain becomes fully exposed when these loops are removed.
FIGURE 5.

Exposure of the C4F6 epitope correlates with elevated SOD1-mediated microglial activation. Primary murine microglia were incubated with the indicated SOD1 protein (2.5 μm) for 48 h prior to analysis. A, immunofluorescence imaging with an anti-α-tubulin antibody reveals the activation status of microglia (active, closed arrows; inactive, open arrows). Preincubation with anti-CD14 (+ anti-CD14) attenuated microglial activation. Scale bar, 50 microns. B, media harvested from cells treated as in A were probed for TNFα by Western analysis (n = 4); the highest signal was observed for WT SOD1ΔIV/VII-treated cells. TNFα secretion was attenuated for the condition with anti-CD14. SOD1 proteins added to the media were assessed by Western analysis with a pan-SOD1 antibody (bottom panel) to ensure that wells were exposed to equal amounts of protein; this antibody was less reactive for the loopless constructs. C, densitometry analysis of B, all SOD1 variants are significantly increased compared with WT SOD1 (p < 0.05) for the condition without anti-CD14; statistically significant comparisons with SOD1ΔIV/VII (*, p < 0.05) for the condition without anti-CD14 are indicated; SOD1 variants that exhibit a significant attenuation in microglia activation in the presence of CD14 compared with the condition without anti-CD14 are indicated (#, p < 0.05).
Enhancing the Metal Occupancy of SOD1 Reduces Exposure of the C4F6 Epitope
To gain further insight into the relationship between the functional loops of SOD1 and the C4F6 epitope, we assessed the effect of SOD1 metal binding on C4F6 reactivity. Inductively coupled plasma optical emission spectroscopy was used to quantify the metallation status of our recombinant SOD1 proteins (Fig. 3). The WT SOD1 dimer contained ∼1.9 equivalents of zinc and only 0.2 equivalents of copper, consistent with previous measurements (25). SOD1 variants exhibited a relatively reduced metallation status with respect to zinc, especially SOD1 G85R (25) (Fig. 3). The dynamics within loops IV and VII are severely perturbed in metal-free SOD1 variants (21, 38), whereas coordination of copper and especially zinc stabilizes the tertiary and quaternary structures of SOD1 (11, 39–41). Because the metallation status of SOD1 correlates with its propensity to misfold, it may also influence C4F6 reactivity.
In the absence of excess copper and zinc, a native Western analysis of as-isolated recombinant SOD1 variants demonstrate C4F6 reactivity in the following rank order: SOD1 G85R> G93A> A4V ∼ SOD1ox (Fig. 6A, left panel). The relatively weak reactivity of C4F6 for SOD1ox and A4V is consistent with the binding affinities measured by the Octet, which showed a relatively tight binding interaction for the C4F6 antigen, SOD1 G93A (Table 1). The strong reactivity for SOD1 G85R may stem from the low metallation status of this protein (Fig. 3), because an apo-form of SOD1 G93A was used to create C4F6 (17). Although the binding of C4F6 for SOD1ox and A4V is weaker than SOD1 G93A and G85R, this antibody can still be used to detect these misfolded species with selectivity over the normally folded WT SOD1 protein (Table 1 and Figs. 1 and 6). In addition, C4F6 may exhibit enhanced reactivity for SOD1 A4V when this protein adopts a self-associated or aggregated form (18, 42).
FIGURE 6.
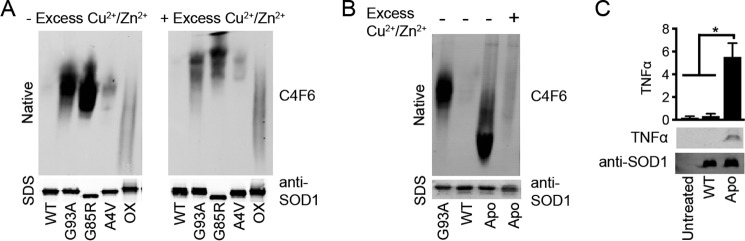
The metallation status of SOD1 correlates with C4F6 reactivity. SOD1 was incubated for 24 h without (−) or with (+) 4-fold molar excess copper and zinc (Cu2+/Zn2+) and subjected to a native PAGE Western analysis with the C4F6 antibody. A denaturing Western blot probed with a pan-SOD1 antibody (bottom panels) serves as a loading control. A, C4F6 reactivity is reduced for SOD1 G93A, G85R, and A4V when these proteins are incubated with excess metals. B, removing metals from WT SOD1, thereby generating apo-SOD1, confers reactivity with C4F6. C4F6 reactivity is attenuated when apo-SOD1 is incubated with excess Cu2+/Zn2+. C, apo-WT SOD1 activates primary microglia as indicated by the TNFα Western analysis (gels, bottom panels; quantification, top panels; n = 2, performed as described in Fig. 5). Statistically significant comparisons are indicated (*, p < 0.05).
Upon incubation with excess copper chloride (II) and zinc sulfate (Cu2+/Zn2+) (43), a change in the migration patterns of SOD1 variants (G93A, G85R, and A4V) was observed, consistent with an increased positive charge and change in conformation for these proteins (44). A robust decrease in C4F6 reactivity was also observed by native Western analyses when misfolded SOD1 species were incubated with metals (Fig. 6A, right panel). The addition of excess metals did not have an effect on SOD1ox, and therefore an alternative strategy is necessary to “refold” this protein.
As expected, C4F6 did not recognize WT SOD1 in the absence or presence of excess Cu2+/Zn2+. However, the C4F6 epitope was exposed in full-length WT SOD1 when this protein was demetallated (Fig. 6B). As observed for the loopless constructs (Fig. 4), exposure of the C4F6 epitope in apo WT SOD1 correlated with microglia activation (Fig. 6C). C4F6 reactivity was attenuated when apo-SOD1 was incubated with excess metals (Fig. 6B); however, this attenuation was not sufficient to abolish SOD1-induced microglia activation (data not shown). Because the microglia assay is sensitive, even residual amounts of misfolded SOD1 that may have escaped remetallation are expected to induce microglia activation. An additional technical caveat of this experiment was that higher molarities of copper and zinc were used to remetallate apo-SOD1 for the microglia assay compared with the native gel analysis, potentially causing toxicity to the cells and/or causing aberrant modifications to the SOD1 protein itself (45–47). Nonetheless, the behavior of apo WT SOD1 in these experiments suggests that both C4F6 reactivity and SOD1-induced microglia activation can be modulated to some extent by the metallation status of SOD1.
Identification of Key SOD1 Residues Required for C4F6 Binding
The results of our cross-linking experiments revealed an important role for loops IV and VII in masking the C4F6 epitope, although the exact amino acids that comprise this epitope remained elusive. Therefore, a comprehensive site-directed mutagenesis strategy was employed to identify key residues within SOD1 that are involved in C4F6 binding. A series of amino acid substitutions were engineered throughout and proximal to the region corresponding to exon 4, which was suggested to contain the C4F6 epitope in a previous study (4). Experiments were initially performed in the SOD1ΔIV/ΔVII background, because this protein exhibits the strongest interaction with C4F6 (Fig. 4C and Table 1). Partially purified recombinant SOD1 mutants were assessed for C4F6 binding by native and denaturing (i.e. SDS) Western analyses with pan-SOD1 and C4F6 antibodies (Fig. 7). Of interest were those constructs that exhibited reactivity with the pan-SOD1 antibody, but little or no reactivity with C4F6. Initially, multiple mutations were engineered to efficiently target the region within SOD1 that mediated C4F6 binding. The triple mutations V97A/I99A/D101A, D90A/D92A/V94A, and D96A/S98A/D101A abolished C4F6 reactivity under both denaturing and native conditions (Fig. 7A). Next, single mutations were engineered into SOD1ΔIV/ΔVII to pinpoint residues that are required for the SOD1-C4F6 interaction. D92A and D96A mutations prevented C4F6 binding under both denaturing and native conditions for SOD1ΔIV/ΔVII, whereas V97A prevented binding only under native conditions (Fig. 7B). However, CD spectroscopy indicated that the V97A mutation severely altered the secondary structure of SOD1ΔIV/ΔVII (Fig. 7C), and thus the loss of C4F6 reactivity likely reflects a significant structural perturbation arising from this mutation. Conversely, the secondary structures as determined by CD were comparable for SOD1ΔIV/ΔVII WT, D92A, and D96A (Fig. 7C).
The D92A and D96A mutations also prevented C4F6 binding under both denaturing and native conditions in the context of full-length ALS-linked SOD1 variants (G93A, G85R, and A4V) (Fig. 7D). That these mutations abolish the interaction between C4F6 and mutant SOD1 under denaturing conditions suggests that residues Asp92 and Asp96 are directly involved in the binding between these proteins. For example, aspartic acid residues are charged and therefore have the potential to form ionic bonds between SOD1 and C4F6, whereas these bonds are eliminated by alanine substitutions. To assess the role of these residues in SOD1-induced microglia activation, the D96A mutation was introduced into ALS-linked SOD1 (G93A, G85R, and A4V) proteins. All of the Asp96-containing variants maintained the ability to activate primary microglia (Fig. 7E), indicating there is still a misfolded conformation within SOD1, despite abolishing the binding interaction between SOD1 and C4F6. In fact, the D96A mutation may worsen the misfolded nature of mutant SOD1 as indicated by the higher levels of TNFα for ALS-linked proteins with this mutation (Fig. 7E). Although native SOD1 must be misfolded for C4F6 to bind, a loss of C4F6 binding does not require SOD1 to be properly folded. These data demonstrate that a single amino acid substitution is not sufficient to restore a wild type-like conformation in an ALS-linked SOD1 protein. Alternatively, strategies aimed at developing chemical chaperones to stabilize misfolded SOD1 and/or mask the C4F6 epitope could be pursued.
DISCUSSION
The structural conformation(s) within misfolded SOD1 that trigger toxic events in ALS have not been defined. Based on the following observations, we posit that the C4F6 epitope represents a misfolded conformation that confers toxicity. First, C4F6 reactivity has been demonstrated in both SALS (4) and FALS human spinal cord sections (18). Second, C4F6 preferentially binds both FALS-linked SOD1 (4, 17–19) and aberrantly modified WT SOD1 (4) proteins (Table 1). Third, C4F6 blocks the toxic effect of SOD1 derived from SALS human spinal cord tissues in an axonal transport assay (4). Fourth, C4F6 reactivity within spinal cord sections of SOD1 G93A transgenic mice directly correlates with disease progression (18).
In the present study, we quantified the disassociation constants between C4F6 and FALS-linked SOD1 mutants and demonstrated binding for all FALS-linked SOD1 variants tested. Although C4F6 exhibited the lowest relative affinity for SOD1 A4V (Table 1 and Fig. 6), this antibody is still able to detect this variant, which is both aggressive and common in North America, with clear selectivity over normally folded WT SOD1. Further, Brotherton et al. (18) detected C4F6-positive inclusions in human FALS spinal cord tissues expressing SOD1 A4V, suggesting that C4F6 reactivity is enhanced for aggregated forms of SOD1 A4V.
Information regarding the residues and structural elements that comprise the C4F6 epitope have been lacking (4). Our chemical cross-linking and mass spectrometry strategy developed here was employed to define the C4F6 epitope, leading us first to loops IV and VII within SOD1 (Fig. 2). To our surprise, SOD1ΔIV/ΔVII and all loopless constructs tested herein exhibited significantly enhanced binding affinity for C4F6 relative to their full-length SOD1 counterparts (Fig. 4C). These results demonstrate that DSP and EDC report on SOD1 residues proximal to, but not directly in, the C4F6 epitope. Nonetheless, using site-directed mutagenesis we were able to pin-point residues Asp92 and Asp96 as being critical for the SOD1-C4F6 binding interaction (Fig. 7). These residues are likely engaged in chemical bonding with C4F6, such that their mutagenesis precludes a C4F6-SOD1 interaction but yet retains the misfolded nature of ALS-linked SOD1. The model that emerges from these studies is one where loops IV and VII modulate the exposure of the C4F6 epitope. FALS-linked mutations, oxidation, and demetallation (Figs. 1, 2, and 6) alter the stability and conformation of these loops (21, 38). These events contribute to the formation of the C4F6 epitope, and removal of these loops allows full exposure to this region (Fig. 4).
Although the toxicity of misfolded SOD1 proteins is well documented in the context of different assays and model systems (2), the current study provides insight into one region within misfolded SOD1 that may confer toxicity. We demonstrate a direct correlation between exposure of the C4F6 epitope and SOD1-induced microglial activation (Fig. 5). Chronic microglia activation is a hallmark of ALS pathogenesis, and SOD1-induced microglia activation leads to motor neuron death (24). We also note that elements of β-strands 5 and 6, including residue Asp96, comprise the C4F6 epitope (Figs. 2B and 7F). Exposure of β-strands 5 and 6 mediate oligomerization of SOD1 and similar β-strands have been shown to mediate aberrant protein-interactions (38, 48, 49). This is primarily because edge strands are poised and readily available to interact with other exposed edge strands (38, 48). Thus, these strands may contribute to the mechanism of toxicity induced by the C4F6 epitope. Other conformation specific antibodies with SOD1 epitopes overlapping with and unique to C4F6 have been described, indicative of additional misfolded regions within ALS-linked SOD1 proteins (12–16, 50, 51). Thus, it is plausible that other misfolded regions within SOD1 confer toxicity as well. It will be important to determine which epitope is most relevant to the toxic ALS-phenotypes induced by misfolded SOD1 in vivo.
Importantly, we show it is possible to convert the properly folded WT SOD1 to a microglia-activating, misfolded conformation recognized by C4F6 by stripping SOD1 of both copper and zinc (Fig. 6). We further show the ability to revert the misfolded conformation of SOD1 to a more “WT-like” structure, as demonstrated by the reduced accessibility of the C4F6 epitope in misfolded SOD1 upon incubation with excess metals (Fig. 6). A similar outcome could potentially be achieved with chemical chaperones, which have been used to mitigate the misfolding and toxicity of other amyloidogenic proteins (52). Either with small molecules or alternative approaches, our results advocate for concealing the C4F6 epitope and/or stabilizing loops IV and VII as rational therapeutic strategies for SOD1-mediated ALS.
Acknowledgments
We are grateful to Dr. J. Zitzewitz (University of Massachusetts Medical School) for the pET3d WT SOD1 and pET3d SOD1ΔIV/ΔII-AS plasmids; Drs. CR Matthews (University of Massachusetts Medical School) and J. Zitzewitz for use of the CD instruments; Dr. T. Broering and N. Boatright (Mass Biologics, University of Massachusetts Medical School) for help with Octet measurements; Dr. K. Kanekura (Washington University) for training in microglia purification; Dr. J. P. Julien (Laval University) for the C4F6 hybridoma cell line; Dr. S. Gulati (University of Massachusetts Medical School) for training in culturing hybridoma cells; Dr. G. Babcock (Mass Biologics, University of Massachusetts Medical School) and Dr. J Leszyk (University of Massachusetts Medical School) for assistance with C4F6 sequencing; Dr. K. Boggio and all members of the Bosco lab for helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant R01NS067206. This work was also supported by funds from the ALS Therapy Alliance/CVS Pharmacy and the ALS Association (to D. A. B.).
- ALS
- amyotrophic lateral sclerosis
- FALS
- familial ALS
- SALS
- sporadic ALS
- SOD1
- Cu,Zn-superoxide dismutase
- DSP
- dithiobis[succinimidyl propionate]
- EDC
- 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride
- SOD1ΔIV/ΔVII
- SOD1 lacking loops IV and VII.
REFERENCES
- 1. Sreedharan J., Brown R. H., Jr. (2013) Amyotrophic lateral sclerosis: problems and prospects. Ann. Neurol. 74, 309–316 [DOI] [PubMed] [Google Scholar]
- 2. Rotunno M. S., Bosco D. A. (2013) An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 7, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Auclair J. R., Johnson J. L., Liu Q., Salisbury J. P., Rotunno M. S., Petsko G. A., Ringe D., Brown R. H., Jr., Bosco D. A., Agar J. N. (2013) Post-translational modification by cysteine protects Cu/Zn-superoxide dismutase from oxidative damage. Biochemistry 52, 6137–6144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J. P., Brady S. T., Brown R. H., Jr. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 6. Chen X., Shang H., Qiu X., Fujiwara N., Cui L., Li X. M., Gao T. M., Kong J. (2012) Oxidative modification of cysteine 111 promotes disulfide bond-independent aggregation of SOD1. Neurochem. Res. 37, 835–845 [DOI] [PubMed] [Google Scholar]
- 7. Ezzi S. A., Urushitani M., Julien J. P. (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 102, 170–178 [DOI] [PubMed] [Google Scholar]
- 8. Banci L., Bertini I., Boca M., Calderone V., Cantini F., Girotto S., Vieru M. (2009) Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc. Natl. Acad. Sci. U.S.A. 106, 6980–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ding F., Dokholyan N. V. (2008) Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc. Natl. Acad. Sci. U.S.A. 105, 19696–19701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Durazo A., Shaw B. F., Chattopadhyay M., Faull K. F., Nersissian A. M., Valentine J. S., Whitelegge J. P. (2009) Metal-free superoxide dismutase-1 and three different ALS variants share a similar partially unfolded β-barrel at physiological temperature. J. Biol. Chem. 284, 34382–34389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arnesano F., Banci L., Bertini I., Martinelli M., Furukawa Y., O'Halloran T. V. (2004) The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J. Biol. Chem. 279, 47998–48003 [DOI] [PubMed] [Google Scholar]
- 12. Forsberg K., Jonsson P. A., Andersen P. M., Bergemalm D., Graffmo K. S., Hultdin M., Jacobsson J., Rosquist R., Marklund S. L., Brännström T. (2010) Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5, e11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gros-Louis F., Soucy G., Larivière R., Julien J. P. (2010) Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J. Neurochem. 113, 1188–1199 [DOI] [PubMed] [Google Scholar]
- 14. Vande Velde C., Miller T. M., Cashman N. R., Cleveland D. W. (2008) Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 4022–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujisawa T., Homma K., Yamaguchi N., Kadowaki H., Tsuburaya N., Naguro I., Matsuzawa A., Takeda K., Takahashi Y., Goto J., Tsuji S., Nishitoh H., Ichijo H. (2012) A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann. Neurol. 72, 739–749 [DOI] [PubMed] [Google Scholar]
- 16. Rakhit R., Robertson J., Vande Velde C., Horne P., Ruth D. M., Griffin J., Cleveland D. W., Cashman N. R., Chakrabartty A. (2007) An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat. Med. 13, 754–759 [DOI] [PubMed] [Google Scholar]
- 17. Urushitani M., Ezzi S. A., Julien J. P. (2007) Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 104, 2495–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brotherton T. E., Li Y., Cooper D., Gearing M., Julien J. P., Rothstein J. D., Boylan K., Glass J. D. (2012) Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl. Acad. Sci. U.S.A. 109, 5505–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prudencio M., Borchelt D. R. (2011) Superoxide dismutase 1 encoding mutations linked to ALS adopts a spectrum of misfolded states. Mol. Neurodegener. 6, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shipp E. L., Cantini F., Bertini I., Valentine J. S., Banci L. (2003) Dynamic properties of the G93A mutant of copper-zinc superoxide dismutase as detected by NMR spectroscopy: implications for the pathology of familial amyotrophic lateral sclerosis. Biochemistry 42, 1890–1899 [DOI] [PubMed] [Google Scholar]
- 21. Molnar K. S., Karabacak N. M., Johnson J. L., Wang Q., Tiwari A., Hayward L. J., Coales S. J., Hamuro Y., Agar J. N. (2009) A common property of amyotrophic lateral sclerosis-associated variants: destabilization of the Cu/Zn superoxide dismutase electrostatic loop. J. Biol. Chem. 284, 30965–30973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Danielsson J., Awad W., Saraboji K., Kurnik M., Lang L., Leinartaite L., Marklund S. L., Logan D. T., Oliveberg M. (2013) Global structural motions from the strain of a single hydrogen bond. Proc. Natl. Acad. Sci. U.S.A. 110, 3829–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Danielsson J., Kurnik M., Lang L., Oliveberg M. (2011) Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric β-barrels. J. Biol. Chem. 286, 33070–33083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao W., Beers D. R., Henkel J. S., Zhang W., Urushitani M., Julien J. P., Appel S. H. (2010) Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 58, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayward L. J., Rodriguez J. A., Kim J. W., Tiwari A., Goto J. J., Cabelli D. E., Valentine J. S., Brown R. H., Jr. (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277, 15923–15931 [DOI] [PubMed] [Google Scholar]
- 26. Svensson A. K., Bilsel O., Kondrashkina E., Zitzewitz J. A., Matthews C. R. (2006) Mapping the folding free energy surface for metal-free human Cu,Zn superoxide dismutase. J. Mol. Biol. 364, 1084–1102 [DOI] [PubMed] [Google Scholar]
- 27. Taylor D. M., Gibbs B. F., Kabashi E., Minotti S., Durham H. D., Agar J. N. (2007) Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 282, 16329–16335 [DOI] [PubMed] [Google Scholar]
- 28. Broering T. J., Wang H., Boatright N. K., Wang Y., Baptista K., Shayan G., Garrity K. A., Kayatekin C., Bosco D. A., Matthews C. R., Ambrosino D. M., Xu Z., Babcock G. J. (2013) Identification of human monoclonal antibodies specific for human SOD1 recognizing distinct epitopes and forms of SOD1. PLoS One 8, e61210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 30. Panchaud A., Singh P., Shaffer S. A., Goodlett D. R. (2010) xComb: a cross-linked peptide database approach to protein-protein interaction analysis. J. Proteome Res. 9, 2508–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suzumura A., Mezitis S. G., Gonatas N. K., Silberberg D. H. (1987) MHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by γ-interferon. J. Neuroimmunol. 15, 263–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sasabe J., Chiba T., Yamada M., Okamoto K., Nishimoto I., Matsuoka M., Aiso S. (2007) d-Serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J. 26, 4149–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turner M. R., Cagnin A., Turkheimer F. E., Miller C. C., Shaw C. E., Brooks D. J., Leigh P. N., Banati R. B. (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 15, 601–609 [DOI] [PubMed] [Google Scholar]
- 34. Liao B., Zhao W., Beers D. R., Henkel J. S., Appel S. H. (2012) Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Block M. L., Zecca L., Hong J. S. (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 [DOI] [PubMed] [Google Scholar]
- 36. Graeber M. B. (2010) Changing face of microglia. Science 330, 783–788 [DOI] [PubMed] [Google Scholar]
- 37. Cao X., Antonyuk S. V., Seetharaman S. V., Whitson L. J., Taylor A. B., Holloway S. P., Strange R. W., Doucette P. A., Valentine J. S., Tiwari A., Hayward L. J., Padua S., Cohlberg J. A., Hasnain S. S., Hart P. J. (2008) Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 16169–16177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elam J. S., Taylor A. B., Strange R., Antonyuk S., Doucette P. A., Rodriguez J. A., Hasnain S. S., Hayward L. J., Valentine J. S., Yeates T. O., Hart P. J. (2003) Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 10, 461–467 [DOI] [PubMed] [Google Scholar]
- 39. Banci L., Bertini I., Cramaro F., Del Conte R., Viezzoli M. S. (2003) Solution structure of Apo Cu,Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry 42, 9543–9553 [DOI] [PubMed] [Google Scholar]
- 40. Stathopulos P. B., Rumfeldt J. A., Scholz G. A., Irani R. A., Frey H. E., Hallewell R. A., Lepock J. R., Meiering E. M. (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 7021–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lynch S. M., Boswell S. A., Colón W. (2004) Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: implications for ALS. Biochemistry 43, 16525–16531 [DOI] [PubMed] [Google Scholar]
- 42. Redler R. L., Fee L., Fay J. M., Caplow M., Dokholyan N. V. (2014) Non-native soluble oligomers of Cu/Zn superoxide dismutase (SOD1) contain a conformational epitope linked to cytotoxicity in amyotrophic lateral sclerosis (ALS). Biochemistry 53, 2423–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Auclair J. R., Boggio K. J., Petsko G. A., Ringe D., Agar J. N. (2010) Strategies for stabilizing superoxide dismutase (SOD1), the protein destabilized in the most common form of familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 107, 21394–21399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tiwari A., Liba A., Sohn S. H., Seetharaman S. V., Bilsel O., Matthews C. R., Hart P. J., Valentine J. S., Hayward L. J. (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 284, 27746–27758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Steinebach O. M., Wolterbeek H. T. (1993) Effects of zinc on rat hepatoma HTC cells and primary cultured rat hepatocytes. Toxicol. Appl. Pharmacol. 118, 245–254 [DOI] [PubMed] [Google Scholar]
- 46. Lemire J., Mailloux R., Appanna V. D. (2008) Zinc toxicity alters mitochondrial metabolism and leads to decreased ATP production in hepatocytes. J. Appl. Toxicol. 28, 175–182 [DOI] [PubMed] [Google Scholar]
- 47. Rakhit R., Cunningham P., Furtos-Matei A., Dahan S., Qi X. F., Crow J. P., Cashman N. R., Kondejewski L. H., Chakrabartty A. (2002) Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 277, 47551–47556 [DOI] [PubMed] [Google Scholar]
- 48. Richardson J. S., Richardson D. C. (2002) Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. U.S.A. 99, 2754–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valentine J. S., Hart P. J. (2003) Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 100, 3617–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pickles S., Destroismaisons L., Peyrard S. L., Cadot S., Rouleau G. A., Brown R. H., Jr., Julien J. P., Arbour N., Vande Velde C. (2013) Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 22, 3947–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pokrishevsky E., Grad L. I., Yousefi M., Wang J., Mackenzie I. R., Cashman N. R. (2012) Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7, e35050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coelho T., Maia L. F., da Silva A. M., Cruz M. W., Planté-Bordeneuve V., Suhr O. B., Conceiçao I., Schmidt H. H., Trigo P., Kelly J. W., Labaudinière R., Chan J., Packman J., Grogan D. R. (2013) Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 260, 2802–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]