

Background: HIV-1 Nef interacts with Src-family kinases to enhance viral replication and immune escape of infected cells.
Results: The crystal structure of HIV-1 Nef bound to the Hck SH3-SH2 region was solved to 1.86 Å resolution.
Conclusion: SH3-SH2 binding stabilizes a compact Nef dimer interface and exposes Nef Asp123 for effector recruitment.
Significance: Src-family kinase binding may stabilize a functional Nef dimer conformation.
Keywords: Human Immunodeficiency Virus (HIV), Src, Src Homology 2 Domain (SH2 Domain), Src Homology 3 Domain (SH3 Domain), X-ray Crystallography, Hck Kinase
Abstract
HIV-1 Nef supports high titer viral replication in vivo and is essential for AIDS progression. Nef function depends on interactions with multiple host cell effectors, including Hck and other Src-family kinases. Here we describe the x-ray crystal structure of Nef in complex with the Hck SH3-SH2 regulatory region to a resolution of 1.86 Å. The complex crystallized as a dimer of complexes, with the conserved Nef PXXPXR motif engaging the Hck SH3 domain. A new intercomplex contact was found between SH3 Glu-93, and Nef Arg-105. Mutagenesis of Hck SH3 Glu-93 interfered with Nef·Hck complex formation and kinase activation in cells. The Hck SH2 domains impinge on the N-terminal region of Nef to stabilize a dimer conformation that exposes Asp-123, a residue critical for Nef function. Our results suggest that in addition to serving as a kinase effector for Nef, Hck binding may reorganize the Nef dimer for functional interaction with other signaling partners.
Introduction
Nef is a lentiviral virulence factor required for the high titer replication of both human and simian immunodeficiency viruses in vivo and is essential for the development of AIDS-like disease in non-human primates (1–4). Viral strains defective for Nef have been found in patients with long term, non-progressive HIV infection, implicating Nef as a critical virulence factor for human AIDS (5, 6). Expression of Nef in CD4+ T-cells and macrophages causes AIDS-like disease in transgenic mice, supporting a key role for Nef in HIV-1 pathogenesis (7, 8).
Nef interacts with multiple host cell proteins, including viral (CD4, CXCR4, CCR5) and immune (MHC-I, T-cell receptor) receptors, trafficking proteins, guanine nucleotide exchange factors, and protein kinases (9). Interactions of Nef with members of the Src kinase family are among the best understood in molecular terms. Nef was first shown to bind to the SH33 domain of the macrophage Src-family member Hck (10, 11), resulting in constitutive kinase activation both in vitro and in cells (12, 13). Activation of Hck, Lyn, and c-Src is a shared property of representative Nef proteins derived from all major and minor subtypes of HIV-1 (14, 15). Each of these Nef subtypes activates endogenous Src-family kinase activity in HIV-infected cells, and inhibition of this pathway blocks Nef-dependent enhancement of HIV replication, infectivity (14, 16), and MHC-I down-regulation (17).
One critical determinant of Nef·SH3 interaction is the PXXPXR motif (18), which is highly conserved among primary HIV isolates (19, 20). Mutagenesis of this Nef sequence prevents high titer HIV replication in primary cells (11) and completely reverts the AIDS-like phenotype in the Nef-transgenic mouse model (22). The PXXPXR motif is also required for activation of Hck and other Src-family kinases (13, 15, 23) as well as down-regulation of MHC-I (24, 25) and CCR5/CXCR4 (26, 27). Nef-induced Src-family kinase activation is an essential early step in the MHC-I down-regulation pathway, which contributes to immune escape of HIV-infected cells (17, 28).
Structural studies of HIV-1 Nef have provided important insight regarding the mechanism of Src-family kinase activation as well as viral and immune receptor down-regulation (18, 30–33). Early NMR and crystal structures of Nef revealed the mechanism of Src-family kinase SH3 domain binding as well as a Nef dimer interface, although structural details of the N-terminal anchor domain and internal flexible loop were absent (18, 30, 31). A more recent structure of full-length Nef fused to an MHC-I peptide in complex with the clathrin adaptor AP-1 μ1 subunit, which models a late step in the MHC-I down-regulation pathway, revealed a larger portion of the Nef N-terminal anchor domain (32). This structure includes the putative N-terminal amphipathic α-helix and the acidic cluster, which are observed to interact with the second helix in the Nef core domain and μ1 subunit, respectively (32). Very recently, a crystal structure of Nef in complex with the AP2 α-σ2 hemicomplex, an interaction critical for CD4 down-regulation, revealed the first biologically relevant conformation of the Nef C-terminal flexible loop (33). These findings underscore the principle that interaction with larger protein ligands provides additional stabilizing contacts for flexible Nef regions.
To better understand the mechanism of Nef-dependent Src-family kinase activation and identify additional regions of contact between Nef and Hck, we determined the x-ray crystal structure of the HIV-1 Nef core domain in complex with the SH3-SH2 tandem regulatory domains of human Hck. Remarkably, the structure of this Nef complex reveals previously unrecognized contacts at the Nef·SH3 interface, contacts between Nef and the SH2 domain, and a novel Nef dimer interface. Cellular studies demonstrate that these interactions are essential for stable association of Nef with full-length Hck in cells and kinase activation. Our findings suggest that Nef interaction with Src-family kinases not only leads to kinase activation but also results in structural remodeling of Nef consistent with recruitment of the AP-1 machinery essential for MHC-I down-regulation.
EXPERIMENTAL PROCEDURES
Bacterial Expression Vector Construction
The coding region for the human Hck SH3-SH2 regulatory region (residues 72–242; numbering was based on the structure of human c-Src (34)) was PCR-amplified and subcloned via the NdeI and XhoI restriction sites of the bacterial expression vector pET-21a(+) (EMD Millipore) to yield a C-terminal His6-tagged Hck32 coding sequence. The coding region of full-length and the core domain of HIV-1 Nef (SF2 allele residues 1–205 and 58–205, respectively; numbering was based on the crystal structure of Nef NL4–3 (18)) was PCR-amplified and subcloned via the NdeI and XhoI restriction sites into pET-21b(+), yielding untagged Nef coding sequences. The Hck SH3-SH2 (E93A) mutant was created via site-directed mutagenesis using the Hck SH3-SH2 bacterial expression vector described above and the QuikChange II XL site-directed mutagenesis kit (Stratagene). Coding sequences for Hck32, Hck32(E93A), and Nef in the final bacterial expression plasmids were confirmed by DNA sequencing.
Expression and Purification of the Recombinant Nef·Hck32 Complex for Crystallography
Escherichia coli strain Rosetta2(DE3) pLysS (EMD Millipore) was transformed with each of the pET-based Hck32 and Nef SF2 core domain expression plasmids, and single colonies were used to inoculate starter cultures of LB medium and grown for 12 h at 37 °C. Starter cultures were diluted 100-fold into fresh LB (1 liter) and grown at 37 °C to an A600 of 0.6. Cultures were then cooled to 25 °C over 30 min followed by the addition of isopropyl 1-thio-β-d-galactopyranoside (IPTG) to a final concentration of 0.4 mm IPTG to induce protein expression for 4 h at 25 °C. After induction, cells were collected by centrifugation, snap-frozen on dry ice, and stored at −80 °C.
To purify the Nef·Hck32 complex, cell pellets from the Nef core domain (4 g) and Hck32 (3 g) were thawed on ice and resuspended in 50 ml of Ni-IMAC (nickel-immobilized metal affinity column) binding buffer (25 mm Tris-HCl, pH 8.3, 0.3 m NaCl, 20 mm imidazole, 10% (v/v) glycerol, 2 mm 2-mercaptoethanol). Cell suspensions were mixed together and combined with a protease inhibitor mixture for histidine-tagged proteins according to the manufacturer's protocol (Sigma) and passed through a microfluidizer (Microfluidics) 10 times at 4 °C. The cell lysate was incubated at 4 °C with gentle rocking for 1 h to promote Nef·Hck32 complex formation followed by centrifugation at 50,000 rpm for 1 h at 4 °C. The clarified lysate was loaded onto a 5-ml HIS-TrapHP column (GE Healthcare) at 2.0 ml/min pre-equilibrated with Ni-IMAC binding buffer. Bound protein was eluted using a 170-ml linear gradient of 20 mm to 500 mm imidazole using Ni-IMAC elution buffer (binding buffer containing 500 mm imidazole). Fractions containing both Nef and Hck32 proteins by SDS-PAGE were pooled and concentrated to a volume of 1 ml using an Amicon 50-ml stirred-cell concentrator with a 10-kDa molecular mass cutoff membrane (Millipore). The concentrated Nef·Hck32 protein complex was buffer-exchanged twice with gel filtration buffer (25 mm Tris-HCl, pH 8.0, 200 mm NaCl, 10% glycerol, 2 mm TCEP) followed by centrifugation at 14,000 rpm for 10 min at 4 °C. The soluble protein complex was loaded onto a Hi-Load 16/60 Superdex 75 gel filtration column (GE Healthcare) equilibrated with gel filtration buffer at a flow rate of 0.5 ml/min. Fractions containing both proteins were pooled, concentrated, and buffer-exchanged with 20 mm Tris-HCl, pH 8.3, containing 150 mm NaCl, 10% glycerol, and 2 mm TCEP. The purified Nef·Hck32 complex was concentrated to 20.5 mg/ml (537 μm) and stored at −80 °C.
Expression and Purification of Individual Hck32 and Nef Proteins
Individual Nef and Hck32 proteins were expressed in the E. coli strain Rosetta2(DE3)pLysS (EMD Millipore) using the same procedure described above for Nef·Hck32 complex.
Bacterial cell pellets of full-length Nef and the Nef core domain were thawed on ice and resuspended in 50 ml of anion-exchange buffer A (25 mm Tris-HCl, pH 8.0, 0.05 m NaCl, 1 mm EDTA, 10% (v/v) glycerol, 2 mm 2-mercaptoethanol). Protease inhibitor mixture (Sigma) was added, and the cell suspensions were passed through a microfluidizer (Microfluidics) 10 times at 4 °C. The cell lysates were clarified by centrifugation at 50,000 rpm for 1 h at 4 °C and loaded onto a 5-ml Hi-TrapQ HP column (GE Healthcare) at 2.0 ml/min pre-equilibrated with anion-exchange buffer A. Bound proteins were eluted with a 170-ml linear gradient of 0.05 m to 0.5 m NaCl using anion exchange buffer B (buffer A containing 1 m NaCl). Fractions containing full-length Nef by SDS-PAGE were pooled and concentrated to 10 ml using an Amicon 50-ml stirred-cell concentrator with a (10-kDa molecular mass cutoff; EMD Millipore). The concentrated protein was diluted to 100 ml in cation-exchange buffer A (25 mm HEPES, pH 7.5, 1 mm EDTA, 10% (v/v) glycerol, 1 mm DTT). The full-length Nef protein was loaded onto a Hi-TrapSP HP column (GE Healthcare) at 2.0 ml/min pre-equilibrated with cation exchange buffer A. Nef was eluted with a 170-ml linear gradient of 0 m to 0.5 m NaCl using cation-exchange buffer B (buffer A containing 1 m NaCl). For both full-length and Nef core domain, fractions containing Nef by SDS-PAGE were pooled and concentrated to a volume of 4 ml using an Amicon 50-ml stirred-cell concentrator with a 10-kDa molecular mass cutoff membrane (Millipore). The concentrated Nef proteins were buffer-exchanged with gel filtration buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10% (v/v) glycerol, 2 mm TCEP) followed by centrifugation at 14,000 rpm for 10 min at 4 °C. The soluble Nef proteins were loaded onto a Hi-Load 16/60 Superdex 75 gel filtration column (GE Healthcare) pre-equilibrated with gel filtration buffer at a flow rate of 0.5 ml/min. Fractions containing Nef proteins were pooled and concentrated. The purified full-length Nef and Nef core domain proteins were concentrated to 12.5 and 10.0 mg/ml, respectively, and stored at −80 °C.
Bacterial cell pellets from the wild-type and E93A mutant Hck32 proteins were thawed on ice and resuspended in 50 ml of Ni-IMAC binding buffer (25 mm Tris-HCl, pH 8.3, 0.5 m NaCl, 20 mm imidazole, 10% (v/v) glycerol, 2 mm 2-mercaptoethanol). Protease inhibitor mixture for histidine-tagged proteins (Sigma) was added, and each cell suspension was passed through a microfluidizer (Microfluidics) 10 times at 4 °C. The cell lysates were clarified by centrifugation at 50,000 rpm for 1 h at 4 °C and loaded onto a 5-ml His-TrapHP column (GE Healthcare) at 4.0 ml/min pre-equilibrated with Ni-IMAC binding buffer. Bound proteins were eluted using a 170-ml linear gradient of 20 mm to 500 mm imidazole using Ni-IMAC elution buffer (binding buffer containing 500 mm imidazole). Fractions containing Hck32 proteins by SDS-PAGE were pooled and concentrated to 10 ml using an Amicon 50-ml stirred-cell concentrator (10-kDa molecular mass cutoff membrane). The concentrated proteins were diluted to 200 ml with anion-exchange buffer A (25 mm Tris-HCl, pH 8.0, 0.05 m NaCl, 1 mm EDTA, 10% (v/v) glycerol, 1 mm TCEP). Hck32 proteins were then loaded onto a 5-ml Hi-TrapQ HP column at 4.0 ml/min pre-equilibrated with anion-exchange buffer A. Bound proteins were eluted with a linear NaCl gradient, and fractions containing the Hck32 proteins were pooled and concentrated to a volume of 3 ml as described above. The concentrated Hck32 proteins were exchanged with gel filtration buffer (25 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10% (v/v) glycerol, 1 mm TCEP) followed by centrifugation at 14,000 rpm for 10 min at 4 °C. Soluble Hck32 proteins were loaded onto a Hi-Load 16/60 Superdex 75 gel filtration column pre-equilibrated with gel filtration buffer at a flow rate of 0.5 ml/min. Fractions containing Hck32 proteins were pooled and concentrated. The purified Hck32 and Hck32(E93A) proteins were concentrated to 25.6 and 14.5 mg/ml, respectively, and stored at −80 °C.
Crystallization
Diffraction quality crystals were grown by sitting-drop vapor diffusion at 4 °C by mixing the Nef·Hck32 complex (10.2 mg/ml final) in a 1:1 ratio with 0.09 m N-(2-acetamido)-iminodiacetic acid (ADA), pH 6.5, 10.8% (v/v) 2-methyl-2,4-pentanediol, and 100 mm NaI. Large single crystals (∼0.20 × 0.1 × 0.01 mm) of the complex grew after 60 days. Before x-ray data collection, crystals were cryoprotected by transfer to mother liquor supplemented with 2-methyl-2,4-pentanediol (20% v/v) and flash-cooled in liquid nitrogen.
X-ray Data Collection and Processing
X-ray diffraction data were collected at SER-CAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. Crystals of the Nef·Hck32 complex diffracted to a resolution of 1.86 Å (Table 1). Data integration and scaling was carried out using HKL2000 (35). Diffraction data are consistent with the triclinic P1 space group. Solvent content analysis suggests 53.90% solvent and a Mathew's coefficient (Vm) of 2.67 Å3/Da, which is consistent with the presence of two complexes in the asymmetric unit.
TABLE 1.
X-Ray data collection and structure refinement statistics for the Nef·Hck32 complex
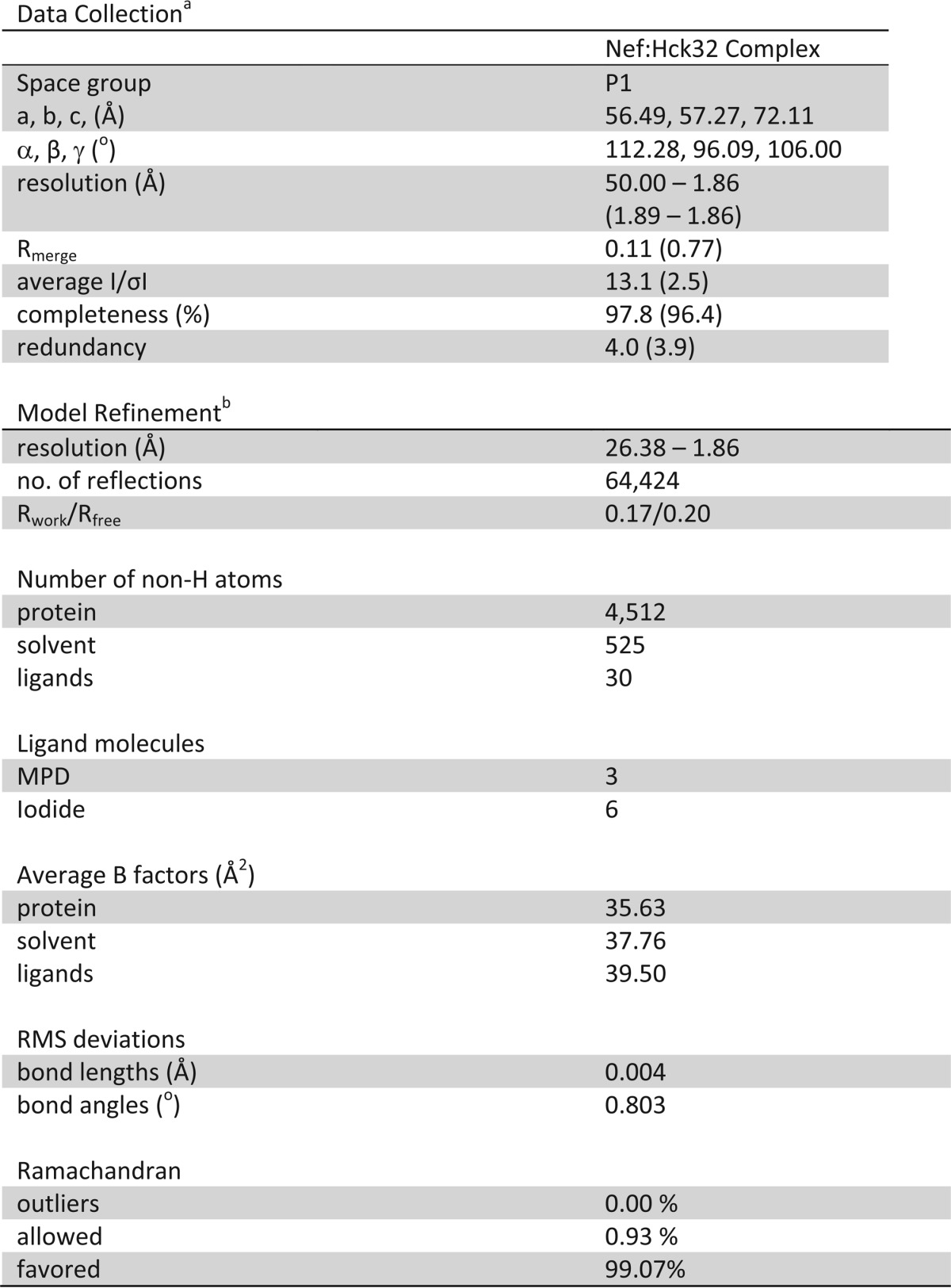
a Values in parenthesis are for the highest resolution shell.
b Data cutoff for refinement was F/σF ≥ 2.
Structure Determination and Refinement
The structure factor data were analyzed using the program PHENIX (36), and no twinning was detected. Phasing and structure solution were completed by molecular replacement with the program PHASER (36) using the structure coordinates of the individual SH3 (residues 83–140) and SH2 (residues 152–246) domains of the Hck SH3-SH2-linker structure (PDB code 3NHN (37)) and residues 72–151/185–208 of the HIV-1 Nef-SF2 core structure (PDB code 3RBB (38)) as independent search models. Iterative molecular replacement was carried out using found solutions as fixed models in combination with search models. Although no information about the SH3·Nef interaction or Nef dimer was included in the molecular replacement procedure, the top molecular replacement solution generated a dimer of Nef·Hck32 complexes in the asymmetric unit with the major dimer contacts occurring via the Nef proteins and putative Nef·SH3 interactions observed. In addition, despite excluding structure coordinates for the Hck connector region that links the SH3 and SH2 domains during the molecular replacement procedure, clear differences in electron densities were observed for the connector region after rigid-body refinement. This top molecular replacement solution generating a dimer of Nef·Hck32 complexes in the asymmetric unit was consistently observed from individual crystals grown in the presence and absence of NaI, for which x-ray diffraction data were collected.
The molecular replacement solution was refined using rigid-body, individual coordinate, individual isotropic b-factor, occupancy, simulated annealing, and TLS refinement using the program phenix.refine (36). After the last cycle of refinement and model building, water molecules were added using phenix.refine. A final round of refinement was conducted using all atoms, and final refinement statistics are summarized in Table 1. Model building was carried using the program Coot (39). The stereochemical quality of the final refined model was evaluated using Procheck (40) and MolProbity (41). Models of x-ray structures and electron density were produced using PyMOL (Schrodinger). The final structure includes Nef-SF2core residues 72–161 and 183–208 for chain A, residues 71–154 and 182–208 for chain C, Hck32 residues 83–246 for chain B, and residues 83–176 and 182–246 for chain D.
Yeast Expression Vectors
The coding sequence for human wild-type Hck (p59 isoform with YEEI tail) and HIV-1 Nef (SF2 allele) was PCR-amplified and subcloned downstream of the Gal1 and Gal10 promoters in the yeast expression vectors pYC2/CT-Ura (Invitrogen) and pESC-Trp (Stratagene), respectively. The Hck SH3 domain mutant E93A was created via site-directed mutagenesis using the QuikChange II XL site-directed mutagenesis kit and the manufacturer's protocol (Stratagene).
Yeast Assay for Nef-mediated Hck Activation
Saccharomyces cerevisiae strain YPH 499 (Stratagene) was transformed via electroporation (Bio-Rad GenePulser II) with pESC-Trp and pYC2/CT-Ura expression plasmids for Hck-YEEI and Nef as described elsewhere (42). Transformed colonies were grown for 3 days at 30 °C on synthetic drop-out agar plates lacking uracil and tryptophan with glucose as the sole carbon source to repress protein expression. Colonies were then cultured in liquid medium with glucose for 18 h at 30 °C. Culture densities were normalized to an A600 of 0.2 and spotted onto synthetic drop-out agar plates lacking uracil and tryptophan with galactose as the sole carbon source to induce protein expression. Plates were incubated for 4 days at 30 °C and imaged on a scanner. Yeast colonies appear as dark spots against the translucent agar background.
Transformed yeast colonies were also cultured in liquid synthetic drop-out medium lacking uracil and tryptophan plus galactose for 18 h at 30 °C to induce protein expression. Culture densities were normalized to an A600 of 0.2, and cells were pelleted and lysed with 0.1 n NaOH for 5 min at room temperature. Lysates were separated via SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed for protein phosphotyrosine content by immunoblotting with the anti-phosphotyrosine antibody, PY99 (Santa Cruz Biotechnology). Protein expression was verified by immunoblotting with antibodies to Hck (N-30, Santa Cruz Biotechnology) and Nef (mouse monoclonal EH1, National Institutes of Health AIDS Research and Reference Reagent Program). Actin levels were probed as a loading control (monoclonal Ab Clone C4, Millipore).
Mammalian Expression Vectors for Bimolecular Fluorescence Complementation (BiFC)
The C-terminal coding sequence of the Venus variant (29) of the YFP protein (residues Ala-154–Lys-238) was PCR-amplified and subcloned into the mammalian expression vector, pcDNA3.1(−) (Invitrogen). The coding regions of wild-type human Hck SH3 domain as well as full-length p59 Hck were also amplified and subcloned upstream and in-frame with this Venus fragment for expression of the SH3-VC and Hck-VC fusion protein, respectively. The SH3 domain mutation (E93A) was then introduced into these vectors via site-directed mutagenesis using the QuikChange II XL site-directed mutagenesis kit (Stratagene). Construction of the complementary BiFC expression vector for HIV-1 Nef (Nef-VN) has been described elsewhere (43).
BiFC Assay
Human 293T cells were transfected with BiFC expression vectors using the calcium phosphate method as described (43). Eighteen hours post-transfection, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and immunostained with primary antibodies against Hck (N-30, rabbit polyclonal, Santa Cruz Biotechnology) and Nef (Hyb 6.1, mouse monoclonal, NIH AIDS Research and Reference Reagent Program). Secondary antibodies used were goat anti-rabbit IgG (H+L)-Texas Red (Southern Biotech) and goat anti-mouse IgG (H+L)-Pacific Blue (Invitrogen). Cells were imaged by confocal microscopy (Fluoview FV1000, Olympus), and images were analyzed by calculating cell-to-cell mean pixel intensities of the BiFC and immunofluorescence signals using ImageJ (imagej.nih.gov) and computing the mean BiFC to immunofluorescence signal ratios.
Multiangle Light Scattering
Size-exclusion chromatography/multiangle light scattering data were obtained at room temperature using an analytical Superdex200 column (10 × 300 mm, GE Healthcare) with in-line multiangle light scattering (HELEOS, Wyatt Technology), variable wavelength UV (Agilent 1100, Agilent Technologies), and refractive index (Optilab rEX, Wyatt Technology) detectors. Purified Hck32 (18.5 mg/ml) and the Nef·Hck32 complex (10.3 mg/ml), each in a volume of 100 μl, were loaded onto the column pre-equilibrated with 20 mm Tris-HCl, pH 8.3, 150 mm NaCl, 0.02% NaN3, and 5% (v/v) glycerol at a flow rate of 0.5 ml/min. For analysis of the purified Nef SF2 core domain, 100 μl of protein at 10.0 mg/ml was injected onto an analytical Superdex75 column (10 × 300 mm, GE Healthcare) with in-line light scattering, UV, and refractive index detectors. The molecular masses of eluted protein species were determined using the ASTRA V5.3.4 software (Wyatt Technologies).
Surface Plasmon Resonance (SPR)
SPR analyses were performed on a BIAcore T100 instrument (GE Healthcare) using a four-channel CM5 biosensor chip. The full-length Nef SF2 protein in HBS-EP buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.05% v/v P20 surfactant) was covalently attached to the CM5 chip via standard amine coupling chemistry (44, 45). Hck32 and Hck32(E93A) proteins in HBS-EPD buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.05% v/v P20 surfactant, 1 mm DTT) were injected over a range of concentrations and flowed past the immobilized Nef protein channel and a reference channel at a flow rate of 10 μl/min for 5 min. The initial binding reaction was followed by a 5-min dissociation period in the HBS-EPD running buffer. Regeneration of the chip surface was carried out with HBS-EPD buffer at a flow rate of 40 μl/min for 8 min. The resulting sensorgrams were corrected for buffer effects and fitted using the BIAcore T100 evaluation software suite (v2.0.4).
RESULTS AND DISCUSSION
Interaction with the Hck32 Region Stabilizes Nef Dimers
We first characterized the recombinant Nef and Hck32 proteins individually and after complex formation using size-exclusion chromatography coupled to multiangle light scattering analysis (Fig. 1). The Hck32 protein eluted as a single peak of 20.7 kDa consistent with a monomer. The Nef protein used in this study is derived from the B-clade allele SF2 and consists of residues 58–205 (numbering based on the crystal structure of Nef NL4–3 (18)). This Nef protein lacks the flexible N-terminal anchor domain but retains the complete structured core used in previous studies. The recombinant Nef protein eluted as two peaks of 38.7 and 17.4 kDa, indicative of monomeric and dimeric forms. In contrast, the complex of Nef with Hck32 (referred to hereafter as the Nef·Hck32 complex) yielded a single peak of 76.3 kDa, which is consistent with a dimer of Nef·Hck32 protein complexes. This analysis shows that interaction with the Hck32 region stabilizes Nef as a dimer in solution. As described in the next section, interaction with Hck32 induces a novel Nef dimer interface as determined by x-ray crystallography.
FIGURE 1.
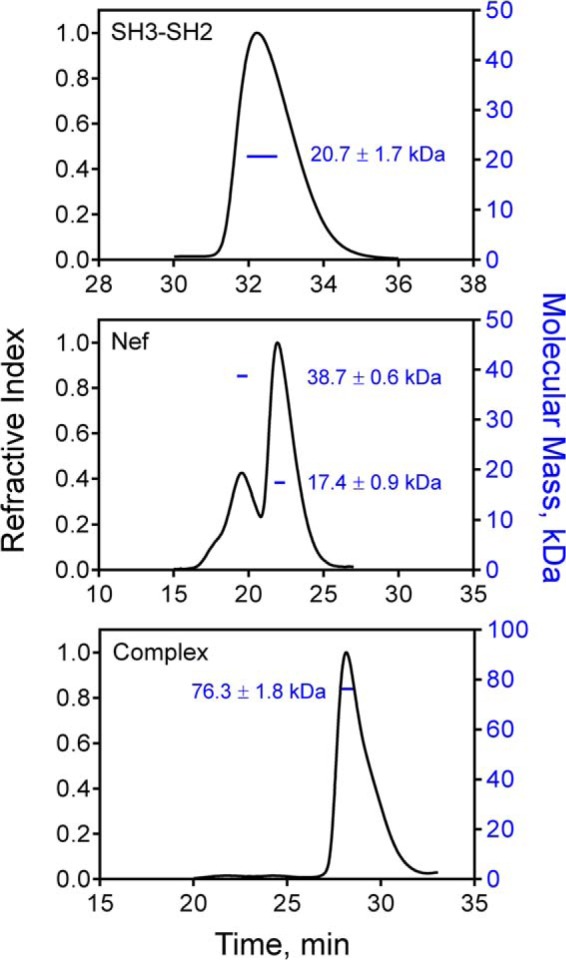
The Nef·Hck32 complex forms a dimer of complexes in solution. Size-exclusion chromatography/multiangle light scattering elution profiles of the purified Hck32 protein (top), the Nef core (middle), and the Nef·Hck32 complex (bottom) are shown, with the refractive index trace in black (left y axes). The resulting weight-averaged molecular masses obtained from the elution peak of each protein or complex are plotted as the blue lines (right y axes).
Overview of the Nef·Hck32 Complex Structure
To better understand the impact of Src-family kinase binding on HIV-1 Nef, we determined the structure of Nef in complex with the Hck SH3-SH2 region to 1.86 Å resolution (Table 1). Two Nef·Hck32 complexes pack together via the Nef proteins to form a larger dimer of complexes with a total buried surface area of 10,520 Å2 (Fig. 2A). The individual Nef, SH3, and SH2 protein structures are nearly identical in each half of the dimer, with root mean square deviations (r.m.s.d.) of 0.41, 0.17, and 0.59 Å after superposition, respectively. In addition, the individual protein structures that form the Nef·Hck32 complex are nearly identical to previous structures of Nef and the Hck SH3 and SH2 domains both alone and in the context of near-full-length Hck (Table 2).
FIGURE 2.

Overview of the Nef·Hck32 structure. A, the Nef·Hck32 complex crystallized as a dimer of complexes. For complex A, Nef is colored light purple, the SH3 domain is red, and the SH2 domain is blue; in complex B, Nef is rendered in green, the SH3 domain is pink, and the SH2 domain is light blue. B, superposition of the Nef structures in the A and B complexes reveals alternative positioning of the SH2 domains. The positions of the SH3-SH2 connector regions are indicated in the superposition on the left (arrow).
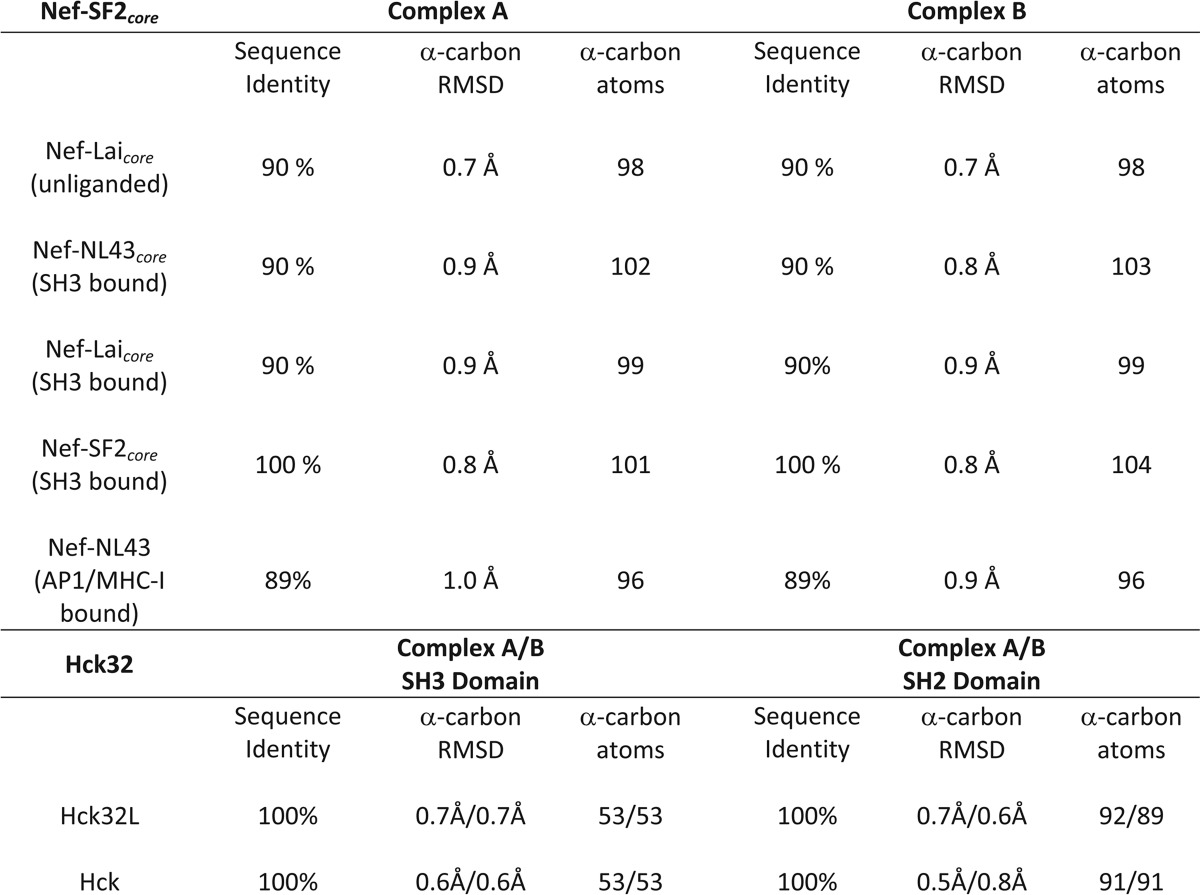
TABLE 2.
Sequence and structural comparison of individual Nef·Hck32 complex components with other Nef and Hck structures
Sequence identity and superposition RMSD values were calculated using the Nef core domains Nef-Lai (PDB ID 1AVV; unliganded), Nef-NL43 (PDB ID 1EFN; SH3 bound), Nef-Lai (PDB ID 1AVZ; SH3 bound), Nef-SF2 (PDB ID 3RBB; SH3 bound), full-length Nef-NL43 (PDB ID 4EN2; AP1/MHC-I peptide bound). The SH2 and SH3 domains from the Nef:Hck32 complex were compared with previous structures of Hck32L (PDB ID 3NHN; SH3-SH2-linker fragment) and down-regulated, near full length Hck (PDB ID 1QCF). Superpositions were calculated using secondary structure matching in Coot and the number of α-carbon atoms used in the alignment are indicated.
Although the structures of the individual proteins making up the dimeric Nef·Hck32 complex are nearly identical, the relative orientation of the SH2 domains in each of the two hemi-complexes are distinct (Fig. 2B). Superposition of the Nef proteins in each Nef·Hck32 complex results in nearly superimposed SH3 domains by virtue of direct SH3 binding to Nef. However, the SH2 domains are oriented 116° away from each other based on the angle of the axes passing through the center of mass of each domain. This difference in orientation is due to structural differences in the SH3-SH2 connector regions (Figs. 2B and 3A). All Src-family kinases possess a connector region of approximately eight residues that joins the SH3 and SH2 domains. Conserved structural elements observed in the SH3-SH2 connectors include an N-terminal β-turn followed by a 310-helix (34, 37, 46–51). In the connector regions of both Hck SH3-SH2 units found in the dimeric Nef·Hck32 structure, the β-turns (Hck residues 139–142) remain intact. However, although a 310-helix (Hck residues 143–146) is present in the connector of Nef·Hck32 complex A, this complex B connector region assumes an extended non-helical conformation (Fig. 3A). In the complex A connector, Hck Leu-143 makes hydrophobic contacts with SH2 domain residues Trp-148, Phe-149, Tyr-184, and Leu-223. In contrast, the 310-helix in the complex B connector lacks main-chain hydrogen bonding, which extends the connector by 2.8 Å and reorients the Leu-143 side chain toward the SH3 domain. As a result, the SH2 domain rotates around the α-carbon atom of connector B residue Ser-142, resulting in the distinct SH2 domain orientations shown in Fig. 2B.
FIGURE 3.
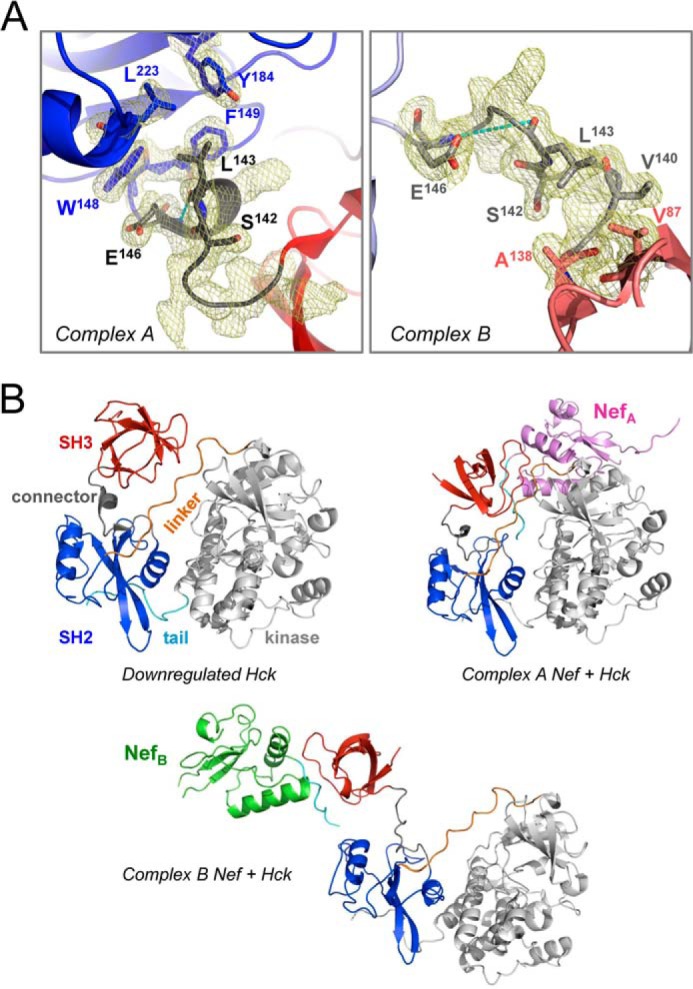
Nef·Hck32 complexes A and B have different SH3-SH2 connectors. A, side-by-side comparison of the SH3-SH2 connectors from Nef·Hck32 complexes A (left) and B (right). The connector regions are rendered in black and gray for complexes A and B, respectively, and have well defined conformations as shown by the 2Fo − Fc electron density maps (mesh; contoured at 1σ). The 310 helix in the complex A connector is stabilized by a main-chain H-bond involving Leu-143 and Glu-146 of 2.9 Å (cyan dashes); this contact is lost in connector B as the interatomic distance is now increased to 5.7 Å (cyan dashes). For clarity, not all connector side chains are shown in the electron density. B, superposition of the Nef·Hck32 complexes onto the SH2 domain of down-regulated near-full-length Hck (PDB code 1QCF (48)) supports SH3 domain displacement from the Hck SH2 kinase linker after Nef binding. The structure of down-regulated near-full-length Hck is shown on the top left and includes the SH3 domain (red), SH3-SH2 connector (gray), SH2 domain (blue), SH2-kinase linker (orange), kinase domain (gray), and C-terminal tail (cyan). The result of SH2-based superposition of Hck with Nef·Hck32 complexes A and B is shown on the top right and below, respectively. The Nef cores are colored as per Fig. 1 with the PXXPXR motif in cyan.
Variability in SH3-SH2 connector secondary structure has been reported in previous structures of the Hck, Lck, and Fyn SH3-SH2 regions. For both the unliganded Hck32 and phosphotyrosyl peptide-bound Lck32 regulatory domain structures, the connector β-turns are completely disrupted, whereas the connector secondary structure in the isolated Fyn32 structure is preserved (37, 46, 47). Interestingly, the relative orientations of the SH3 and SH2 domains in our new structure are unique compared with those observed in these earlier SH3-SH2 structures (37, 46, 47), highlighting the variability in interdomain positioning among the Src-family kinases. In addition, 15N NMR relaxation data show differences in correlation times between the SH3 and SH2 domains of Fyn, Lck, and c-Abl when these domains are joined (46, 52–54). Finally, NMR studies of Nef interaction with a Hck SH3-SH2-linker protein demonstrated significant chemical shift perturbations not only for residues in the Hck SH3 domain but also in the connector and N-terminal region of the SH2 domain upon Nef binding (55).
Modeling of Active Nef·Hck Complexes
To relate our dimeric Nef·Hck32 complex structure to the activation mechanism of full-length Hck by Nef, we constructed a series of models using the structure of down-regulated near-full-length human Hck (PDB code 1QCF) (48). Superposition of the SH2 domains common to both structures resulted in two alternative models based on each of the Nef·Hck32 complexes with different SH3-SH2 connectors (complexes A and B). These models are presented in Fig. 3B and are discussed in more detail below.
In the down-regulated Hck structure (PDB code 1QCF), the SH2 kinase linker adopts a polyproline type II helical conformation and makes internal contacts with the SH3 domain to suppress kinase activity. The SH2 kinase linker is a suboptimal SH3 ligand (37, 56), allowing transactivation of the kinase by cellular SH3 ligands that displace the linker from the SH3 domain. HIV-1 Nef is an excellent example of such a ligand, binding to and activating Hck via this SH3 domain displacement mechanism (12). Superposition of the Nef·Hck32 complexes onto the SH2 domain of down-regulated, near full-length Hck results in significant SH3 domain displacement from the linker, supporting the SH3 domain displacement model (Fig. 3B) (12). In the down-regulated state of Hck, SH2 kinase linker residues Pro-250 and Pro-253 make stabilizing hydrophobic contacts with SH3 domain residues Tyr-90, Trp-118, and Tyr-136 (48, 49) (Fig. 4). Linker Pro-250 interacts with SH3 Tyr-90 and Tyr-136 at distances of 3.7 and 5.0 Å, respectively, whereas Pro-253 is positioned 3.3 Å from Trp-118. These regulatory hydrophobic interactions are clearly disrupted in both SH2 superposition models. In the model based on Nef·Hck32 complex A, Pro-250 is repositioned 9.7 Å and 27.7Å away from Tyr-90 and Tyr-136, respectively, with Pro-253 positioned 15.2 Å away from Trp-118. These distances are even greater in complex B, with Pro-250 now 12.4 Å and 30.9 Å away from Tyr-90 and Tyr-136, respectively, whereas Pro-253 is repositioned nearly 40 Å away from Trp-118 (Fig. 4).
FIGURE 4.
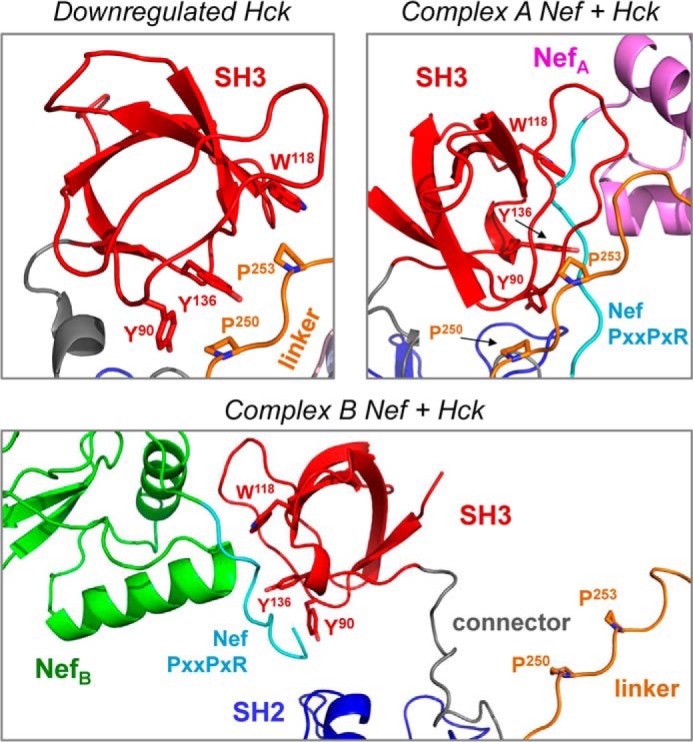
Molecular models of active Nef·Hck complexes based on the Nef·Hck32 structure predict displacement of SH3 from SH2 kinase linker. Upper left, close-up view of the SH3:Linker interface in the down-regulated structure of Hck (PDB code 1QCF). In the absence of Nef, SH3 domain residues Tyr-90, Trp-118, and Tyr-136 make stabilizing hydrophobic contacts with linker residues Pro-250 and Pro-253. Upper right, SH2-based superposition model of Hck with the structure of Nef·Hck32 complex A. In this model the presence of Nef repositions linker Pro-250 9.7 Å and 27.7 Å away from SH3 Tyr-90 and Tyr-136, respectively, whereas linker Pro-253 is repositioned 15.2 Å away from SH3 Trp-118. Lower panel, superposition model of Hck with the structure of Nef·Hck32 complex B. SH3-linker displacement is even more dramatic in this case, with linker Pro-250 now 12.4 Å and 30.9 Å away from SH3 Tyr-90 and Tyr-136, respectively, whereas linker Pro-253 is repositioned nearly 40 Å away from SH3 Trp-118.
The active Nef·Hck complexes modeled in Fig. 3B highlight the importance of the SH3-SH2 connector region in the SH3 domain displacement mechanism for Nef-mediated Hck activation. Functional roles for this connector are also supported by previous biochemical studies and molecular dynamics simulations of near full-length human Hck and c-Src activation. In c-Src, dynamic coupling between the SH3 and SH2 domains in the down-regulated state is dependent on a structured connector region. Simulations show that upon activation, the connector β-turn and 310-helix adopt a more flexible conformation (57). In this same study, replacement of c-Src connector residues with glycines induced kinase activation, supporting a key role for the connector in maintenance of the down-regulated conformation. Molecular dynamics studies also support an important regulatory role for the Hck SH3-SH2 connector showing that connector modification influences the conformation of the kinase domain activation loop (58, 59). Finally, molecular dynamics simulations combined with small angle x-ray scattering have shown that multiple SH3 domain orientations are possible in response to binding of an SH3 domain peptide ligand to near full-length Hck (60). Taken together, these data suggest that the two SH3-SH2 conformations captured in our Nef·Hck32 crystal structure as well as other intermediate states may be present during the activation of Hck by Nef in solution.
Hck SH3-SH2 Binding Stabilizes the Nef Dimer Interface
Previous crystal structures of Nef in complex with SH3 alone revealed a dimer of Nef·SH3 complexes in which the Nef αB-helices form the dimer interface (18, 30). The Nef αB-helices also form the dimer interface in our new Nef·Hck32 structure (Fig. 5A). However, the Nef monomers in the Nef·Hck32 complex are dramatically reoriented relative to the previous Nef·SH3 complex (Fig. 5B). To illustrate this point, Nef monomers from each of the dimer complexes were superimposed as shown in Fig. 5C. The non-superimposed Nef monomer is rotated 82° around the z axis and 87° around the y axis from the center of mass. The resulting structure is much more compact and involves multiple new dimer interactions as described below.
FIGURE 5.

Hck SH3-SH2 binding stabilizes a compact Nef dimer. Comparison of the Nef dimer in the Nef·Hck32 complex (A; Nef monomers are green and purple) with the Nef dimer from a previous Nef·SH3 complex (B; Nef monomers are light and dark orange; PDB code 1EFN (18)). C, superposition of Nef monomers from the structures in A and B. The resulting rotations along the y and z axes for the non-superimposed Nef monomer are shown. D, hydrophobic dimer interface of residue Val-70 with a pocket formed by αB-helix residues Leu-112 and Tyr-115 and C-terminal loop residues Phe-121 and Pro-122. E, hydrophobic dimer interface formed by interweaving of αB-helix residues Ile-109, Leu-112, Trp-113, and His-116. F, Nef residues Ile-109, Leu-112, Tyr-115, His-116, Phe-121, and Pro-122 also participate in the dimer interface of the Nef·SH3 structure. G, Nef dimer interface involving hydrophobic interactions between the side chains of residues Leu-100 and Arg-106 and a network of hydrogen-bonding contacts between the main-chain carbonyl atoms of Gly-95 and Gly-96 and side chains of Ser-103 and Arg-106. H, close up view of the dimer interface from a complex of Nef with an SH3 domain showing the role of Asp-123–Arg-105 interaction in dimerization. I, Nef Asp-123 is repositioned away from the dimer interface to the surface of the structure more than 22 Å away from Nef Arg-105 in the Nef·Hck32 structure. Arg-105 makes a new contact with Glu-93 in the SH3 domain in this reoriented dimer (see Fig. 6).
The unique Nef dimer observed in our Nef·Hck32 complex structure is stabilized by four distinct dimer interfaces. Three of these interfaces are composed of hydrophobic contacts, whereas the fourth involves a mixture of hydrophobic contacts and hydrogen bonds. Two identical small hydrophobic dimer interfaces (each burying 226.6 Å2 of surface area) are formed by the insertion of N-terminal residue Nef Val-70 into a pocket formed by Nef αB-helix residues Leu-112 and Tyr-115 and Nef C-terminal loop residues Phe-121 and Pro-122; these dimer interfaces appear to stabilize the region N-terminal to the PXXPXR motifs, which is absent in earlier structures (Fig. 5D). The third hydrophobic dimer interface is larger (burying 559.1 Å2 of surface area) and is formed by an interweaving of Nef αB-helix residues Ile-109, Leu-112, Trp-113, and His-116 (Fig. 5E). Interestingly, these three hydrophobic dimer interfaces possess “moonlighting” residues: Leu-112 participates in both dimer interfaces, whereas Trp-113 also contributes to the formation of the αA/αB-helix hydrophobic pocket that cradles the Hck SH3 domain RT-loop residue, Ile-95. Although the Nef·Hck32 complex dimer involves three distinct Nef·Nef interfaces, many of the residues participating in these interfaces also contribute to the Nef dimer interface present in previous Nef·SH3 structures (18, 30), with the exceptions of Nef Val-70 and Trp-113. These residues include Ile-109, Leu-112, Tyr-115, His-116, Phe-121, and Pro-122, and their positions in the dimer interface of a previous Nef·SH3 structure are shown in Fig. 5F for comparison.
A fourth Nef dimer interface unique to the Nef·Hck32 complex is located in a region that encompasses the Nef αA-helix C terminus, the αB-helix N terminus, and the αA-αB loop. This region involves hydrophobic interactions between the side chains of Nef residues Leu-100 and Arg-106 and a network of hydrogen-bonding contacts between the main-chain carbonyl atoms of Gly-95 and Gly-96 and the side chains of Ser-103 and Arg-106 (Fig. 5G). This interface buries 462.7 Å2 of surface area and is quite distinct from the reciprocal ionic Nef dimer contacts involving the side chains of Nef Arg-105 and Asp-123 found in the Nef·SH3 structures (18, 30) (Fig. 5H). Interestingly, Nef residues Arg-105 and Asp-123 do not contribute to the Nef dimer interface in the Nef·Hck32 complex. Instead, they are now repositioned 22 Å away from each other (Fig. 5I), with Arg-105 participating in a previously unrecognized intermolecular-intercomplex salt bridge with the SH3 domain RT-loop residue Glu-93 (described below). As a result, Nef Asp-123 is now solvent-exposed and potentially available to participate in interactions with other proteins, such as the AP-1 μ1 clathrin adaptor protein critical to MHC-I down-regulation (32, 61).
Previous structural and biological studies support the biological relevance of these newly described Nef dimer interfaces in the Nef·Hck32 complex and argue that they are not crystallographic anomalies. In NMR solution studies, chemical shift changes and line broadening have been detected for all of the Nef residues participating in these four dimer interfaces in response to Hck SH3-SH2 binding (55), with the exceptions of Nef Tyr-115 and Pro-122. Moreover, cell-based bimolecular fluorescence complementation studies have shown that Nef dimer formation is compromised by mutagenesis of Leu-112, Tyr-115, Phe-121, and Arg-105/Arg-106, providing direct evidence for a contribution of these residues to dimerization in vivo (43). Mutagenesis of Nef Leu-112, Tyr-115, and Arg-105/Arg-106 also inhibits receptor down-regulation and enhancement of HIV replication by Nef, linking dimerization with biological activity (43, 61). A recent report describes a small molecule Nef antagonist that binds to the dimerization interface, identifying this structural feature as a critical point for possible therapeutic intervention (16).
Unique Intermolecular Nef·SH3 and Nef·SH2 Interactions
Previous structural and functional studies have identified two HIV-1 Nef regions that are critical for Src-family kinase SH3 domain engagement and kinase activation (10–13, 18, 30, 62). The first of these involves a highly conserved Nef PXXPXR motif which forms a polyproline type II helix that interacts with the RT- and N-Src-loops of the SH3 domain. In addition, a single RT-loop isoleucine residue unique to the Hck and Lyn SH3 domains engages a hydrophobic pocket formed by the first two α-helices of the Nef core (10). Both of these interactions are present in the structure of the Nef·Hck32 complex (Fig. 6). In addition, the Nef PXXPXR motif is extended to include an additional proline residue (Nef Pro-69) as well as Nef Pro-72, Val-74, Pro-75, and Arg-77. The PXXPXR motif intercalates with Hck SH3 surface residues Tyr-90, Tyr-92, Trp-118, Pro-133, and Tyr-136. This interface involves additional main-chain hydrogen bonds (SH3 residues Tyr-136 and Trp-118 with Nef P72 and Val-74) plus a salt bridge between the side chains of SH3 D99 and the conserved arginine in the Nef PXXPXR motif (Arg-77). SH3 RT-loop residue Ile-95 inserts into a hydrophobic pocket formed by Nef core residues Ile-87, Phe-90, Trp-113, and Tyr-120 from the Nef αA and αB helices.
FIGURE 6.
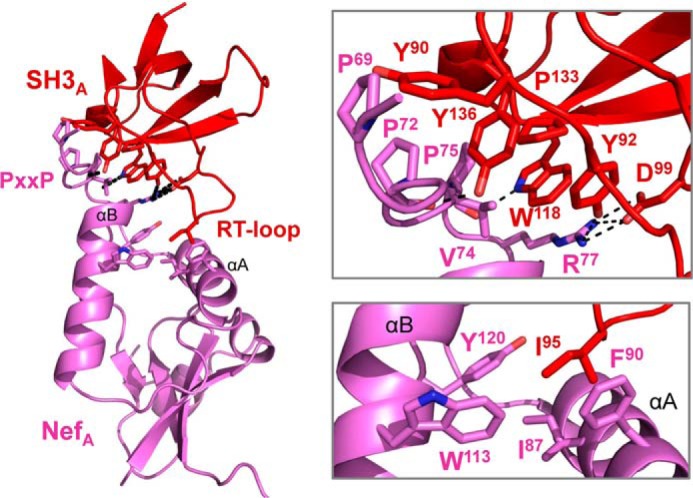
Conserved features of the Nef·SH3 interface in the Nef·Hck32 complex. An overview of the Nef·SH3 structure from complex A is shown on the left, with the interface areas enlarged on the right. The Nef PXXPXR motif interacts with the hydrophobic residues on the SH3 domain RT and N-Src loops shown (top). A hydrophobic pocket formed by Nef residues Ile-87, Phe-90, Trp-113, and Tyr-120 interacts with the SH3 RT loop residue, Ile-95 (bottom).
In addition to these known Nef·SH3 contacts, a unique intermolecular-intercomplex interaction was discovered in our dimeric Nef·Hck32 structure. This interaction involves a salt bridge between Nef Arg-105 from one complex and Glu-93 in the SH3 RT-loop from the opposite complex (Fig. 7). Two reciprocal intercomplex Arg-105–Glu-93 salt bridges were observed within the dimeric Nef·Hck32 complex structure, and these interactions were not influenced by crystal lattice contacts. This salt bridge provides an additional intermolecular contact between Nef and the SH3 domain RT-loop near residue Ile-95, supporting a stabilizing influence in the context of full-length Hck. The residues that form this contact are conserved across diverse Nef alleles and are present in each of the Src family members known to interact directly with Nef (15). As described in more detail below, mutagenesis studies show that this previously unrecognized interaction is critical for stable Nef·Hck complex formation as well as kinase activation.
FIGURE 7.
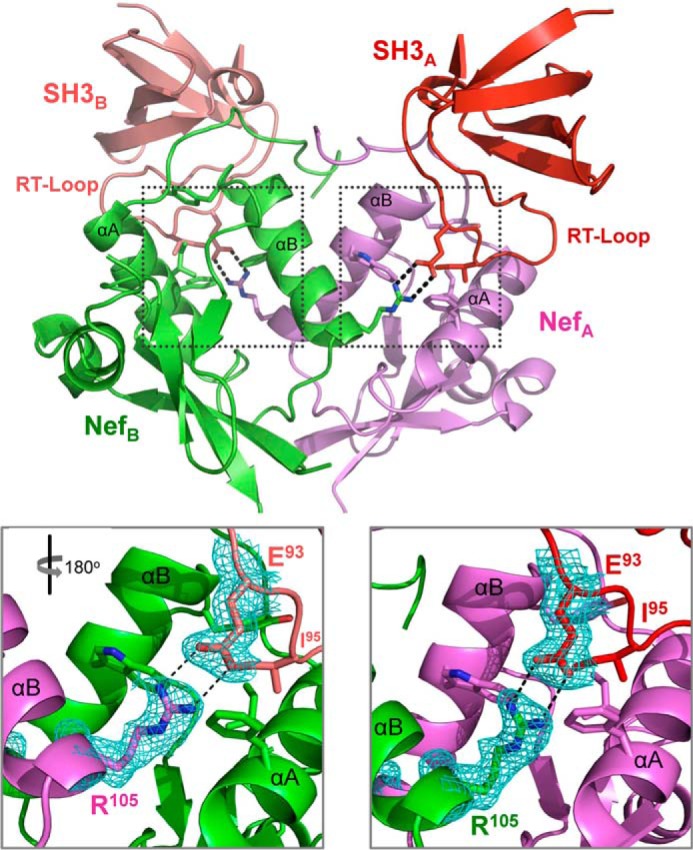
Unique Nef·SH3 interactions in the Nef·Hck32 complex. The dimer of Nef·SH3 complexes A and B is shown at the top, with the unique intercomplex ionic contacts between Nef Arg-105 and SH3 Glu-93 shown as sticks. Close-up view of each ionic interaction is enlarged below and shows well ordered 2Fo − Fc electron density (cyan mesh; contoured at 1σ). The expanded view on the left is rotated 180° with respect to the overall view above to maintain the same orientation as the view on the right.
A precedent for intercomplex Nef·SH3 contacts comes from the work of Horenkamp et al. (38), in which the x-ray crystal structure of the Nef core domain was determined in complex with an engineered high affinity Hck SH3 domain. This structure, referred to as Nef·Hck SH3B6, also crystallized as a dimer of complexes with the Nef C-terminal loop making contacts with a hydrophobic crevice adjacent to the SH3 RT-loop recognition site. Hck SH3 binding affinity for Nef was increased 6-fold by mutation of six RT-loop residues. Two of these residues (Tyr-90 and Pro-92) make intercomplex contacts with the Nef C-terminal loop of the opposing Nef·SH3 complex. Analogous intercomplex interactions were also observed in our Nef·Hck32 complex structure as described above. The SH3 Glu-93 residue in our Nef·Hck32 complex assumes a nearly equivalent position as the RT-loop Tyr-90 in the Horenkamp Nef·Hck SH3B6 structure upon superposition of the Nef proteins in both complex structures (not shown). Together, our Nef·Hck32 and the Nef·Hck SH3B6 dimeric complex structures support the importance of intercomplex interactions between residues in the SH3 RT-loop and Nef for high affinity binding.
Our Nef·Hck32 complex structure is the first to include the SH3-SH2 connector and the SH2 domain. As shown in Fig. 8, each SH2 domain makes an extensive network of Van der Waals contacts with both of the Nef molecules present in the complex. These contacts involve loops connecting the central β-sheets and α-helices of each SH2 domain with Nef residues from the distal end of the N-terminal anchor domain and helix αB. Because the SH2 domains are in different orientations relative to one another in the complex, the SH2 residues contacting Nef are distinct on each side of the Nef dimer. However, there is significant overlap in the Nef regions involved, with Nef residues Phe-68, Pro-69, Leu-76, and Tyr-115 from both Nef monomers making contact with each of the SH2 domains. These Nef·SH2 interactions may help to position the PXXPXR motif for interaction with the SH3 domain and may also stabilize a functionally important Nef dimer conformation as described in more detail below. All of the residues involved in the Nef·SH2 interface are listed in Table 3.
FIGURE 8.
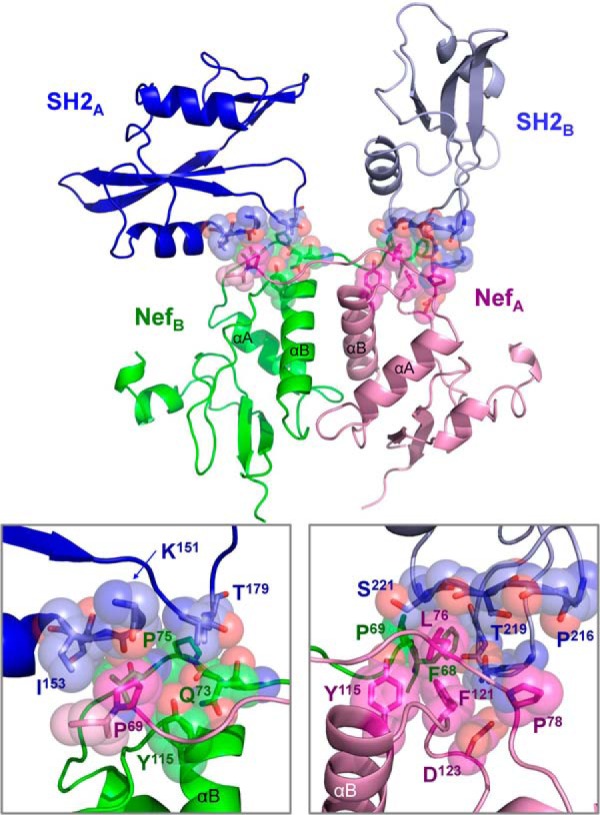
The Hck SH2 domain contacts the Nef core. The overall structure is shown at the top, with the SH3 domains hidden to provide a clearer view of each SH2·Nef interface. The SH2 domains and Nef subunits are colored as per Fig. 1. The interfaces of Nef with SH2 from complex A (SH2-A) and complex B (SH2-B) are enlarged on the lower left and right panels, respectively. Side and main chain atoms making Van der Waals interactions are shown as sticks enveloped by Van der Waals spheres, and selected residues are labeled for orientation. A list of all residues contributing to these interfaces is presented in Table 3.
TABLE 3.
Van Der Waals interactions between SH2 domain and Nef residues in the Nef·Hck32 complex
SH2 domain and Nef residues making Van Der Waals interactions with distances of 3.8–5.5 Å are shown. Hck SH2 domain residues are numbered as per the c-Src crystal structure (PDB code 2SRC). Nef residues are numbered as per the Nef·SH3 crystal structure (PDB code 1EFN). Note that residues in the PDB file from the Nef·Hck32 complex presented in this paper are numbered as per the Nef-SF2 sequence; due to an internal insertion, the Nef-SF2 residues are offset by +4 relative to the Nef-NL43 sequence numbering used in the table.
| Complex A SH2 domain residues | Complex B Nef residues |
|---|---|
| Ser-154 (side/main chain) | Leu-76 (side chain) |
| Glu-178 (main chain) | Gln-73 (side chain) |
| Glu-178 (side chain) | Tyr-115 (side chain) |
| Thr-179 (side chain) | Gln-73 (side/main chain) |
| Thr-179 (side chain) | Val-74 (main chain) |
| Thr-179 (side chain) | Pro-75 (side chain) |
| Thr-179 (side chain) | Leu-76 (main chain) |
| Complex A SH2 domain residues | Complex A Nef residues |
| Lys-151 (main chain) | Pro-69 (side chain) |
| Gly-152 (main chain) | Pro-69 (side chain) |
| Ile-153 (side/main chain) | Pro-69 (side chain) |
| Ile-153 (side chain) | Phe-68 (main chain) |
| Complex B SH2 domain residues | Complex A Nef residues |
| Asn-209 (side/main chain) | Phe-121 (side chain) |
| Asn-209 (side/main chain) | Asp-123 (side chain) |
| Pro-216 (main chain) | Pro-78 (side chain) |
| Arg-217 (main chain) | Pro-78 (side chain) |
| Ser-218 (main chain) | Leu-76 (main chain) |
| Thr-219 (side/main chain) | Leu-76 (side/main chain) |
| Phe-220 (main chain) | Leu-76 (side chain) |
| Ser-221 (side/main chain) | Leu-76 (side chain) |
| Ser-221 (side chain) | Tyr-115 (side chain) |
| Complex B SH2 domain residues | Complex B Nef residues |
| Asp-208 (main chain) | Phe-68 (side chain) |
| Asn-209 (main chain) | Phe-68 (side/main chain) |
| Gly-210 (main chain) | Phe-68 (side chain) |
| Ser-221 (side/main chain) | Pro-69 (side chain) |
SH3 Glu-93 Is Required for High Affinity Binding of Nef to Hck32 in Vitro
The structure of Nef in complex with the Hck SH3-SH2 region revealed a new interaction at the Nef·SH3 interface not present in previous structures of Nef with the SH3 domain alone (Fig. 7). To explore the contribution of this contact to Nef·Hck32 complex assembly, we measured the kinetics and affinity of the interaction by surface plasmon resonance. As shown in Fig. 9, wild-type Hck32 protein (as analyte) bound tightly and reversibly to Nef on the SPR chip surface in a concentration-dependent manner, yielding a KD value of 2.63 μm. This value is comparable with those reported in previous studies of Nef interactions with the isolated SH3 domain by this technique (63). We then repeated the experiment using Hck32 protein with an Glu-93 to alanine (E93A) mutation. This Hck32 single point mutant bound to Nef more slowly, as reflected in the smaller association rate constant, and yielded a KD value of 5.73 μm. This observation demonstrates that the Glu-93-Arg-105 contact contributes to high affinity complex formation between Hck and Nef in vitro.
FIGURE 9.
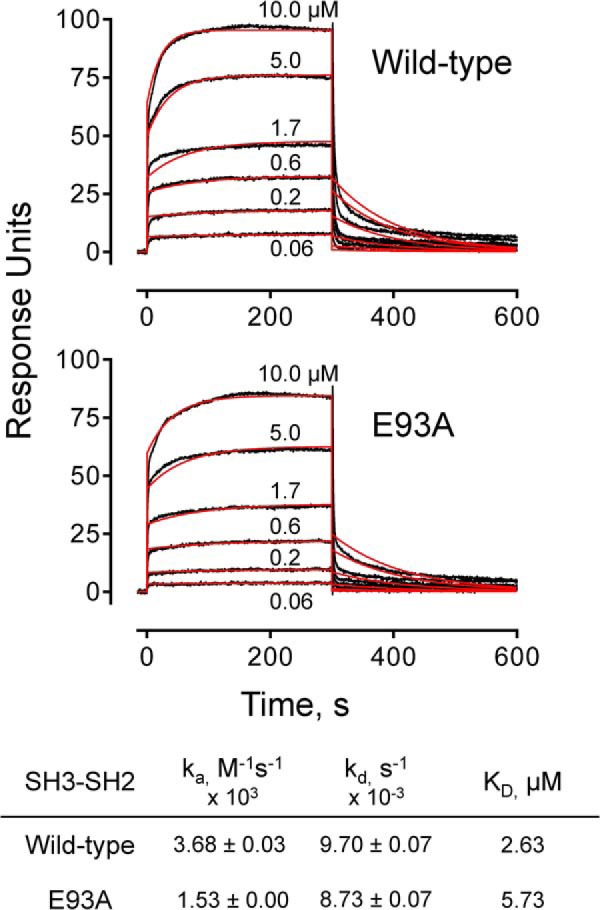
SPR analysis of Nef·Hck32 interaction. SPR analyses were conducted with recombinant purified wild-type Hck32 (top) and the Hck32 SH3 E93A mutant (bottom). Recombinant purified Nef was covalently attached to the SPR chip surface, and Hck32 proteins were injected over the range of concentrations shown. The kinetics and affinities of binding were determined by fitting the sensorgram data (black traces) to a 1:1 binding model (red curves) using the Biacore T100 evaluation (2.0.4) software. The resulting binding constants are summarized below the graphs.
Interaction of Nef Arg-105 with SH3 Glu-93 Is Required for Complex Formation with Full-length Hck in a Cell-based Assay
To evaluate the functional importance of Hck SH3 Glu-93 to complex formation in vivo, we performed cell-based BiFC analysis (43, 64). For these studies, Nef was fused to a non-fluorescent N-terminal fragment of the Venus variant of YFP (Nef-VN) (29), whereas the N-terminal portion of Hck including the SH3 domain as well as full-length Hck were fused to the complementary C-terminal Venus fragment (SH3-VC; Hck-VC). Nef-VN was then co-expressed with either SH3-VC or full-length Hck-VC in 293T cells followed by confocal microscopy. As shown in Fig. 10, a bright fluorescent signal was observed in the BiFC channel indicative of Nef interaction with both the truncated and full-length Hck proteins and subsequent complementation of the Venus fluorophore. This experiment was then repeated using SH3-VC and Hck-VC constructs in which SH3 Glu-93 was substituted with alanine. This substitution did not impact the interaction of Nef with the shorter Hck construct consisting of only the unique and SH3 domains, consistent with previous Nef·SH3 crystal structures in which the SH3 Glu-93·Nef Arg-105 contact is not present (18, 30). However, when Nef-VN was expressed with full-length Hck-VC, the BiFC signal was reduced by more than 90%, indicating that this additional SH3 domain contact is important for stable interaction of full-length Hck with Nef in cells.
FIGURE 10.
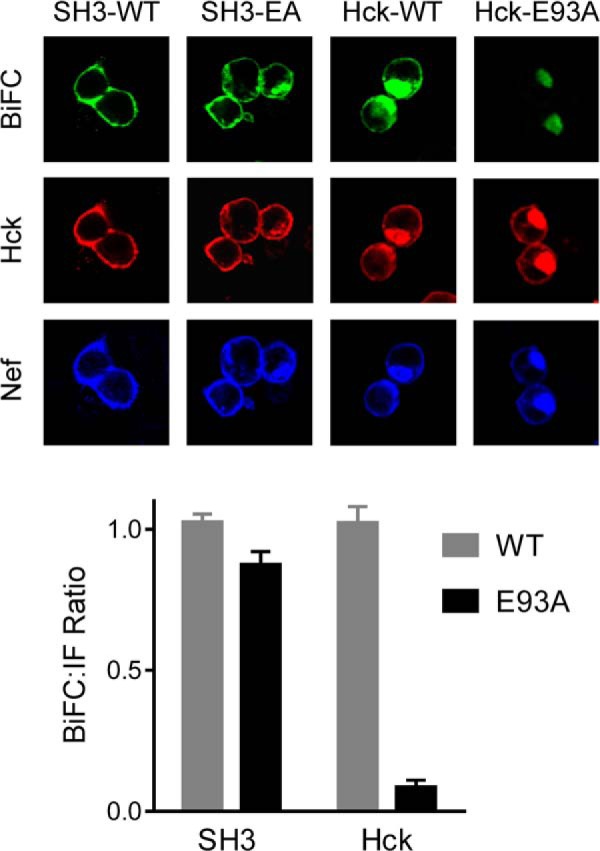
Interaction of Nef Arg-105 with Hck SH3 Glu-93 is required for complex formation in vivo. Interaction of Nef with truncated and full-length Hck proteins was assessed using a cell-based BiFC assay. Human 293T cells were transfected with expression plasmids for fusion proteins of Nef with an N-terminal fragment of the YFP variant, Venus, and either the N-terminal region of Hck including the SH3 domain or full-length Hck fused to the complementary C-terminal Venus fragment. Parallel experiments were conducted with wild-type (WT) and SH3 domain mutants in which Glu-93 was replaced with alanine (E93A). Eighteen hours after transfection cells were fixed and stained with antibodies to the Hck N-terminal region and Nef. Three-color confocal images were recorded for BiFC (green), which is indicative of Nef·Hck interaction as well as Hck and Nef protein expression. Mean pixel intensities of the BiFC and immunofluorescent (IF) signals were determined for at least 50 cells per condition using ImageJ. BiFC:immunofluorescent signal ratios were calculated and are presented in the bar graph as the mean ratio ± S.E.
Hck SH3 Glu-93 Is Required for Nef-mediated Kinase Activation
In a final series of experiments we evaluated the effect of the Hck SH3 domain Glu-93 mutation on Nef-mediated kinase activation. For these studies we turned to a yeast-based co-expression system previously used in our laboratory to study Nef-mediated activation of Src-family kinases (14, 15). This system uses a modified form of Hck in which the C-terminal tail sequence is modified to Tyr(P)-Glu-Glu-Ile (Hck-YEEI, for short). This modification enhances tail interaction with the SH2 domain and controls Hck activity in the absence of Csk, the “tail kinase” and master regulator of Src-family kinases that is absent in yeast. However, expression of Nef with Hck-YEEI causes constitutive activation of Hck via an SH3 domain displacement mechanism, leading to growth arrest in yeast. Note that the structural basis of Hck-YEEI activation by Nef in yeast is indistinguishable from that observed in mammalian cells (42).
To use the yeast system to explore the effect of the SH3 Glu-93 mutant on Nef-induced Hck activation, the wild-type and E93A forms of Hck-YEEI were expressed alone or in the presence of Nef. The cultures were grown to equal densities and then plated on galactose agar over a range of dilutions to induce protein expression. Parallel cultures were grown in liquid medium containing galactose, and cell lysates were immunoblotted for Hck and Nef protein expression as well as protein-tyrosine phosphorylation of yeast cell proteins. As shown in Fig. 11A, co-expression of wild-type Hck-YEEI with Nef caused almost complete growth arrest in yeast. This effect correlated with strong phosphorylation of cellular proteins on tyrosine, consistent with our previous work (Fig. 11B) (14, 15). In contrast, co-expression of Nef with the Hck-YEEI E93A mutant resulted in only a partial induction of growth arrest and a modest increase in phosphorylation of yeast cell proteins. These results support an essential role for Hck SH3 Glu-93 in the formation of the active Nef·Hck complex.
FIGURE 11.

Hck activation by Nef requires SH3 domain residue Glu-93. S. cerevisiae was transformed with galactose-inducible expression plasmids for wild-type Hck (WT), the SH3 domain mutant (E93A), and HIV-1 Nef in the combinations shown. The form of Hck used in these studies has a modified C-terminal tail to enable down-regulation in the absence of Csk (see “Results” for details). A, transformed colonies were cultured in glucose medium to equal densities and spotted onto agar plates containing galactose as the sole carbon source to induce protein expression. Plates were incubated for 4 days at 30 °C and imaged on a scanner. Growing yeast patches appear as dark spots against the translucent agar background. B, transformed yeast colonies were also cultured in liquid medium plus galactose to induce protein expression. Cell lysates were separated via SDS-PAGE followed by immunoblotting for protein phosphotyrosine content (pTyr) as well as Nef and Hck expression along with actin as a loading control. This experiment was repeated in triplicate with comparable results; a representative example is shown.
Conclusions
Here we present the x-ray crystal structure of HIV-1 Nef in complex with the regulatory apparatus (tandem SH3-SH2 unit) of its binding partner and kinase effector, Hck. Although key elements of previous Nef structures with isolated SH3 domains are present, the addition of the SH2 domain and SH3-SH2 connector resulted in several remarkable differences. These include a new intercomplex salt bridge between Nef Arg-105 and Glu-93 in the RT loop of the SH3 domain, shown by mutagenesis studies to be critical to both interaction and function. The Nef dimer interface, although still involving the Nef αB helices, was completely reorganized relative to previous Nef·SH3 structures and stabilized by contacts with the SH2 domain. Models based on the Nef·Hck32 structure provide fresh insight as to possible conformations for the active Nef·Hck complex, which is essential for Nef function and represents a druggable target for HIV-1 (14, 16, 17, 21). Finally, the Nef dimer that results from Hck SH3-SH2 binding allows for surface display of Nef residue Asp-123, which plays a critical role in multiple Nef functions including CD4 and MHC-I down-regulation (32, 43, 61) as well as enhancement of viral replication (43). These results support the idea that interaction with Nef not only promotes kinase activation but also stabilizes a Nef conformation compatible with downstream effector binding and function.
Acknowledgments
We acknowledge the National Institutes of Health AIDS Research and Reference Reagent Program for generously providing antibodies for this project. We also thank Dr. John R. Engen (Northeastern University) and Dr. Gary Thomas (University of Pittsburgh) for critical evaluation of the manuscript and Chang Byeon (Department of Structural Biology, University of Pittsburgh) for help with the SEC-MALS experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants AI057083 and AI102724. Initial phases of this work were supported by a pilot grant from the University of Pittsburgh Center for HIV Protein Interactions (National Institutes of Health P50 GM082251).
- SH3
- Src homology 3
- Ni-IMAC
- nickel-immobilized metal affinity column
- TCEP
- Tris(2-carboxyethyl)phosphine
- BiFC
- bimolecular fluorescence complementation
- SPR
- surface plasmon resonance
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Renkema G. H., Saksela K. (2000) Interactions of HIV-1 NEF with cellular signal transducing proteins. Front. Biosci. 5, D268–D283 [DOI] [PubMed] [Google Scholar]
- 2. Geyer M., Fackler O. T., Peterlin B. M. (2001) Structure-function relationships in HIV-1 Nef. EMBO Rep. 2, 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arold S. T., Baur A. S. (2001) Dynamic Nef and Nef dynamics: how structure could explain the complex activities of this small HIV protein. Trends Biochem. Sci. 26, 356–363 [DOI] [PubMed] [Google Scholar]
- 4. Kestler H. W., 3rd, Ringler D. J., Mori K., Panicali D. L., Sehgal P. K., Daniel M. D., Desrosiers R. C. (1991) Importance of the nef gene for maintenance of high viral loads and for development of AIDS. Cell 65, 651–662 [DOI] [PubMed] [Google Scholar]
- 5. Kirchhoff F., Greenough T. C., Brettler D. B., Sullivan J. L., Desrosiers R. C. (1995) Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332, 228–232 [DOI] [PubMed] [Google Scholar]
- 6. Deacon N. J., Tsykin A., Solomon A., Smith K., Ludford-Menting M., Hooker D. J., McPhee D. A., Greenway A. L., Ellett A., Chatfield C., Lawson V. A., Crowe S., Maerz A., Sonza S., Learmont J., Sullivan J. S., Cunningham A., Dwyer D., Dowton D., Mills J. (1995) Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270, 988–991 [DOI] [PubMed] [Google Scholar]
- 7. Jolicoeur P. (2011) The CD4C/HIV(Nef)transgenic model of AIDS. Curr. HIV. Res. 9, 524–530 [DOI] [PubMed] [Google Scholar]
- 8. Hanna Z., Kay D. G., Rebai N., Guimond A., Jothy S., Jolicoeur P. (1998) Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 95, 163–175 [DOI] [PubMed] [Google Scholar]
- 9. Saksela K. (2011) Interactions of the HIV/SIV pathogenicity factor Nef with SH3 domain-containing host cell proteins. Curr. HIV. Res. 9, 531–542 [DOI] [PubMed] [Google Scholar]
- 10. Lee C. H., Leung B., Lemmon M. A., Zheng J., Cowburn D., Kuriyan J., Saksela K. (1995) A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 14, 5006–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saksela K., Cheng G., Baltimore D. (1995) Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 14, 484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moarefi I., LaFevre-Bernt M., Sicheri F., Huse M., Lee C. H., Kuriyan J., Miller W. T. (1997) Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650–653 [DOI] [PubMed] [Google Scholar]
- 13. Briggs S. D., Sharkey M., Stevenson M., Smithgall T. E. (1997) SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 272, 17899–17902 [DOI] [PubMed] [Google Scholar]
- 14. Narute P. S., Smithgall T. E. (2012) Nef alleles from all major HIV-1 clades activate Src-family kinases and enhance HIV-1 replication in an inhibitor-sensitive manner. PLoS ONE 7, e32561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trible R. P., Emert-Sedlak L., Smithgall T. E. (2006) HIV-1 Nef selectively activates SRC family kinases HCK, LYN, and c-SRC through direct SH3 domain interaction. J. Biol. Chem. 281, 27029–27038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Emert-Sedlak L. A., Narute P., Shu S. T., Poe J. A., Shi H., Yanamala N., Alvarado J. J., Lazo J. S., Yeh J. I., Johnston P. A., Smithgall T. E. (2013) Effector kinase coupling enables high-throughput screens for direct HIV-1 Nef antagonists with antiretroviral activity. Chem. Biol. 20, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dikeakos J. D., Atkins K. M., Thomas L., Emert-Sedlak L., Byeon I. J., Jung J., Ahn J., Wortman M. D., Kukull B., Saito M., Koizumi H., Williamson D. M., Hiyoshi M., Barklis E., Takiguchi M., Suzu S., Gronenborn A. M., Smithgall T. E., Thomas G. (2010) Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol. Biol. Cell 21, 3279–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee C. H., Saksela K., Mirza U. A., Chait B. T., Kuriyan J. (1996) Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 85, 931–942 [DOI] [PubMed] [Google Scholar]
- 19. Shugars D. C., Smith M. S., Glueck D. H., Nantermet P. V., Seillier-Moiseiwitsch F., Swanstrom R. (1993) Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 67, 4639–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geyer M., Peterlin B. M. (2001) Domain assembly, surface accessibility, and sequence conservation in full-length HIV-1 Nef. FEBS Lett. 496, 91–95 [DOI] [PubMed] [Google Scholar]
- 21. Emert-Sedlak L., Kodama T., Lerner E. C., Dai W., Foster C., Day B. W., Lazo J. S., Smithgall T. E. (2009) Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem. Biol. 4, 939–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanna Z., Weng X., Kay D. G., Poudrier J., Lowell C., Jolicoeur P. (2001) The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J. Virol. 75, 9378–9392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lerner E. C., Smithgall T. E. (2002) SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat. Struct. Biol. 9, 365–369 [DOI] [PubMed] [Google Scholar]
- 24. Collins K. L., Chen B. K., Kalams S. A., Walker B. D., Baltimore D. (1998) HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 [DOI] [PubMed] [Google Scholar]
- 25. Collins K. L., Baltimore D. (1999) HIV's evasion of the cellular immune response. Immunol. Rev. 168, 65–74 [DOI] [PubMed] [Google Scholar]
- 26. Michel N., Allespach I., Venzke S., Fackler O. T., Keppler O. T. (2005) The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to down-regulate cell-surface CCR5 and CD4. Curr. Biol. 15, 714–723 [DOI] [PubMed] [Google Scholar]
- 27. Venzke S., Michel N., Allespach I., Fackler O. T., Keppler O. T. (2006) Expression of Nef down-regulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surface of target cells and thereby enhances resistance to superinfection. J. Virol. 80, 11141–11152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hung C. H., Thomas L., Ruby C. E., Atkins K. M., Morris N. P., Knight Z. A., Scholz I., Barklis E., Weinberg A. D., Shokat K. M., Thomas G. (2007) HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to down-regulate cell-surface MHC-I. Cell Host Microbe 1, 121–133 [DOI] [PubMed] [Google Scholar]
- 29. Rekas A., Alattia J. R., Nagai T., Miyawaki A., Ikura M. (2002) Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J. Biol. Chem. 277, 50573–50578 [DOI] [PubMed] [Google Scholar]
- 30. Arold S., Franken P., Strub M. P., Hoh F., Benichou S., Benarous R., Dumas C. (1997) The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure 5, 1361–1372 [DOI] [PubMed] [Google Scholar]
- 31. Grzesiek S., Bax A., Clore G. M., Gronenborn A. M., Hu J.-S., Kaufman J., Palmer I., Stahl S. J., Wingfield P. T. (1996) The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat. Struct. Biol. 3, 340–345 [DOI] [PubMed] [Google Scholar]
- 32. Jia X., Singh R., Homann S., Yang H., Guatelli J., Xiong Y. (2012) Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 19, 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren X., Park S. Y., Bonifacino J. S., Hurley J. H. (2014) How HIV-1 Nef hijacks the AP-2 clathrin adaptor to down-regulate CD4. Elife 3, e01754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu W., Doshi A., Lei M., Eck M. J., Harrison S. C. (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638 [DOI] [PubMed] [Google Scholar]
- 35. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 36. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alvarado J. J., Betts L., Moroco J. A., Smithgall T. E., Yeh J. I. (2010) Crystal structure of the Src-family kinase Hck SH3-SH2-linker regulatory region supports an SH3-dominant activation mechanism. J. Biol. Chem. 285, 35455–35461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horenkamp F. A., Breuer S., Schulte A., Lülf S., Weyand M., Saksela K., Geyer M. (2011) Conformation of the dileucine-based sorting motif in HIV-1 Nef revealed by intermolecular domain assembly. Traffic 12, 867–877 [DOI] [PubMed] [Google Scholar]
- 39. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 41. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by Cα geometry: φ, ψ, and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 42. Trible R. P., Narute P., Emert-Sedlak L. A., Alvarado J. J., Atkins K., Thomas L., Kodama T., Yanamala N., Korotchenko V., Day B. W., Thomas G., Smithgall T. E. (2013) Discovery of a diaminoquinoxaline benzenesulfonamide antagonist of HIV-1 Nef function using a yeast-based phenotypic screen. Retrovirology 10, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Poe J. A., Smithgall T. E. (2009) HIV-1 Nef dimerization is required for Nef-mediated receptor down-regulation and viral replication. J. Mol. Biol. 394, 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murphy M., Jason-Moller L., Bruno J. (2006) Using Biacore to measure the binding kinetics of an antibody-antigen interaction. Curr. Protoc. Protein Sci. 19, 14. [DOI] [PubMed] [Google Scholar]
- 45. Jason-Moller L., Murphy M., Bruno J. (2006) Overview of Biacore systems and their applications. Curr. Protoc. Protein Sci. 19, 13. [DOI] [PubMed] [Google Scholar]
- 46. Arold S. T., Ulmer T. S., Mulhern T. D., Werner J. M., Ladbury J. E., Campbell I. D., Noble M. E. (2001) The role of the Src homology 3-Src homology 2 interface in the regulation of Src kinases. J. Biol. Chem. 276, 17199–17205 [DOI] [PubMed] [Google Scholar]
- 47. Eck M. J., Atwell S. K., Shoelson S. E., Harrison S. C. (1994) Structure of the regulatory domains of the Src-family tyrosine kinase Lck. Nature 368, 764–769 [DOI] [PubMed] [Google Scholar]
- 48. Schindler T., Sicheri F., Pico A., Gazit A., Levitzki A., Kuriyan J. (1999) Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol. Cell 3, 639–648 [DOI] [PubMed] [Google Scholar]
- 49. Sicheri F., Moarefi I., Kuriyan J. (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385, 602–609 [DOI] [PubMed] [Google Scholar]
- 50. Williams J. C., Weijland A., Gonfloni S., Thompson A., Courtneidge S. A., Superti-Furga G., Wierenga R. K. (1997) The 2.35 Å crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J. Mol. Biol. 274, 757–775 [DOI] [PubMed] [Google Scholar]
- 51. Xu W., Harrison S. C., Eck M. J. (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602 [DOI] [PubMed] [Google Scholar]
- 52. Fushman D., Xu R., Cowburn D. (1999) Direct determination of changes of interdomain orientation on ligation: use of the orientational dependence of 15N NMR relaxation in Abl SH(32). Biochemistry 38, 10225–10230 [DOI] [PubMed] [Google Scholar]
- 53. Hofmann G., Schweimer K., Kiessling A., Hofinger E., Bauer F., Hoffmann S., Rösch P., Campbell I. D., Werner J. M., Sticht H. (2005) Binding, domain orientation, and dynamics of the Lck SH3-SH2 domain pair and comparison with other Src-family kinases. Biochemistry 44, 13043–13050 [DOI] [PubMed] [Google Scholar]
- 54. Ulmer T. S., Werner J. M., Campbell I. D. (2002) SH3-SH2 domain orientation in Src kinases: NMR studies of Fyn. Structure. 10, 901–911 [DOI] [PubMed] [Google Scholar]
- 55. Jung J., Byeon I. J., Ahn J., Gronenborn A. M. (2011) Structure, dynamics, and Hck interaction of full-length HIV-1 Nef. Proteins 79, 1609–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hochrein J. M., Lerner E. C., Schiavone A. P., Smithgall T. E., Engen J. R. (2006) An examination of dynamics crosstalk between SH2 and SH3 domains by hydrogen/deuterium exchange and mass spectrometry. Protein Sci. 15, 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Young M. A., Gonfloni S., Superti-Furga G., Roux B., Kuriyan J. (2001) Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 105, 115–126 [DOI] [PubMed] [Google Scholar]
- 58. Meiselbach H., Sticht H. (2011) Effect of the SH3-SH2 domain linker sequence on the structure of Hck kinase. J. Mol. Model. 17, 1927–1934 [DOI] [PubMed] [Google Scholar]
- 59. Faraldo-Gómez J. D., Roux B. (2007) On the importance of a funneled energy landscape for the assembly and regulation of multidomain Src tyrosine kinases. Proc. Natl. Acad. Sci. U.S.A. 104, 13643–13648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang S., Blachowicz L., Makowski L., Roux B. (2010) Multidomain assembled states of Hck tyrosine kinase in solution. Proc. Natl. Acad. Sci. U.S.A. 107, 15757–15762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu L. X., Heveker N., Fackler O. T., Arold S., Le Gall S., Janvier K., Peterlin B. M., Dumas C., Schwartz O., Benichou S., Benarous R. (2000) Mutation of a conserved residue (Asp-123) required for oligomerization of human immunodeficiency virus type 1 Nef protein abolishes interaction with human thioesterase and results in impairment of Nef biological functions. J. Virol. 74, 5310–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Choi H. J., Smithgall T. E. (2004) Conserved residues in the HIV-1 Nef hydrophobic pocket are essential for recruitment and activation of the Hck tyrosine kinase. J. Mol. Biol. 343, 1255–1268 [DOI] [PubMed] [Google Scholar]
- 63. Pene-Dumitrescu T., Shu S. T., Wales T. E., Alvarado J. J., Shi H., Narute P., Moroco J. A., Yeh J. I., Engen J. R., Smithgall T. E. (2012) HIV-1 Nef interaction influences the ATP-binding site of the Src-family kinase, Hck. BMC Chem. Biol. 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kerppola T. K. (2008) Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 37, 465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]