

Background: The gating mechanism of transient receptor potential melastatin 8 (TRPM8) channels remains elusive.
Results: Mutations neighboring the C-end region of the TRPM8 channel inner gate modulate allosteric coupling.
Conclusion: The region adjacent to the inner gate in TRPM8 channels is pivotal for allosteric channel activation.
Significance: These findings increase our understanding of the allosteric mechanism of TRPM8 channel gating.
Keywords: Allosteric Regulation, Gating, Ion Channel, Receptor Structure-function, Signal Transduction, TRP Channels, Conductance, Free Energy
Abstract
The ability of transient receptor potential (TRP) channels to sense and respond to environmental and endogenous cues is crucial in animal sensory physiology. The molecular mechanism of channel gating is yet elusive. The TRP box, a conserved region in the N-end of the C terminus domain, has been signaled as pivotal for allosteric activation in TRP channels. Here, we have examined the role of the linker region between the TRPM8 inner gate and the TRP box (referred to as the S6-TRP box linker) to identify structural determinants of channel gating. Stepwise substitutions of segments in the S6-TRP box linker of TRPM8 channel with the cognate TRPV1 channel sequences produced functional chimeric channels, and identified Tyr981 as a central molecular determinant of channel function. Additionally, mutations in the 986–990 region had a profound impact on channel gating by voltage and menthol, as evidenced by the modulation of the conductance-to-voltage (G-V) relationships. Simulation of G-V curves using an allosteric model for channel activation revealed that these mutations altered the allosteric constants that couple stimuli sensing to pore opening. A molecular model of TRPM8, based on the recently reported TRPV1 structural model, showed that Tyr981 may lie in a hydrophobic pocket at the end of the S6 transmembrane segment and is involved in inter-subunit interactions with residues from neighbor subunits. The 986–990 region holds intrasubunit interactions between the TRP domain and the S4–S5 linker. These findings substantiate a gating mechanism whereby the TRP domain acts as a coupling domain for efficient channel opening. Furthermore, they imply that protein-protein interactions of the TRP domain may be targets for channel modulation and drug intervention.
Introduction
Translation of physical and chemical signals from the cellular milieu into the electrical cues used by the peripheral nervous system depends on specialized receptors at the terminals of sensory neurons. These receptors include the transient receptor potential (TRP)3 family of nonselective cation channels, which are involved in the response to a variety of environmental and endogenous compounds, as well as physical stimuli including temperature and pressure (1). Most TRP channels are involved in several physiological processes ranging from sensing temperatures, osmolarity, body temperature regulation, pro-algesic agents, and pain (2). Particularly, TRPM8 is capital for mild cold sensing in mammals (3–5) and alteration of its normal function is related to disorders such as prostate cancer (6), overactive bladder (7), and ocular dryness (8).
The functional TRPM8 is a homotetramer that allows the flow of cations in response to temperatures below 24 °C, the presence of cooling compounds such as menthol or icilin, as well as strong depolarizing voltages (9–11). Each subunit comprises a cytosolic N and C terminus and six membrane-spanning segments (S1–S6). The S5–S6 transmembrane domain from each subunit folds into the ion-conducting pore. The gating of TRPM8 is also regulated by phosphoinositides, phosphatidylinositol 4,5-bisphosphate being essential for channel function and desensitization (12–14). The ability of the channel to sense distinct stimuli depends on protein regions in each subunit of the structure-sensitive sensors. For instance, menthol interacts with a hydrophobic pocket within the S1–S4 transmembrane bundle, and causes conformational rearrangements that lead to gate opening (15, 16). Voltage and cold responses depend on residues located in the S4–S5 loop and the C terminus, respectively (17, 18). As observed in other TRP channels, there is interplay between menthol, cold, and voltage responses. Indeed, in the presence of menthol or cold temperatures, the G-V curves shift toward more physiological membrane potentials (11). Notwithstanding this cross-talk, these stimuli appear to be independently coupled to the gate through an allosteric mechanism (19, 20).
A hallmark of most TRP channels is the presence of the so-called TRP domain in the C terminus region (Fig. 1), adjacent to the S6 transmembrane segment that structures the channel inner gate. The TRP domain is a moderately conserved region of about 30 amino acids that contains the highly conserved 6-mer segment referred to as the TRP box (Fig. 1). This protein domain is a pivotal molecular determinant of TRP channel structure and activity, as it contributes to their tetrameric assembly and to regulate channel responses to all activating stimuli (12, 21–26). A recent study in TRPV1 has shown that the TRP domain is primarily involved in the allosteric activation of the gate of the channel (27). Noteworthy, a peptide patterned after the TRP domain of TRPV1 selectively blocked TRPV1 activity (28), supporting the tenet that the TRP domain is involved in protein-protein interactions that are central for coupling stimuli sensing to gating (15). Moreover, in TRPM8, the TRP domain appears directly involved in phosphatidylinositol 4,5-bisphosphate-mediated regulation (12). The recent proposition of a high resolution structural model for TRPV1 in the absence and presence of activating ligands reveals a central role of this channel region as a determinant of the conformational change needed to activate the receptor (29).
FIGURE 1.

Experimental strategy to explore the role of the S6-TRP box linker region of TRPM8. A, TRPM8 model depicting in red the S6-TRP box linker region. B, the schematic depicts a topological model of a TRPM8 subunit. Below the model are shown the amino acid sequences of the TRP domain proximal to S6 transmembrane segment of TRPM8 (blue) and TRPV1 (red). The protein regions in the S6-TRP box linker of TRPM8 replaced by the cognate of TRPV1 are indicated in red giving rise to chimeras ChA (Gly980-Ile985), ChB (Gly980-Asn990), and ChC (Gly980-Val993).
To further understand the functional role of the TRP domain in channel gating and unveil molecular determinants of this process, we have investigated the contribution of the S6-TRP box linker region in TRPM8 channel activation (Fig. 1). We used a chimeric approach that substituted 5-mer regions in the S6-TRP box linker of TRPM8 by the cognate sequences of TRPV1 (Fig. 1). Notably, our results indicate that the linker region of TRPV1 can replace that of TRPM8 without a major impact in channel expression and channel responsiveness. Analysis of the phenotype of the chimeras identified Tyr981 in this region as a central structural determinant of TRPM8 gating. Mutation of Tyr981 to Glu and Lys (Y981E and Y981K) rendered constitutively active channels. Noteworthy, additional mutation of segment 986–990 in the S6-TRP box linker restored voltage-dependent and menthol sensitivity to the Y981E mutant. Simulation of G-V relationships of chimeric and mutant channels with an allosteric model of gating suggests that these residues and, in general the S6-TRP box linker, are important for the allosteric coupling of stimuli sensing to channel activation. Our findings provide novel insights on the mechanism of channel gating in the TRP channel family, and substantiate the importance of the TRP domain in transmitting the energy of stimuli recognition to gate opening.
EXPERIMENTAL PROCEDURES
TRPM8 Mutagenesis
Mutations were introduced in the rat TRPM8 construct (from D. Julius) using site-directed mutagenesis (QuikChange II, Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. Mutants were confirmed by DNA sequencing.
Cell Culture and Transfection
Human embryonic kidney cells (HEK293 cells) were cultured in DMEM-Glutamax (Invitrogen) supplemented with 10% FBS, and 1% penicillin-streptomycin (Sigma) at 37 °C and 5% CO2. For patch clamp HEK293 cells were seeded in 12-mm l-polylysine (Sigma)-coated coverslips. For protein expression analysis the cells were seeded in 6-well plates. Constructs encoding the mutants were transfected using the Lipofectamine 2000 (Invitrogen) when the culture was at ≈50% confluence for patch clamp or ≈90% for protein analysis. Cells were used up to 48 h after transfection in patch clamp, and 48 h after transfection in biotinylation and Western blot assays.
Total Protein Expression and Surface Labeling
To obtain total extracts, cells were washed with phosphate-buffered saline (PBS), suspended in 2× sample buffer (0.125 m Tris-HCl, pH 6.8, 4% SDS, 200 ml/liter of glycerol, 0.2 g/liter of bromphenol blue, 0.1 m dithiothreitol; all from Sigma) and boiled at 95 °C for 5 min.
The biotinylation reaction was carried out in the culture plates at 4 °C. After two washing steps with cold PBS, cells were incubated for 1 h with 0.9 mg/ml of biotin (Thermo Scientific). An equivalent volume of Tris-buffered saline (10 mm Tris, pH 7.4, 154 mm NaCl) was added to halt the reaction. After 30 min, cells were recovered from the plates and washed twice with cold PBS. Thereafter, biotinylated cells were lysed using RIPA buffer (50 mm Hepes, pH 7.4, 140 mm NaCl, 10% glycerol, 1% (v/v) Triton X-100, 1 mm EDTA, 2 mm EGTA, 0.5% deoxycholate, 10 mm PMSF and protease inhibitor mixture (Sigma)) under gentle agitation for 1 h at room temperature. The soluble fraction was quantified using the BCA method (Thermo Scientific, Pierce). Streptavidin resin (Sigma) was added to equal amounts of extracts and incubated overnight at 4 °C. After washing, biotinylated proteins were eluted with sample buffer 2 times, and analyzed by SDS-PAGE and Western blot.
SDS-PAGE Electrophoresis and Western Blots
Samples were electrophoresed in a 10% gels and then blotted onto a nitrocellulose membrane (0.2 μm, Bio-Rad) using a standard transfer buffer (30 mm Tris base, 190 mm glycine, and 20% methanol). Membranes were stained with Ponceau Red (Sigma) to assess the amount of protein loaded. After blocking at room temperature in TBS-T with 2% ECL Advance Blocking agent (GE Healthcare Europe), membranes were incubated overnight with anti-TRPM8 (dilution 1:5,000, gift from Dr. F. Viana) or actin (dilution 1:10,000, Sigma) at 4 °C. After washing, membranes were incubated with the secondary antibody for 1 h (1:50,000 goat anti-rabbit IgG-horseradish peroxidase conjugate (Sigma)). Finally, the immunoreactive bands were visualized using ECL advanced and Amersham Biosciences HyperfilmTM ECL (GE Healthcare) according to the manufacturer's instructions.
Patch Clamp Measurements
For patch clamp experiments, cells were co-transfected with the different channel species and eYFP. Currents from channels were recorded in whole cell configuration (EPC10 amplifier with Pulse software, HEKA Elektronik, Germany). Patch pipettes were prepared from thin-walled borosilicate glass capillaries (Harvard Apparatus, Holliston, MA), pulled with a P-97 horizontal puller (Sutter Instruments, Novato, CA) to have a tip resistance of 2–4 MΩ. Pipette solution contained (in mm): 150 NaCl, 5 MgCl2, 5 EGTA, and 10 Hepes, pH 7.2, with NaOH. The bath solution contained (in mm): 150 NaCl, 3 MgCl2, 2 CaCl2, and 10 Hepes, pH 7.4, with NaOH. The buffer temperature was kept at 30 °C (Automatic Temperature Controller TC-324B, Warner Instruments).
For chemical stimulation of TRPM8, 1 mm menthol was applied in the external solution. For voltage stimulation a step protocol consisting of depolarizing pulses from −100 to 240 mV in 10-mV steps was used. The holding potential was −60 mV, the time interval between each pulse was 5 s, and the duration of the pulses was 50 ms. Data were sampled at 10 kHz and the series resistance was compensated to ≈80%.
The G-V curves were obtained by converting the steady-state current values from the voltage step protocol to conductance using the relationship G = I/(V − VR), where G is the conductance, I is the current at steady-state, V is the command pulse potential, and VR is the reversal potential of the ionic current obtained from I-V curves. Normalized G-V curves were fitted to the Boltzmann equation,
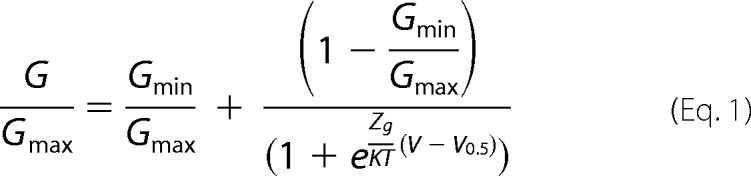 |
where Gmax is the true maximal conductance of the channel species obtained in the presence of 1 mm menthol at depolarized potentials, Gmin is the minimal conductance at hyperpolarized potentials (≤−100 mV), V0.5 is the voltage required to activate the half-maximal conductance, and zg is the apparent gating valence. The true Gmax value was estimated from the fitting of the G-V curves in the presence of 1 mm menthol for wild type and each mutant (27).
For voltage-dependent gating, the free energy difference between the closed and open states at 0 mV at 30 °C for a two-state model (ΔG) was calculated using ΔGo(V) = zgFV0.5, were F is the Faraday constant (0.023 kcal/mol mV) (22).
Molecular Model Building
The molecular model for TRPM8 was modeled using the structures of the TRPV1 ion channel in the closed (Protein Data Bank code 3J5P) and open state (Protein Data Bank code 3J5R) determined by electron microscopy at 3.4-Å resolution (28). Sequence alignment between rat TRPV1 and TRPM8 was performed with ClustalO (30) from the European Bioinformatic Institute (EBI). After visual inspection, the transmembrane alignments were adjusted manually. The visualization and editing of the molecules were done with Yasara (31, 32). The homology modeling was performed with the standard homology modeling protocol implemented in Yasara (version 13.9.8). In this process only small loops were modeled, avoiding the modeling of S2–S3 or S5-pore helix loops. After side chain construction, optimization, and fine-tuning, all new modeled parts were subjected to a combination of steepest descent and simulated annealing minimization, keeping the backbone atoms fixed to avoid molecule damage. Finally, a full-unrestrained simulated annealing minimization was run for the entire model, obtaining a satisfactory −1.597 quality Z-score for dihedrals, and −2.992 for the overall model.
The protein design algorithm and force-field FoldX (33, 34) from CRG were used for protein mutagenesis, and theoretical energy measurements. The interaction energy between different parts of the molecule were calculated by unfolding the selected regions and determining the stability energy of the separated molecules, and then subtracting the sum of individual energies from the global energy of the complex. Such parameters as the atomic contact map, the accessibility of the atoms and residues, the backbone dihedral angles, the hydrogen bonds, and the electrostatic networks of the protein were assessed with FoldX. In addition, the model was evaluated using PROCHECK to show the residues in the allowed regions of the Ramachandran plots (35). The final molecular graphic representations were created using PyMOL version 1.4.1.
RESULTS
Mutations in the S6-TRP Box Linker Region of TRPM8 Modulate Channel Gating
To investigate the contribution of the linker region between the cytosolic end of the S6 and the TRP box in TRPM8 channels (amino acids 980–992 in rat TRPM8, Fig. 1A), we used a chimeric approach whereby short fragments of TRPM8 were cumulatively replaced by the cognate region of TRPV1 (Fig. 1B). Using this approach, we generated three chimeras, namely chimera A (ChA, Gly980-Ile985), chimera B (ChB, Gly980-Asn990), and chimera C (ChC, Gly980-Val993) (Fig. 1B). Chimeric channels were expressed in HEK293 cells for functional characterization by patch clamp. As depicted in Fig. 2A, application of a family of 50-ms voltage pulses from −100 to +240 mV in 10-mV steps in the absence of menthol evoked ionic currents in ChB and ChC, but not in ChA. A similar result was obtained when the depolarizing voltages were applied in the presence of 1 mm menthol. ChB exhibited ionic currents similar to TRPM8, whereas ChC displayed significantly lower responses than wild type and ChB channels. To analyze channel gating, we obtained the normalized G-V curves. Because voltage is a partial activator of the channel, we normalized the G-V curves with the Gmax value obtained in the presence of 1.0 mm menthol (20). For wild type channels, the normalized G-V curve in the absence of menthol revealed a V0.5 of 206 ± 4 mV, a zg of 0.71 ± 0.06, and saturation of the curve at 60% of the Gmax in agreement with partial activation induced by voltage of TRPM8 in the absence of menthol (20) (Fig. 2A, right panel, and Table 1). The free energy of channel gating, assuming a simple two-state model indicates that activation of TRPM8 channels requires 3.3 ± 0.2 kcal/mol (Table 1). Notice that our V0.5 and ΔG values are significantly higher than those reported by other groups (20). Considering that gating of thermo-TRP channels is highly sensitive to the recording temperature, this difference is probably because we have obtained the G-V curve at 30 °C.
FIGURE 2.

Chimeric receptors are functional channels. A, representative currents elicited by a voltage step protocol (inset) in the presence or absence of 1 mm menthol recorded from HEK293 cells expressing TRPM8 and chimeras ChA, ChB, and ChC. Normalized G-V curves for TRPM8, ChB, and ChC in the absence and presence of menthol are displayed at the right. Conductances for each channel species were normalized with respect to the maximal conductance obtained in the presence of 1 mm menthol (denoted as Gmax). Solid lines depict the best fit to a Boltzmann distribution. Parameters derived from the fit are reported in Table 1. B, representative ion current form ChA channels evoked at −60 mV upon washing out DD01050 in the extracellular medium. DD01050 was used at 10 μm. C, Western blot of the total and surface expression of TRPM8, ChA, ChB, and ChC expressed in HEK293 cells grown in the presence of 10 μm DD01050. Surface expression was obtained by protein biotinylation. D, quantitative assessment of the surface/total and total/actin ratios of TRPM8 and ChA, ChB, and ChC from Western blots. Data are given as mean ± S.E.; n ≥ 3.
TABLE 1.
Values of biophysical parameters obtained from the fitting of the G-V curves to a Boltzmann distribution in the absence (control) and presence of 1 mm menthol
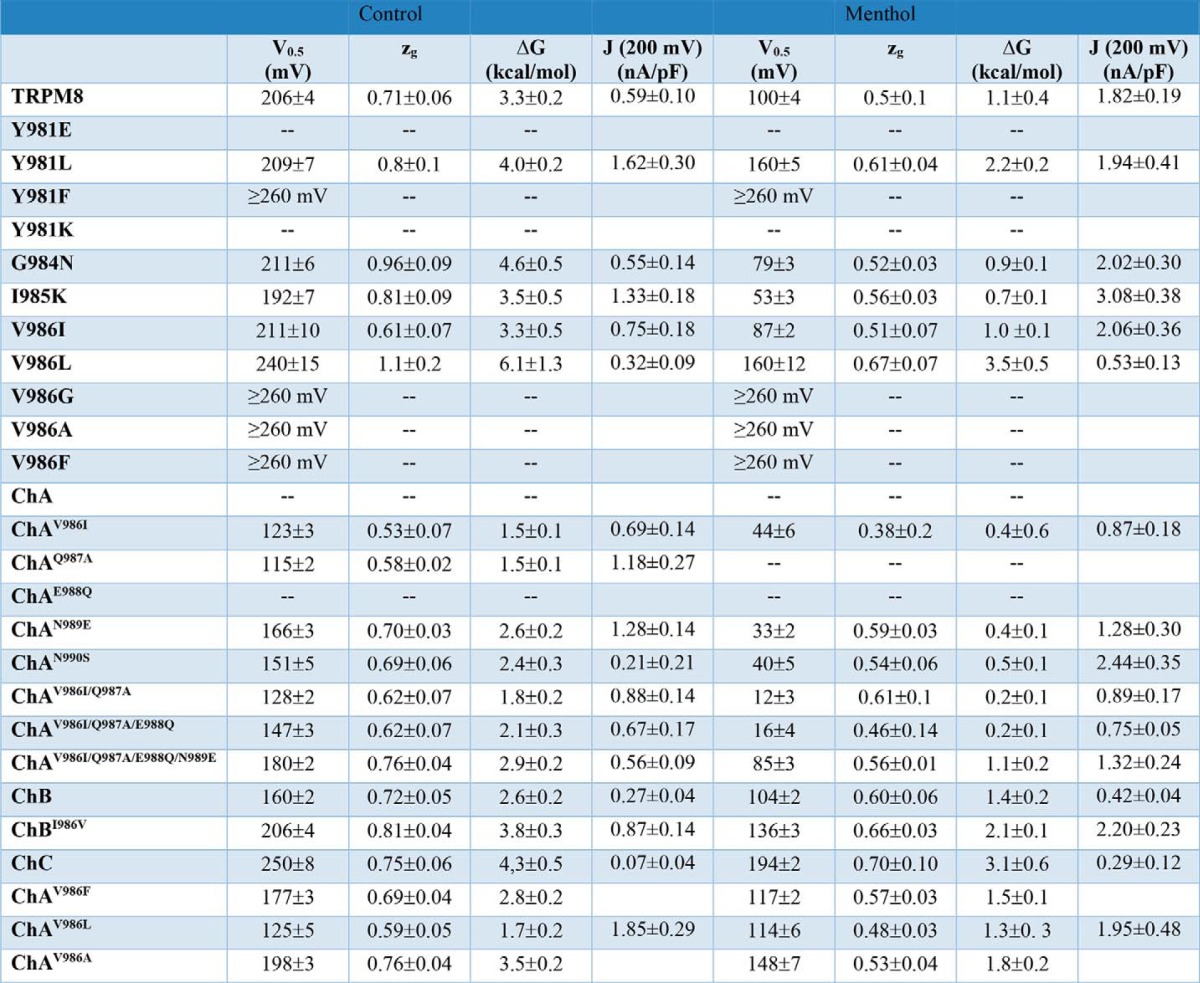
In the absence of menthol, ChB displayed a G-V relationship with a lower V0.5 (160 ± 2 mV), and similar conductance saturation (70% of Gmax) than wild type channels. In contrast, ChC showed larger V0.5 of 250 ± 8 mV than TRPM8 and ChB, but similar conductance saturation (70% of Gmax). The zg of both chimeras was nearly identical to TRPM8 (Fig. 2A, Table 1). These values resulted in free energies of activation of 2.6 ± 0.3 and 4.3 ± 0.5 kcal/mol for ChB and ChC, respectively. Accordingly, our data imply that the S6-TRP box linker of TRPV1 is structurally compatible with the gating structure of TRPM8, and indicate that substitution of the S6-TRP box linker region of TRPM8 by TRPV1 primarily affects the energetic of voltage-dependent channel opening.
In the presence of menthol, TRPM8 was activated at less depolarized potentials (V0.5 = 100 ± 4 mV) with a lower zg = 0.5 ± 0.1, which resulted in a free energy of 1.1 ± 0.4 kcal/mol. ChB exhibited V0.5, zg, and ΔG similar to wild type channels, namely V0.5 = 104 ± 2 mV, zg = 0.60 ± 0.05, and ΔG = 1.4 ± 0.2 kcal/mol, whereas ChC displayed much higher values (V0.5 = 194 ± 2 mV; zg = 0.7 ± 0.1; ΔG = 3.1 ± 0.6 kcal/mol) consistent with the lower responsiveness of this chimera (Fig. 2A, Table 1). Taken together, these data indicate that replacement of linker Gly980-Ile993 in TRPM8 has an impact in channel function, particularly the region encompassing residues Gly980-Ile985 that resulted in apparently channels that did not respond to voltage or menthol.
During characterization of the chimeric channels, we observed that HEK293 cells transfected with ChA exhibited a large extent of cell death (≥70% of transfected cells), as compared with cell death induced by wild type channels, and chimeras ChB and ChC (≈10%). This observation suggested that ChA may induce cell death because it is a constitutively active channel, rather than non-functional as indicated by the lack of voltage and menthol-evoked responses. Thus, to test this hypothesis we grew transfected cells in the presence of the cell impermeable TRP channel blocker DD01050 whose binding site is extracellular (36, 37). In the presence of 10 μm DD01050, we observed a significant decrease in cell death in ChA-transfected HEK293 cells (≤20% of dead cells). Wild type, ChB, and ChC expressing cells were not affected by the presence of the blocker. Furthermore, patch-clamped ChA-transfected cells exhibited reversible ionic currents at −60 mV when the channel blocker was washed out (Fig. 2B). The same experimental paradigm did not elicit responses in wild type channels. Analysis of surface expressed protein revealed that, in the presence of DD01050, ChA reached the plasma membrane in a similar percentage than TRPM8, ChB, and ChC channels (Fig. 2, C and D). Collectively, these findings indicate that replacement of Gly980-Ile985 in TRPM8 by the cognate TRPV1 produces constitutively active channels. A larger substitution (Gly980-Asn990) restores voltage- and menthol-regulated ion channel activity. Therefore, our results are consistent with the existence of structural determinants of TRPM8 gating in the S6-TRP box region.
Residue Tyr981 Adjacent to the S6 Transmembrane Segment Is a Molecular Determinant of Gating
Inspection of segment Gly980-Ile985 reveals only a three-amino acid difference between TRPM8 and TRPV1 that constitute the mutations in ChA, namely Y980E, G984N, and I985K (Fig. 1). To unveil the amino acid responsible for the constitutive activity of ChA, we carried out site-specific mutagenesis in TRPM8 wild type protein. As illustrated in Fig. 3A, mutation of Y981E resulted in constitutively active channels whose activity was blocked by 10 μm DD01050. In the absence of the blocker, transfection of this mutant also resulted in conspicuous cell death (≥70% of transfected cells). Conversely, neither G984N nor I985K mutant channels showed this phenotype, Instead, they evoked ionic currents in response to stimulation with voltage and menthol (Fig. 3B). In the absence of menthol, G984N and I985K exhibited a V0.5 of 211 ± 6 and 192 ± 7 mV, respectively. The zg and the free energy required for opening of the channels, calculated according to the two-state model, were 0.96 ± 0.09 and 4.6 ± 0.5 kcal/mol for G984N mutant and 0.81 ± 0.09 and 3.5 ± 0.5 kcal/mol for I985K (Fig. 3, D and E, Table 1). In the presence of menthol, the V0.5 was smaller than that of TRPM8 (Fig. 3C, Table 1), giving rise to a lower free energy of activation (Fig. 3E, Table 1). Therefore, these results imply that the constitutive activity of ChA is due to mutation of Y981E.
FIGURE 3.

Mutation Y981E, but not of G984N and I985K, in TRPM8 produce constitutively active channels. A, representative inward ionic currents of Y981E, G984N, and I985K mutants at −60 mV upon washing out blocker DD01050. Cells expressing these mutants were grown in the presence of 10 μm DD01050. B, representative currents elicited by a voltage step protocol (inset) in the presence or absence of 1 mm menthol recorded from HEK293 cells expressing TRPM8 and mutants G984N and I985K. Normalized G-V curves for G984N and I985K mutants in the absence and presence of menthol are displayed at the right. Conductance for each channel species were normalized with respect to the maximal conductance obtained in the presence of 1 mm menthol (denoted as Gmax). Solid lines depict the best fit to a Boltzmann distribution. C–E, values of V0.5, zg, and ΔG derived from the fit to the Boltzmann distribution. Data are given as mean ± S.E.; n = 7.
Because substitution of Tyr981 with an acidic residue (Y981E) rendered constitutively active channels, we assessed the effect of mutating Tyr981 into the basic residue (Y981K). Akin to mutant Y981E, the expression of Y981K channels was toxic for the cells unless the DD01050 blocker was present in the growth medium. In addition, inward ionic currents were observed when the blocker was removed from the bath (Fig. 4A). This result suggests that a charged residue at position Tyr981 destabilized the channel closed state, giving rise to constitutively active channels.
FIGURE 4.

Effect of mutations Y981K, Y981L, and Y981F on channel activity. A, representative ionic currents of Y981K mutants at −60 mV upon washing out 10 μm DD01050. B, representative ionic currents of Y981F mutants at −60 mV upon washing out 10 μm DD01050. C, family of ion channel recordings evoked from Y981F mutants according to the voltage-step protocol depicted as an inset. D, family of ion channel recordings evoked from Y981L mutants according to the voltage step protocol depicted as an inset. E, normalized G-V relationship for Y981L mutant. Solid lines depict the best-fit to a Boltzmann distribution to obtain the V0.5 and zg values. F, Western blot of the total and surface expression of TRPM8, Y981F, and Y981L expressed in HEK293 cells. Conductance was normalized with respect to the maximal conductance obtained in the presence of 1 mm menthol (denoted as Gmax). Data are given as mean ± S.E.; n = 6.
We next evaluated the consequences of mutating Tyr981 to phenylalanine. Unexpectedly, Y981F channels were unresponsive to membrane depolarization in the absence and presence of menthol (Fig. 4C). This lack of channel function was not due to death of cells expressing the channel because it did not have constitutive channel activity as removal of DD01050 did not evoke ionic currents. (Fig. 4B). Furthermore, the absence of channel activity did not result from abrogation of protein expression because the mutant channel was expressed at the cell surface at comparable levels to wild type protein (Fig. 4F).
To further examine the role of Tyr981 in channel gating, we also mutated it to the aliphatic hydrophobic amino acid leucine, which exhibits a similar hydrophobic surface to Tyr but reduced volume. At variance with Y981F mutants, substitution of Tyr981 by Leu rendered ion channels that displayed voltage-gated and menthol-elicited channel activity (Fig. 4D). In the absence of menthol, Tyr981 exhibited a V0.5 of 209 ± 7 mV and zg of 0.8 ± 0.1, whereas in the presence of the agonist the values were 160 ± 5 mV and 0.61 ± 0.04 (Table 1). The maximal conductance in response to voltage was similar to that of voltage plus menthol (Fig. 4E). Compared with wild type channels, this result suggests a decrease in the efficacy of menthol in opening the Y981L mutant, as evidenced by its higher free energy for activation (Table 1). Taken together, these findings imply that Tyr981 contributes to define the free energy of pore gating.
Mutation of the 986–990 Region in Chimera A Restores Regulated Channel Gating
Chimera B contains the Y981E mutation and displays regulated channel gating, implying that mutations downstream of residue Ile985, namely V986I, Q987A, E988Q, N989E, and N990S, restore voltage and menthol gating. Thus, we next mutated sequentially and cumulatively these residues in ChA, and evaluated the phenotype of mutants ChAV986I, ChAV986I/Q987A, ChAV986I/Q987A/E988Q, and ChAV986I/Q987A/E988Q/N989E to determine the minimum number of mutations needed to restore regulated gating (Fig. 5).
FIGURE 5.

Functional characterization of ChA cumulative mutations to produce ChB. A–E, representative ionic currents elicited by a voltage step protocol in the absence and presence of 1 mm menthol recorded from HEK293 cells expressing chimeric channels ChAV986I (A), ChAV986I/Q987A (B), ChAV986I/Q987A/E988Q (C), ChAV986I/Q987A/E988Q/N990E (D), and ChBI986V (E). Right panels show the normalized G-V relationship for mutants in the absence and presence of the agonist. Solid lines depict the best fit to a Boltzmann distribution to obtain the V0.5 and zg values. Conductance was normalized with respect to the maximal conductance obtained in the presence of 1 mm menthol (denoted as Gmax). Data are given as mean ± S.E.; n ≥ 5.
At variance with ChA, none of the ChA mutants exhibited a cytotoxic effect and were expressed to similar levels in HEK293 cells in the absence of the blocker DD01050 (data not shown). Voltage-clamped cells responded to membrane depolarization steps in the absence and presence of menthol eliciting non-inactivating currents akin to wild type protein (Fig. 5, A–D). Noteworthy, mutation of Val986 to Ile in the context of ChA sufficed to virtually abolish the constitutive activity of ChA (Fig. 5A). As depicted, ChAV986I responded to voltage changes in the absence of menthol with a V0.5 of 123 ± 3 mV and zg of 0.53 ± 0.07 (Table 1). The V0.5 value for ChAV986I is ≈2-fold lower than that of wild type channels, suggesting a higher propensity of ChAV986I to open (ΔG = 1.5 ± 0.5 kcal/mol). This result was further substantiated by the lower V0.5 (44 ± 6 mV) of ChAV986I than TRPM8 (100 ± 4 mV) in the presence of menthol (Table 1). Furthermore, ChAV986I displayed voltage-independent gating at negative potentials in the presence of the agonist (Fig. 5A, right panel), consistent with a partial coupling of the pore to the menthol sensor. The maximal conductance in the absence of agonist was similar to that in its presence, in accordance with a lower free energy for voltage gating of this mutant.
Additional mutations of ChA (ChAV986I/Q987A, ChAV986I/Q987A/E988Q, and ChAV986I/Q987A/E988Q/N989E) also produced voltage-gated and menthol-sensitive ionic currents (Fig. 5, B–D) that were similar to those recorded for ChAV986I. In the absence of the agonist, analysis of the G-V curves, however, revealed a progressive shift toward more depolarizing potentials with the cumulative mutations, reflected in an increase in the V0.5 (128–180 mV) and ΔG (1.8–2.9 kcal/mol) until reaching the value of the ChB (Table 1). Notably, the cumulative mutations reduced by 40% the maximal conductance evoked by depolarization as compared with the maximum conductance in the presence of menthol (Fig. 5, B–D, and Table 1). The zg values were not significantly affected.
In the presence of menthol, all chimeric mutants displayed a leftward shift in the G-V curve (Fig. 5, B–D, right panels), reflecting a lower free energy for activation (Table 1). In addition, incorporation of these additional mutations in ChA progressively eliminated voltage-independent gating evoked by the agonist, as evidenced by the decrease in the conductance at hyperpolarized potentials (Fig. 5, B–D, right panels).
To further evaluate the role of Val986 reverting the constitutive activity of ChA, we mutated I986V in ChB. As depicted in Fig. 5E, mutant ChBI986V responded to voltage and menthol stimuli. The values of V0.5, zg, and ΔG in control conditions were similar to those of TRPM8 (Table 1). However, the channels exhibited a diminished response to menthol as suggested by the increase of the free energy required to open the channel (1.1 ± 0.4 kcal/mol for TRPM8 versus 2.1 ± 0.1 kcal/mol for ChBI986V). These results imply that, in addition to V986I, other residues in ChB contribute to compensate the constitutive activity of ChA.
We next sought to determine which mutations other than V986I could revert the overactive phenotype of ChA. To this end, mutations Q987A, E988Q, N989E, and N990S were introduced individually in ChA (Fig. 6). Functional analysis of these mutants revealed that ChAQ987A and more conspicuously ChAE988Q exhibited an inward current upon removal of DD01050 (Fig. 6A). This is consistent with the presence of constitutive activity, notably larger for ChAE988Q. We could determine the G-V relationship for ChAQ987A in the absence of menthol, but not in the presence of the agonist because the ionic current did not allow to properly voltage-clamp the cells. Voltage-dependent gating of ChAQ987A rendered V0.5 and ΔG values that were half those of wild type channels (Fig. 6, D–F, Table 1).
FIGURE 6.

Mutation of residues Gln987-Asn990 identify residues in ChA that restore regulated gating. A, representative ionic currents of ChAQ987A and ChAE988Q mutants at −60 mV upon washing out 10 μm DD01050. B and C, representative ionic currents elicited by a voltage step protocol in the absence and presence of 1 mm menthol recorded from HEK293 cells expressing chimeric channels ChAN989E (B) and ChAN990A (C). Right panels show the normalized G-V relationship for mutants in the absence and presence of the agonist. Solid lines depict the best fit to a Boltzmann distribution. D–F, values of V0.5, zg, and ΔG in the absence and presence of menthol, derived from the fit to the Boltzmann distribution. Data are given as mean ± S.E.; n ≥ 5.
Notably, mutations ChAN989E and ChAN990S abolished the constitutive activity of ChA, and the resulting channels were gated by voltage and menthol (Fig. 6, B and C). The V0.5 and ΔG for channel opening in the absence and presence of menthol were significantly smaller than for wild type channels (Fig. 6, D–F, Table 1). These mutants display G-V relationships shifted toward lower voltages, consistent with a facilitated gating mechanism, presumably because of the Y981E mutation. Taken together, these results indicate that the constitutive activity of ChA can be compensated by mutations in the adjacent region (amino acids 986–990), suggesting that this linker region in the TRP domain is pivotal for coupling stimuli sensing to pore opening.
Effect of Hydrophobic Residues at Val986 in Chimera A
A remarkable finding of this study is that substitution of Val986 with a slightly larger hydrophobic residue (Ile) had such an important impact on ChA constitutive activity, restoring regulated channel gating. Thus, to learn more on the molecular requirements of Val986 and in general of the region 986–990, we next studied the effect of introducing larger (ChAV986F), similar (ChAV986L), and smaller (ChAV986A) residues than Ile.
These mutants were expressed well in HEK293 cells without requiring the addition of the channel blocker (Fig. 7). Akin to ChAV986I, they exhibited voltage-dependent responses in the absence and presence of menthol (Fig. 7, A–C). As seen, chimera ChAV986A produced lower ionic currents than the other two chimeras. The smaller responses of ChAV986A were not due to a reduced surface expression because all chimeric species were expressed to a similar level (data not shown). Comparison of the G-V relationships of ChAV986I and ChAV986A showed that introduction of Ala shifted the curve toward depolarizing potentials (Fig. 7A), with a V0.5 and ΔG that were closer to those of wild type channels (198 ± 3 mV and 3.5 ± 0.2 kcal/mol) (Table 1). However, at variance with TRPM8, the maximal conductance in the absence menthol was similar to that in its presence (Fig. 7A). Furthermore, the ChAV986A displayed a lower sensitivity to menthol than ChAV986I and TRPM8, as evidenced by its higher free energy of channel activation in the presence of the agonist (Fig. 7F).
FIGURE 7.

Effect of mutations V986A, V986L, and V986F on ChA constitutive activity. Representative ionic currents elicited by a voltage step protocol in the absence and presence of 1 mm menthol recorded from HEK293 cells expressing chimeric channels ChAV986A (A), ChAV986L (B), and ChAV986F (C). Right panels show the normalized G-V relationship for the mutants in the absence and presence of agonist. Solid lines depict the best fit to a Boltzmann distribution to obtain the V0.5 (D) and zg values (E). Conductance was normalized with respect to the maximal conductance obtained in the presence of 1 mm menthol (denoted as Gmax). F, free energy of channel activation at 0 mV assuming a simple two-state model for gating. Data are given as mean ± S.E.; n ≥ 4. *, p < 0.05 using the Student's t test.
Incorporation of leucine (ChAV986L) rendered ionic channels with phenotype and biophysical properties similar to ChAV986I, except for the lower magnitude of the voltage-independent component at hyperpolarized potentials (Fig. 7B). Accordingly, the energetics of channel gating of ChAV986L was similar to ChAV986I, with V0.5 and ΔG values lower than wild type channels (Fig. 7, D–F, Table 1).
Increasing the volume of residue at Val986 (ChAV986F) yielded ion channels exhibiting gating properties close to TRPM8 channels that resulted from a shift of the G-V curve toward more depolarizing potentials than ChAV986I (Fig. 7, C–F, and Table 1). Taken together, these results indicate a size-dependent effect on channel gating of ChA, whereby a Val residue at position 986 in ChA produces constitutively active ion channels, and lower or higher amino acids restore regulated gating as a function of the residue volume as evidenced for Ala, Ile, Leu, and Phe substitutions. Thus, it appears that amino acid Val986 is located in a hydrophobic pocket that pivotally contributes to define the energetics of channel opening.
Mutations of Val986 in TRPM8 Modulate Gating
Because mutations in Val986 restored regulated channel gating in ChA, we next investigated the impact of mutating this position in wild type channels. Substitution of Val986 by Ala, Gly, and Phe (V986A, V986G, V986F) rendered non-functional channels, as no response could be recorded in the absence and presence of menthol (Figs. 8, A, B, and E). Next, we concentrated on introducing Ile and Leu residues that had a similar effect on ChA. As illustrated in Fig. 8C, replacement of Val986 by Leu (V986L), produced ion channels that displayed a G-V curve displaced to stronger depolarized potentials than wild type channels (Fig. 8, F–H). The maximal conductance for V986L in the presence of menthol had to be estimated from the G-V fit because it was not reached by depolarization up to 240 mV (Fig. 8C). Consequently, the energetic of V986L mutants was significantly larger than that of TRPM8, both in the absence (240 mV for V0.5 and 6.1 kcal/mol for ΔG) and presence of menthol (160 ± 12 mV and ΔG = 3.5 ± 0.5 kcal/mol). Intriguingly, when Ile was introduced at Val986 (V986I) the channels exhibited a similar voltage sensitivity as TRPM8 (Fig. 8D). Analysis of the G-V relationship showed curves that were akin to wild type, including partial activation of the channel in the absence of agonist (60% of Gmax) (Fig. 8D). The biophysical parameters of channel activation were comparable with those of the wild type channels (Fig. 8, F–H, and Table 1). Collectively, these observations further substantiate that the position of Val986 is involved in setting the energetic channel activation, probably by contributing to define the allosteric mechanism of channel activation.
FIGURE 8.

Effect of mutation in position Val986 on TRPM8 activity. Representative ionic currents elicited by a voltage step protocol in the absence and presence of 1 mm menthol recorded from HEK293 cells expressing mutant channels V986G (A), V986A (B), V986L (C), V986I (D), and V986F (E). Right panels show the normalized G-V relationship for mutants in the absence and presence of agonist. Solid lines depict the best-fit to a Boltzmann distribution to obtain the V0.5 (F) and zg values (G). Conductance was normalized with respect to the maximal conductance obtained in the presence of 1 mm menthol (denoted as Gmax). H, free energy of channel activation at 0 mV assuming a simple two-state model for gating. Data are given as mean ± S.E.; n ≥ 5. *, p < 0.05 using the Student's t test.
Mutation of the TRP Domain of TRPM8 Alters Allosteric Coupling for Channel Activation
TRPM8 gating has been described to be a complex allosteric mechanism involving several states that functionally couple the activating stimuli to the channel gate (19, 20, 38). Although a two-state model describes fairly well the energetics of the gating process of our TRPM8 chimeras by voltage and menthol, this represents a simplification of the real mechanism of channel gating. Thus, we also evaluated our functional data using an allosteric model for voltage and menthol gating. Akin to other studies (19, 20, 27), we have assumed the existence of independent sensors for voltage, menthol, and temperature (Fig. 9A). Because all our measurements have been carried out at 30 °C, we considered that the temperature sensor was in its resting state and marginally contributed to voltage and menthol gating. Under these conditions, and accepting that voltage and menthol sensors move simultaneously, an 8-state allosteric model is required for describing the gating mechanism (19, 20, 27) (Fig. 9A). According to this model, the probability of channel gating is given by the relationship,
 |
where J, Q, and L are the equilibrium constants of the voltage sensor, the menthol binding site, and the pore, respectively; D and P are the coupling constants of the voltage and menthol sensors to the pore; and E is the coupling constant between both sensors (Fig. 9A). For the voltage sensor, J = J0 exp(zgFV/RT), where J0 is the equilibrium constant at 0 mV and zg is the gating valence. Similarly, for the menthol sensor Q = [menthol]/KD, where KD is the menthol dissociation constant, which is different from the EC50 obtained from a dose-response curve (20, 27). Notice that, in the absence of ligand, the open probability for voltage is given by Equation 3.
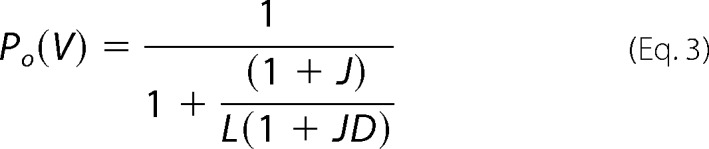 |
We first fitted the normalized G-V curves for voltage activation of TRPM8 wild type to Equation 3 to determine the values of equilibrium constants L and J0. Using a zg of 0.7 (Table 1), the best fit for the G-V at 30 °C was obtained with the following values for the constants: J0 = 0.002, L = 0.015, and D = 107 (Fig. 9B). Notice that the maximum probability of channel gating according to Equation 3 is given by Po,max (V) = 1/(1 + (1/LD)), which renders a value of 0.6 akin to the normalized Gmax obtained in the Boltzmann fit (Fig. 2). We next analyzed the normalized G-V curves for chimeric channels (Fig. 9B). As depicted in Table 2, for chimeras ChB and ChC the values of J0 and L were similar to those of wild type channels, whereas the value of D was higher than wild type for ChB (D = 256) and lower for ChC (D = 40). These values are consistent with the larger and smaller voltage sensitivity of both chimeras.
FIGURE 9.
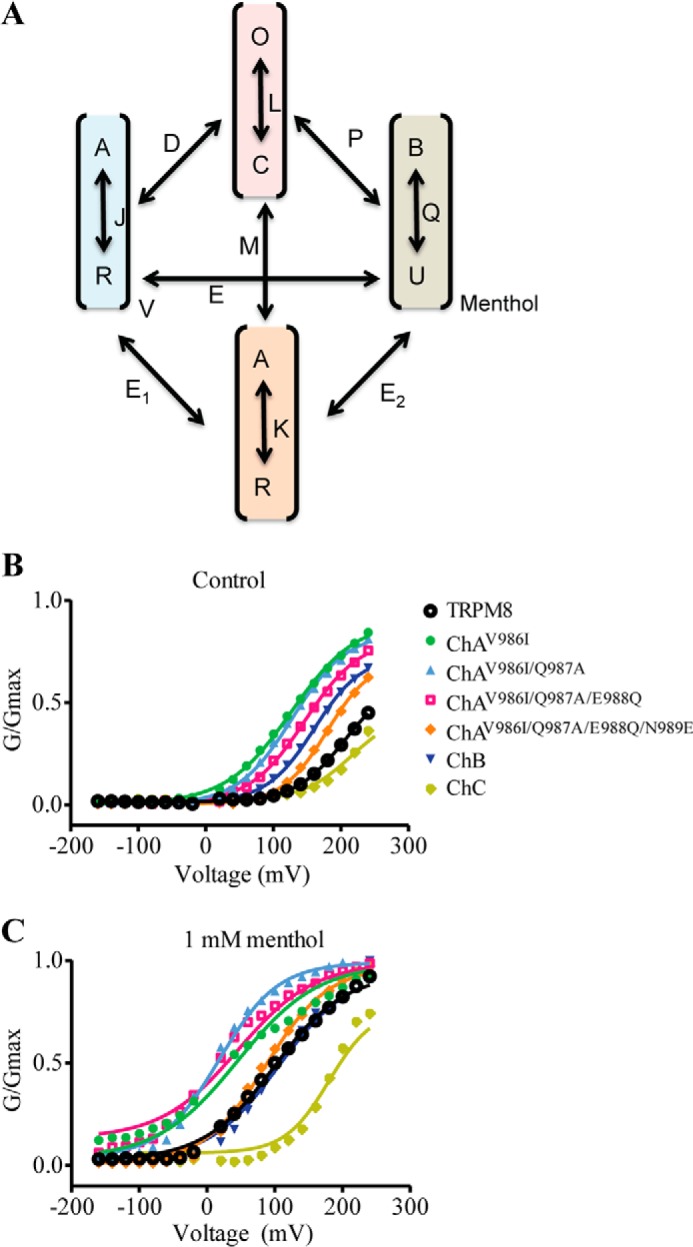
Simulation of G-V curves for chimeric and mutant channels using allosteric gating model. A, proposed 8-state allosteric model of channel activation that assumes the presence of three sensors for the activating stimuli (V, T, and M), and the pore. The voltage and temperature sensors may be in the resting (R) or activated (A) state, the menthol sensor in the unbound (U) or bound (B) state, and the gate in the closed (C) or open (O) state. State transitions are governed by equilibrium constants J, K, Q, and L. Activating sensors are coupled to the channel gate by coupling constants D, M, and P; and to each other by coupling constants E, E1, and E2. The figure depicts the normalized G-V curves obtained for TRPM8 chimeras in the absence (B) and presence (C) of 1 mm menthol. Solid lines depict the best fit to the allosteric models. Normalized G-V curves in the absence of menthol were fitted to Equation 3, and results are reported in Table 2. Normalized G-V curves in the presence of menthol were fitted to Equation 2 fixing the values for the equilibrium constants obtained in the absence of the agonist. The value of E was kept constant to 1.0. The values obtained for allosteric constants P and D and free energy are depicted in Table 2.
TABLE 2.
Values of equilibrium and coupling constants for TRPM8 and mutant channels in the absence (control) and presence of 1 mm menthol obtained from the fitting of the G-V curves to an allosteric model of gating
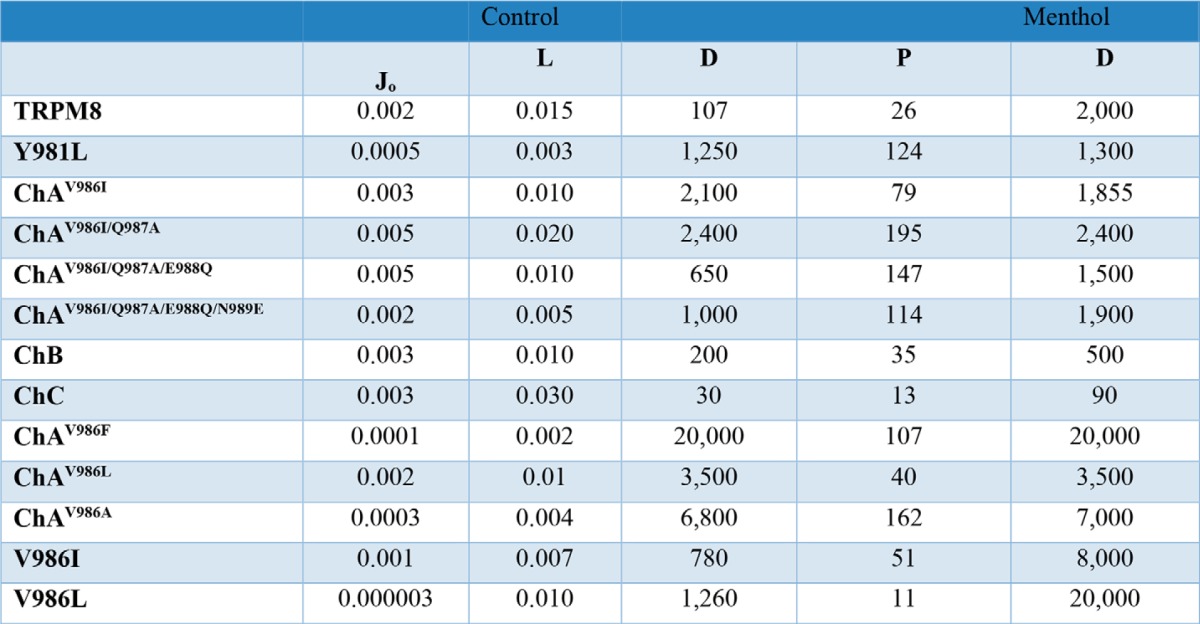
In the absence of ligand, mutations carried out in ChA until reaching the ChB sequence further substantiate a major effect on the allosteric constant D, with more modest alteration of the equilibrium constants (Fig. 9B, Table 2). A comparable result was obtained for mutants Y981L and V986I in TRPM8. Mutation of V986L in TRPM8, which poorly responds to voltage, also displayed an effect on J0 (Table 2), most likely due to the partial activation of this mutant. Similarly, substitution of Val986 in the ChA context mainly affected allosteric coupling constant D, although for mutants ChAV986A and ChAV986F and alteration of J0 is also noticeable, suggesting that this position may be interacting with the voltage sensor. Taken together, these results imply that mutation of the TRP domain primarily affects the allosteric coupling constant between the voltage sensor and the pore, with a modest effect on the equilibrium constant of the voltage sensor.
Simulation of the conductance data in the presence of menthol was carried out using Equation 2, assuming that both sensors activate independently, i.e. E = 1.0. For this analysis, the values of zg, J0, and L were maintained constant and equal to those obtained in the absence of the channel agonist. The value of KD was also kept constant to 10 mm (20). For TRPM8, the addition of 1 mm menthol at 30 °C shifted the G-V toward a hyperpolarized potential (Fig. 2). This change is well described by Equation 2 with the following values zg = 0.5, J0 = 0.002, L = 0.015, P = 26, and D = 2,000, implying that the presence of agonist mainly modified the coupling constant for the voltage sensor (Fig. 9C), consistent with the leftward shift of the G-V curve elicited by the agonist. We also tested the effect of keeping constant the value of D, and estimated the E and P values. Under these conditions, we found P = 18 and E = 55. Thus, it appears that at 30 °C TRPM8 gating in the presence of menthol was primarily driven by the voltage sensor. Consistent with this notion, TRPM8 in the presence of menthol did not exhibit a voltage-independent conductance at hyperpolarized potentials. Therefore, we evaluated the G-V curves of mutants keeping E = 1.0 (Fig. 9B). Inspection of the results (Table 2) showed that, similar to wild type channels, menthol primarily increased the value of the allosteric coupling constant D in the chimeras and mutant channels. Collectively, these findings further indicate that the main impact of mutations in the TRP domain is to modulate allosteric channel activation.
DISCUSSION
The gating process of polymodal thermo-TRP channels such as TRPM8 is governed by a complex mechanism involving allosteric conformational changes that leads to channel gate opening. Cumulative data indicate that the TRP domain, a cytosolic protein region adjacent to the S6 transmembrane segment plays a pivotal role in channel gating. In TRPV1 receptors, the TRP domain was originally identified as an association domain essential for assembling the functional tetrameric channel (26). Furthermore, mutations in this domain in TRPV1 profoundly affected channel gating by all the activating stimuli (22), through a mechanism that involved the modulation of the allosteric coupling between the sensors of the activating stimuli and the channel gate (27). These finding were structurally supported by the publication of a three-dimensional model of TRPV1 at 3.4 Å derived from cryoelectron microscopy images (29, 39). This structural model depicts the TRP domain as an α-helix that runs parallel to the inner leaflet of the plasma membrane, interacting with cytosolic regions of the channel, including the S4–S5 linker (Fig. 1A). This architecture of the TRP domain is consistent with its reported role in TRPV1 gating (22, 26, 27), as it is involved in key intersubunit and intrasubunit interactions that are fundamental for allosteric gating in response to an activating stimulus.
A question that arises is whether the TRP domain plays a similar role in other thermo-TRP channels, thus substantiating its central contribution to allosteric activation in this channel family. The most significant contribution of our study is that the TRP domain of TRPM8 and, in particular, the linker region between the S6 transmembrane segment and the TRP box is essential for allosteric activation of the channel. Our approach consisted of replacing 5-mer segments of TRPM8 by the cognate amino acids of TRPV1 to evaluate the compatibility of both sequences for channel gating. Noteworthy, we found that substitution of the five residues adjacent to the S6 segment (ChA) produced constitutively active TRPM8 channels, whereas incorporation of the next five amino acids (ChB) restored voltage- and menthol-dependent gating. Additional replacements (ChC) sensibly reduced voltage and menthol sensitivity and raised the free energy for channel activation. Simulation of the experimental data with an allosteric model of channel activation suggested that alteration of this region primarily affected the allosteric coupling constants for channel activation. Taken together, these findings imply that: (i) akin to TRPV1 channels, the TRP domain of TRPM8 is pivotal in defining the energetic of channel activation, and (ii) the linker region between the S6 and the TRP box of TRPM8 is structurally compatible and can be readily substituted with that of TRPV1. A fine tuning appears necessary to preserve the voltage- and menthol-dependent gating properties of the wild type TRPM8 channels, suggesting a conserved gating mechanism in thermo-TRP channels.
A closer structure-function analysis of the S6-TRP box linker region revealed that residue Tyr981 is a key molecular determinant of TRPM8 gating. Residues Val986, Asn989, and Asn990 contributed to define the regulated gating of the chimeric channel ChB. For instance, mutation of Y981E is the principal residue contributing the constitutive activity of ChA. Noteworthy, mutation of this residue in TRPM8 channels has profound effects in gating. Indeed, replacement of Tyr981 by Glu or Lys leads to constitutively active TRPM8 channels, whereas substitution by Phe renders non-functional channels, despite that the protein is synthesized and transferred to the plasma membrane in a similar amount as wild type channels. This result is compatible with a complete uncoupling of the channel gate from the voltage and menthol sensors, such that neither stimulus is able to open the channel. However, mutation of Tyr981 to Leu produced functional TRPM8 channels although with lower sensitivity to both activating stimuli, thus displaying higher free energy for channel activation than wild type channels. Taken together, these data support the tenet that Tyr981 is a structural determinant of channel gating that pivotally contributes to define the energetic profile of the inner gate.
It is noticeable that mutation V986I, N989E, and N990S in ChA, partially Q987A, but not E988Q restored regulated gating, implying that these mutations affect channel gating and compensate the constitutive activity of ChA. Notably, single mutation of V986I in ChA restored regulated channel gating, whereas additional mutations in the ChA context until reaching the ChB sequence primarily modulated the energetic of channel gating by shifting the G-V relationships toward more depolarized potentials. Analysis of the hydrophobic amino acid preference at Val986 in ChA revealed that smaller (Ala) and larger (Phe) amino acids than Leu and Ile produced channels with gating properties closer to wild type channels, suggesting that the size of the residue is important for restoring coupling to the ChA chimera. Intriguingly, substitution of Val986 in TRPM8 wild type channels by Ile marginally affected the free energy of channel activation. However, when a Leu was introduced at this position in TRPM8, a significant increase in the free energy of channel gating was observed, indicating a fine-tuning of amino acid at this position for activation of TRPM8 channels.
A central question emerges, is the location of Tyr981 and Val986 in the receptor structure compatible with the functional impact of their mutation? To investigate this question, we built homology models for TRPM8 taking as a template the recently reported TRPV1 models in the absence (closed state) and presence of capsaicin (open state) at 3.4 Å (29, 39). The models encompassed the 654 to 1016 region, including the transmembrane segments and the TRP domain but excluding the S2–S3 linker and the pore loops. Although structural models have to be used with caution, they are valuable tools that may help in understanding the functional consequences of mutations. As seen in Fig. 10, Tyr981 is located at the end of the S6 transmembrane segment, close to the channel internal gate. In the channel closed state, this residue lies in a hydrophobic cavity structured by the four-channel subunits, and could be involved in inter-subunit interactions with Gln987 of neighbor subunits (Fig. 10, A, C, and D). This location appears compatible with a contribution of this residue to define the energy profile of the closed state of the channel.
FIGURE 10.

Tyr981 location in the TRPM8 homology model. Side view of the TRPM8 homology model in the closed (A) and open (B) states based on the structure of TRPV1. The region where Tyr981 resides is delimited by a rectangle and the residue is shown in sticks presentation. The different structural elements are indicated for clarity. C and E, magnification of the rectangle delimited region in A. The residues surrounding Tyr981 are presented in surface representation and colored by chain. Subunits not involved in the interaction were removed for clarity. D and F, rotated view (45° with respect to the plane of the membrane) of C and E, respectively.
In the open state, our model suggests an outward movement of Tyr981 and Gln987 that breaks their interaction, resulting in a widening of the C-end of S6 (Fig. 10, B, E, and F). This conformational change locates Tyr981 in a more hydrophilic environment. This mechanism of gating provides an explanation for the phenotypes of Tyr981 mutants. Hence, incorporation of Glu or Lys residues at Tyr981 would be expected to provoke a conformational change near the C-end of S6 to accommodate the charged side chain in a more hydrophilic environment. This conformation would be presumably similar to that adopted by Tyr981 in the open state, thus disrupting the interaction with Gln987 and leading to the open state (Fig. 10D). Consistent with this hypothesis, substitution of Tyr981 by Phe would strengthen the hydrophobic interactions in the cavity, inducing the stabilization of the channel gate in the closed state, in accordance with the finding that Y981F requires unreachable voltages for activation. Mutation of Tyr981 to the smaller Leu residue also supports this tenet, as this mutant displayed higher free energy of activation than wild type channels, in agreement with the stabilization of the closed state. Further support to this mechanism is provided by the theoretical ΔΔG of the interaction between the amino acid at position 981 and its environment. Compared with wild type (ΔΔG = 0 kcal/mol), the ΔΔG reveals that Y981E increased the ΔΔG by +2.8 kcal/mol, whereas Y981F reduced the free energy of the system to −0.81 kcal/mol, and Y981L did not have a noticeable effect (ΔΔG = −0.18 kcal/mol). Therefore, our findings substantiated the notion that Tyr981 is a key structural determinant of the inner gate energetic profile.
The position of Val986 in the channel structural model appears also compatible with its role in gating. This residue, together with Asn989 and Asn990, lies on a contact region between the TRP domain and the S4–S5 linker (Fig. 10, A–C). In particular, Val986 is located in a hydrophobic contact with the appropriate dimensions to hold a valine. It can also accommodate Ile and less favorably a Leu residue, consistent with the modest effect of V986I and the stronger impact of V986L mutants in TRPM8 gating. Furthermore, this cavity appears too large for Ala and overly small for Phe and, therefore, mutation of Val986 to these residues would be expected to have a profound effect on the free energy of channel activation, as observed when these amino acids were introduced at this position in the context of ChA. In the model predicted open state, Val986 appears to move away from S4–S5 propagating the movement of the voltage and menthol sensors to the pore domain, which favors the opening of the channel inner gate. Note that the putative interaction of Val986 with residues in the S4–S5 segment suggests a key role of this residue in the allosteric coupling of voltage sensor movements to the channel pore. In support to this notion, mutation of Val986 in TRPM8 and ChA strongly affected the allosteric coupling constant of voltage sensing, and for some mutants also reduced the equilibrium constant of the putative voltage sensor, consistent with a direct interaction of this residue with parts of the voltage sensing domain, i.e. the S4–S5 linker.
The query that remains to be answered is how could mutations in Val986, Asn989, and Asn990 rescue regulated gating of ChA that has the Y981E mutation. Considering the available structural model and our functional results, it is tempting to hypothesize that introduction of the larger Ile at Val986 could strengthen the interaction with the S4–S5 linker (through Arg862 and Met863 in our model) compensating partly for the conformational change promoted by Glu at Tyr981. As a result, the closed state of the channel would be partially stabilized. The stronger the interaction of the TRP domain with the S4–S5 linker, the higher the energetic stabilization of the closed state, as seen when Phe is placed at Val986. It follows that smaller amino acids than Val should favor a conformational change that facilitates channel opening. However, the opposite is seen, as replacement of Val986 by Gly in TRPM8 gave rise to unresponsive channels, and mutation V986A in ChA produced channels that require high energy for activation. It is plausible that these small amino acids strengthen interaction of the TRP domain with the S4–S5 linker by partially collapsing the volume of the hydrophobic cavity where position 986 resides. Similarly, the residues at positions 989 and 990 may affect the strength of that interaction, as they also have contacts with the S4–S5 linker. Therefore, our functional data, along with the modeled structure for the channel, substantiate the tenet that interaction of the TRP domain and the S4–S5 loop is pivotal for allosteric coupling of the voltage sensor and the gate of the channel. Nonetheless, additional structure-function data are required to demonstrate this hypothesis.
In conclusion, our findings suggest that the S6-TRP box linker of TRPM8 may contribute to a constellation of inter- and intrasubunit interactions that are essential for TRPM8 gating. Furthermore, they are consistent with a model whereby the TRP domain constrains the channel gate and also serves to couple movements of the S4–S5 loop to the pore domain (Fig. 11). The conformational change provoked by the activating stimuli in the sensors is coupled to that of the S6-TRP box linker, which in turn might break intersubunit interactions in the region of Tyr981, favoring the channel opening. In this process, the interplay of Tyr981 and with the residues that delineate the TRP domain and the S4–S5 loop appears pivotal for a voltage- and menthol-evoked response in TRPM8 channels. Our finding also signal the TRP domain and S4–S5 protein interface, as well as intersubunit interactions at the end of S6 as sites for pharmacological intervention, and provide a potential mechanism for blocking activity of the TRPducins (28).
FIGURE 11.

Putative TRPM8 gating mechanism. Schematic representation of the TRPM8 pore forming segments, TRP domain, and S4–S5 loop. A, at resting, the thermal energy is not sufficient to disrupt TRP domain/S4–S5 linker intrasubunit hydrophobic interactions (green cloud) keeping closed the channel in TRPM8 (red cloud). The released energy upon stimuli recognition disturbs this hydrophobic interaction provoking a conformational change in the TRP domain that, in turn, disrupts intersubunit interactions near the channel pore provoking its opening. B, in ChA, the intersubunit interactions near the gate are significantly debilitated (faint red cloud) by mutation Y981E destabilizing the closed state and the thermal energy suffices to open the channel gate. C, mutation V986I in ChA may strengthen the TRP domain/S4–S5 linker intrasubunit interactions (faint green cloud), which in turn may stabilize intersubunit interactions near Tyr981 stabilizing the closed state of the channel.
Acknowledgments
We thank D. Julius for providing the TRPM8 cDNA and F. Viana for TRPM8 antibody.
This work was supported in part by el Ministerio de Economía y Competitividad Grants BFU2012-39092-C02-01, CONSOLIDER-INGENIO 2010, CSD2008-00005 and la Generalitat Valenciana Grant PROMETEO/2014/011.
- TRP
- transient receptor potential
- ChA
- chimera A
- ChB
- chimera B
- ChC
- chimera C.
REFERENCES
- 1. Clapham D. E. (2003) TRP channels as cellular sensors. Nature 426, 517–524 [DOI] [PubMed] [Google Scholar]
- 2. Julius D. (2013) TRP channels and pain. Annu. Rev. Cell Dev. Biol. 29, 355–384 [DOI] [PubMed] [Google Scholar]
- 3. Bautista D. M., Siemens J., Glazer J. M., Tsuruda P. R., Basbaum A. I., Stucky C. L., Jordt S.-E., Julius D. (2007) The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448, 204–208 [DOI] [PubMed] [Google Scholar]
- 4. Dhaka A., Murray A. N., Mathur J., Earley T. J., Petrus M. J., Patapoutian A. (2007) TRPM8 is required for cold sensation in mice. Neuron 54, 371–378 [DOI] [PubMed] [Google Scholar]
- 5. Colburn R. W., Lubin M. L., Stone D. J., Jr., Wang Y., Lawrence D., D'Andrea M. R., Brandt M. R., Liu Y., Flores C. M., Qin N. (2007) Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379–386 [DOI] [PubMed] [Google Scholar]
- 6. Tsavaler L., Shapero M. H., Morkowski S., Laus R. (2001) Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 61, 3760–3769 [PubMed] [Google Scholar]
- 7. Jun J. H., Kang H. J., Jin M. H., Lee H. Y., Im Y. J., Jung H. J., Han S. W. (2012) Function of the cold receptor (TRPM8) associated with voiding dysfunction in bladder outlet obstruction in rats. Int. Neurourol. J. 16, 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parra A., Madrid R., Echevarria D., del Olmo S., Morenilla-Palao C., Acosta M. C., Gallar J., Dhaka A., Viana F., Belmonte C. (2010) Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat. Med. 16, 1396–1399 [DOI] [PubMed] [Google Scholar]
- 9. McKemy D. D., Neuhausser W. M., Julius D. (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58 [DOI] [PubMed] [Google Scholar]
- 10. Peier A. M., Moqrich A., Hergarden A. C., Reeve A. J., Andersson D. A., Story G. M., Earley T. J., Dragoni I., McIntyre P., Bevan S., Patapoutian A. (2002) A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715 [DOI] [PubMed] [Google Scholar]
- 11. Voets T., Droogmans G., Wissenbach U., Janssens A., Flockerzi V., Nilius B. (2004) The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 430, 748–754 [DOI] [PubMed] [Google Scholar]
- 12. Rohács T., Lopes C. M., Michailidis I., Logothetis D. E. (2005) PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8, 626–634 [DOI] [PubMed] [Google Scholar]
- 13. Daniels R. L., Takashima Y., McKemy D. D. (2009) Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J. Biol. Chem. 284, 1570–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yudin Y., Lukacs V., Cao C., Rohacs T. (2011) Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J. Physiol. 589, 6007–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bandell M., Dubin A. E., Petrus M. J., Orth A., Mathur J., Hwang S. W., Patapoutian A. (2006) High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nat. Neurosci. 9, 493–500 [DOI] [PubMed] [Google Scholar]
- 16. Pedretti A., Labozzetta A., Lo Monte M., Beccari A. R., Moriconi A., Vistoli G. (2011) Exploring the activation mechanism of TRPM8 channel by targeted MD simulations. Biochem. Biophys. Res. Commun. 414, 14–19 [DOI] [PubMed] [Google Scholar]
- 17. Voets T., Owsianik G., Janssens A., Talavera K., Nilius B. (2007) TRPM8 voltage sensor mutants reveal a mechanism for integrating thermal and chemical stimuli. Nat. Chem. Biol. 3, 174–182 [DOI] [PubMed] [Google Scholar]
- 18. Brauchi S., Orta G., Salazar M., Rosenmann E., Latorre R. (2006) A hot-sensing cold receptor: C-terminal domain determines thermosensation in transient receptor potential channels. J. Neurosci. 26, 4835–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brauchi S., Orio P., Latorre R. (2004) Clues to understanding cold sensation: thermodynamics and electrophysiological analysis of the cold receptor TRPM8. Proc. Natl. Acad. Sci. U.S.A. 101, 15494–15499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matta J. A., Ahern G. P. (2007) Voltage is a partial activator of rat thermosensitive TRP channels. J. Physiol. 585, 469–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. García-Sanz N., Valente P., Gomis A., Fernández-Carvajal A., Fernández-Ballester G., Viana F., Belmonte C., Ferrer-Montiel A. (2007) A role of the transient receptor potential domain of vanilloid receptor I in channel gating. J. Neurosci. 27, 11641–11650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valente P., García-Sanz N., Gomis A., Fernández-Carvajal A., Fernández-Ballester G., Viana F., Belmonte C., Ferrer-Montiel A. (2008) Identification of molecular determinants of channel gating in the transient receptor potential box of vanilloid receptor I. FASEB J. 22, 3298–3309 [DOI] [PubMed] [Google Scholar]
- 23. Doerner J. F., Hatt H., Ramsey I. S. (2011) Voltage- and temperature-dependent activation of TRPV3 channels is potentiated by receptor-mediated PI(4,5)P2 hydrolysis. J. Gen. Physiol. 137, 271–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie J., Sun B., Du J., Yang W., Chen H.-C., Overton J. D., Runnels L. W., Yue L. (2011) Phosphatidylinositol 4,5-bisphosphate (PIP2) controls magnesium gatekeeper TRPM6 activity. Sci. Rep. 146, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hirschler-Laszkiewicz I., Tong Q., Waybill K., Conrad K., Keefer K., Zhang W., Chen S. J., Cheung J. Y., Miller B. A. (2011) The transient receptor potential (TRP) channel TRPC3 TRP domain and AMP-activated protein kinase binding site are required for TRPC3 activation by erythropoietin. J. Biol. Chem. 286, 30636–30646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. García-Sanz N., Fernández-Carvajal A., Morenilla-Palao C., Planells-Cases R., Fajardo-Sánchez E., Fernández-Ballester G., Ferrer-Montiel A. (2004) Identification of a tetramerization domain in the C terminus of the vanilloid receptor. J. Neurosci. 24, 5307–5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gregorio-Teruel L., Valente P., González-Ros J. M., Fernández-Ballester G., Ferrer-Montiel A. (2014) Mutation of I696 and W697 in the TRP box of vanilloid receptor subtype I modulates allosteric channel activation. J. Gen. Physiol. 143, 361–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valente P., Fernández-Carvajal A., Camprubí-Robles M., Gomis A., Quirce S., Viana F., Fernández-Ballester G., González-Ros J. M., Belmonte C., Planells-Cases R., Ferrer-Montiel A. (2011) Membrane-tethered peptides patterned after the TRP domain (TRPducins) selectively inhibit TRPV1 channel activity. FASEB J. 25, 1628–1640 [DOI] [PubMed] [Google Scholar]
- 29. Liao M., Cao E., Julius D., Cheng Y. (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krieger E., Darden T., Nabuurs S. B., Finkelstein A., Vriend G. (2004) Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins 57, 678–683 [DOI] [PubMed] [Google Scholar]
- 32. Krieger E., Koraimann G., Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA: a self-parameterizing force field. Proteins 47, 393–402 [DOI] [PubMed] [Google Scholar]
- 33. Guerois R., Nielsen J. E., Serrano L. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387 [DOI] [PubMed] [Google Scholar]
- 34. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. (2005) The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 36. García-Martinez C., Humet M., Planells-Cases R., Gomis A., Caprini M., Viana F., De La Pena E., Sanchez-Baeza F., Carbonell T., De Felipe C., Pérez-Paya E., Belmonte C., Messeguer A., Ferrer-Montiel A. (2002) Attenuation of thermal nociception and hyperalgesia by VR1 blockers. Proc. Natl. Acad. Sci. U.S.A. 99, 2374–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Valero M., Morenilla-Palao C., Belmonte C., Viana F. (2011) Pharmacological and functional properties of TRPM8 channels in prostate tumor cells. Pflugers Arch. 461, 99–114 [DOI] [PubMed] [Google Scholar]
- 38. Voets T. (2012) in Reviews of Physiology, Biochemistry and Pharmacology Reviews of Physiology, Biochemistry and Pharmacology (Nilius B., Amara S. G., Gudermann T., Jahn R., Lill R., Offermanns S., Petersen O. H., eds) pp. 91–119, Springer, Berlin Heidelberg [Google Scholar]
- 39. Cao E., Liao M., Cheng Y., Julius D. (2013) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]