

Background: RhoGDIα regulates activity of Rho family small GTPases, which are recognized as important modulators of ENaC.
Results: RhoGDIα modulates Rac1 activity and partially participates in the effects of EGF on ENaC in the cortical collecting ducts.
Conclusion: RhoGDIα controls ENaC expression and activity via Rac1.
Significance: A novel mechanism for RhoGDIα-dependent regulation of ENaC is presented.
Keywords: Epidermal Growth Factor (EGF), Epithelial Cell, Epithelial Sodium Channel (ENaC), Hypertension, Ion Channel, Kidney, Nephrology, Ras-related C3 Botulinum Toxin Substrate 1 (Rac1), Rho GTPases, Sodium Channel
Abstract
The epithelial sodium channel (ENaC) is expressed in the aldosterone-sensitive distal nephron where it performs sodium reabsorption from the lumen. We have recently shown that ENaC activity contributes to the development of salt-induced hypertension as a result of deficiency of EGF level. Previous studies revealed that Rho GDP-dissociation inhibitor α (RhoGDIα) is involved in the control of salt-sensitive hypertension and renal injury via Rac1, which is one of the small GTPases activating ENaC. Here we investigated the intracellular mechanism mediating the involvement of the RhoGDIα/Rac1 axis in the control of ENaC and the effect of EGF on ENaC in this pathway. We demonstrated that RhoGDIα is highly expressed in the cortical collecting ducts of mice and rats, and its expression is down-regulated in Dahl salt-sensitive rats fed a high salt diet. Knockdown of RhoGDIα in cultured cortical collecting duct principal cells increased ENaC subunits expression and ENaC-mediated sodium reabsorption. Furthermore, RhoGDIα deficiency causes enhanced response to EGF treatment. Patch clamp analysis reveals that RhoGDIα significantly decreases ENaC current density and prevents its up-regulation by RhoA and Rac1. Inhibition of Rho kinase with Y27632 had no effects on ENaC response to EGF either in control or RhoGDIα knocked down cells. However, EGF treatment increased levels of active Rac1, which was further enhanced in RhoGDIα-deficient cells. We conclude that changes in the RhoGDIα-dependent pathway have a permissive role in the Rac1-mediated enhancement of ENaC activity observed in salt-induced hypertension.
Introduction
Epithelial sodium channels (ENaC)3 are responsible for the fine-tuning of Na+ reabsorption in the kidney and are regulated by various stimuli including hormones and growth factors. Among these, epidermal growth factor (EGF) and its family members are recognized as important mediators of ENaC activity involved in maintaining sodium homeostasis in the kidney and particularly in the collecting ducts (1). For instance it was shown that EGF reduces Na+ transport in the isolated and perfused rabbit cortical ducts (2–5) and immortalized renal epithelial cells (6–9).
Previously we demonstrated the biphasic effect of EGF, TGF-α, heparin-binding EGF and amphiregulin on activity of ENaC in the cultured mouse cortical collecting duct (CCD) principal cell line, mpkCCDc14. Basolateral application of these endocrine factors acutely produced a short term increase in ENaC-mediated transepithelial current and then caused long term current reduction (10). A similar observation was reported in A6 amphibian kidney cells; acute treatment with EGF and TGF-α enhanced ENaC activity by increasing channel open probability, whereas continuous treatment resulted in a decrease of the number of channels in the plasma membrane and, therefore, inhibited the current (11). Importantly, we recently demonstrated that the deficiency of renal cortical EGF promotes ENaC-mediated Na+ reabsorption in the CCD and contributes to the development of hypertension in Dahl salt-sensitive (SS) rats (12). However, specific molecular mechanisms mediating effects of EGF on ENaC are not clear yet.
Small GTPases-molecular switches that control the activity of cellular and membrane proteins, including ion channels (13), are well known regulators of ENaC. It was previously reported that several small G proteins in Rho, Ras, and Rab families are able to regulate ENaC activity via different mechanisms (14–21). Small GTPases cycle between inactive GDP-bound and active GTP-bound states; the exchange between G-protein-bound GDP to GTP is catalyzed by guanine nucleotide exchange factors (GEFs), whereas termination of signaling is performed by GTPase-activating proteins which induce GTP hydrolysis (22). Another group of proteins regulating activity of small G proteins is guanine dissociation inhibitors (GDIs), which bind certain G proteins and keep them in an inactive form preventing dissociation of GDP (23). RhoGDIα is one of such factors that binds to the Rho family members RhoA, Rac1, and Cdc42 and keeps them in an inactive state, preventing their activation by GEFs (24, 25). The importance of RhoGDIα for the kidney was demonstrated in mice lacking this protein (Arhgdia−/− mice), where renal abnormalities, including heavy albuminuria and podocyte damage, were associated with increased Rac1 activity and mineralocorticoid receptor signaling (26, 27). Further studies demonstrated the key role of Rac1 in modulating salt susceptibility in mice lacking RhoGDIα. Using SS rats the authors found that a high salt diet caused Rac1 up-regulation in the kidney, whereas Rac1 inhibition prevented hypertension and renal damage (28).
Our previous studies revealed that among Rho family members, Cdc42 has no effect on ENaC current density (29), whereas RhoA and Rac1 were able to increase the channel activity (14, 16, 29–31). Furthermore, our recent findings indicate that EGF had no effect in Rac1-deficient principal cells (32). Thus, it appears that the small GTPase Rac1, which has been shown to be implicated in many renal and cardiovascular diseases (27, 28, 33), is a critical protein in transmission of the signal from EGF to ENaC. However, the mechanistic link between EGF, RhoGDIα, small GTPases in Rho family, and ENaC was unclear.
In this study we investigated if RhoGDΙα is involved in the control of ENaC activity and EGF-dependent regulation of this channel. This hypothesis might explain intracellular processes underlying abnormal ENaC regulation in some pathological conditions such as salt-sensitive hypertension.
EXPERIMENTAL PROCEDURES
Animals
Experiments were performed on C57BL/6J mice and Rapp Dahl salt-sensitive rats (SS/JrHsdMcwi). The mice were obtained from Harlan Laboratories and kept on a standard Harlan Teklad TD.96208 diet until the age of 8 weeks. SS rats have been inbred for >50 generations at the Medical College of Wisconsin. SS rats are a salt-sensitive strain that develops hypertension when fed a high salt diet. To investigate the effect of high salt consumption in these rats, chronic blood pressure monitoring in 8-week-old animals was performed using Data Sciences International (New Brighton, MN) telemetry as previously described (34). After transmitter implantation the rats were allowed to recover for at least 3 days. After an additional 3 days on a 0.4% NaCl diet the salt content of the chow was either maintained at 0.4% NaCl in one group or increased to 4.0% NaCl, and the rats were maintained on these diets for an additional 3 weeks. Both chows were obtained from Dyets Inc. (Bethlehem, PA). Animal use and welfare procedures adhered to the NIH Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee.
Cell Cultures and Plasmids
mCCDcl1 cells (35) were kindly provided by Dr. Bernard Rossier (University of Lausanne, Lausanne, Switzerland) and were maintained as previously described (36, 37) in growth media composed of equal volumes DMEM/Ham's F-12, 100 μg/ml penicillin/streptomycin (Invitrogen), 2% FBS (Invitrogen), and 60 nm Na+ selenate, 5 μg/ml transferrin, 50 nm dexamethasone, 1 nm triiodothyronine, 10 ng/ml EGF, 5 μg/ml insulin (Sigma). Cells were grown in 5%CO2, 95% air atmosphere incubator at 37 °C. M-1 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained under the same conditions.
Chinese hamster ovarian (CHO) cells (ATCC) were maintained under standard culture conditions (DMEM, 10% FBS, 1× penicillin-streptomycin, 37 °C, 5% CO2) and transfected using the Polyfect reagent (Qiagen, Valencia, CA) as previously described (38, 39). For expression of mouse ENaC in CHO cells, subunit cDNA transfection ratios of 1:1:1 were used with 0.3 μg of each cDNA/35-mm culture dish. The plasmids encoding α-, β-, and mouse γ-ENaC have been previously described (15). To define successfully transfected cells, 0.5 μg of a cDNA encoding green fluorescent protein (GFP) was added to the cDNA mix. RhoA, Rac1, and Cdc42 plasmids were obtained from the University of Missouri-Rolla cDNA Resource Center. Myc-tagged RhoGDIα plasmid was kindly provided by Dr. Mark Shapiro (University of Texas Health Science Center, San Antonio, TX).
Generation of Stable RhoGDIα-deficient Cell Lines
To generate a RhoGDIα knockdown in mCCDcl1 and M-1 cells, RhoGDIα short hairpin shRNA pLKO.1 plasmids (catalog no. TRCN0000106160 and TRCN0000106161, respectively) were purchased from OpenBiosystems, Thermo Scientific. After transduction with jetPRIME® reagent (Polyplus-transfection SA, Illkirch, France), a stable cell line expressing the shRNA was isolated via puromycin selection. In short, cells were seeded in 6-well cluster plates (Corning) and grown to ∼90% confluence. 3 μg of plasmid DNA was mixed with 200 μl of jetPRIME buffer and 4 μl of jetPRIME reagent. Cells were overlaid with this DNA mix for another 48 h. Selection of successfully transfected cells was performed by applying 10 μg/ml puromycin to the media.
Transepithelial Current Measurements
mCCDcl1 cells were seeded in the confluence to ∼106 on permeable membranes (Transwell, Costar; 0.4-μm pore, 24-mm diameter) and grown in the media described above. In 7–10 days the cells polarize and form a monolayer with high resistance and avid Na+ flux. Transepithelial Na+ current across the mCCDcl1 cell monolayer was measured by a Millicell Electrical Resistance System with dual Ag+/AgCl pellet electrodes (Millipore Corp., Billerica, MA) and calculated using Ohm's law. 18 h before the experiments growth media was changed to FBS- and supplements-free media. To confirm the net Na+ transport through ENaC, 10 μm amiloride was added to the apical surface of the monolayer at the end of each experiment. EGF (E4127) was obtained from Sigma, and Y27632 (#1254) was from Tocris Bioscience (Bristol, UK).
Patch Clamp Analysis
Whole cell current recordings of mouse ENaC expressed in CHO cells were made under voltage clamp conditions using standard protocols (15, 29). Cells were clamped to a 40-mV holding potential with voltage ramps (500 ms) from 60 mV down to −100 mV used to elicit current. Pipette solution for whole-cell configuration was 120 mm CsCl, 5 mm NaCl, 2 mm MgCl2, 5 mm EGTA, 2 mm Mg-ATP, 0.1 mm GTP, and 10 mm HEPES (pH 7.4). Bath solution was 150 mm NaCl, 1 mm CaCl2, 2 mm MgCl2, and 10 mm HEPES (pH 7.4). ENaC activity was assessed as the amiloride-sensitive current density at −80 mV. Whole-cell capacitance, on average 6–10 picofarads, and series resistances, on average 2–5 megaohms, were compensated.
Cell-attached measurements of single channel ENaC activity were performed on wild type and RhoGDIα-overexpressing mCCDcl1 cells. Cells were transfected with RhoGDIα and GFP encoding plasmids as described above for CHO cells. Individual GFP-positive cells were considered as successfully transfected and used for patch clamp analysis as described earlier (38). Briefly, single-channel signal was registered with an Axopatch amplifier (Molecular Devices, Sunnyvale, CA), filtered with an 8-pole Bessel filter LPF-8 (Warner Inst., Hamden, CT) at 0.2 kHz, and acquired with a Digidata 1440A to a PC running the pClamp 10.2 suite of software (Molecular Devices). Typical bath solution was 150 mm NaCl, 1 mm CaCl2, 2 mm MgCl2, 10 mm HEPES (pH 7.4). Pipette solution for cell attached configuration was 140 mm LiCl, 2 mm MgCl2, and 10 mm HEPES (pH 7.4).
Western Blotting
mCCDcl1 or M-1 cells were washed twice in PBS and lysed in a buffer as described previously (29, 32). Equal amounts (20 μg) of proteins were separated by using 10–20% SDS-PAGE and were then electrophoretically transferred onto nitrocellulose membrane (Millipore), immunoblotted with the appropriate antibody, and visualized by ECL (Amersham Biosciences). Antibodies for RhoGDIα were from Santa Cruz (sc-359), and antibodies for α-, β-, and γ-ENaC (catalog nos. SPC-403D, SPC-404D, and SPC-405D, respectively) were from StressMarq Biosciences Inc. (Victoria, BC, Canada). Active Rac1 detection was performed using the kit purchased from Enzo Life Science (ADI-EKS-450) according to the manufacturer's protocol. The assay uses a GST fusion protein containing the p21 binding domain of human p21-activated kinase 1 (Pak1) to affinity-precipitate active Rac1 (GTP-Rac1) from cell lysates. The GST-Pak binding domain fusion protein (∼35 kDa) was incubated with cell lysate and an immobilized glutathione disc. The pulled down active or GTP-Rac1 was detected by Western blot analysis using a provided specific monoclonal Rac1 antibody.
For renal tissue analysis, rat kidneys were flushed and isolated under isoflurane anesthesia. The approximate apical kidney sections corresponding to cortex were carved (∼1 g) and then diced into small pieces with a razor blade. Samples were pulse-sonicated in gentle lysis buffer with protease inhibitor mixture (Roche Applied Science) for 10 s and spin-cleared at 10,000 × g for 10 min.
Immunohistochemistry
The C57BL/6J mouse and SS rat kidneys were fixed in zinc formalin and processed for paraffin embedding. The kidney sections were cut at 4 μm, dried, and deparaffinized for subsequent labeled streptavidin-biotin immunohistochemistry. After deparaffinization, the slides were treated with a citrate buffer (pH 6) for a total of 35 min. The slides were blocked with a peroxidase block (DAKO), avidin block (Vector Labs), biotin block (Vector Labs), and serum-free protein block (DAKO). Tissue sections were incubated for 90 min in a 1:200 concentration of anti-RhoGDIα (sc-359, Santa Cruz). Mouse kidney sections were also stained with non-immune IgG as a negative control and anti-aquaporin-2 antibodies to demonstrate RhoGDIα localization in the CCDs (sc-2027 and sc-9882, Santa Cruz, respectively). Secondary detection was performed with goat anti-rabbit biotinylated IgG (Biocare) followed by streptavidin horseradish peroxidase (Biocare) and visualized with diaminobenzidine (DAKO). All slides were counterstained with a Mayer hematoxylin (DAKO), dehydrated, and mounted with permanent mounting media (Sakura). RhoGDIα signal intensity was determined in CCDs using ImageJ 1.84D software package. Mean signal intensities in each CCD (background level was subtracted) were averaged for the experimental groups.
Statistics
All summarized data are reported as the mean ± S.E.; statistical analyses were performed using the Mann-Whitney test with Bonferroni correction. Differences were considered statistically significant at p < 0.05 (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
RESULTS
RhoGDIα Abundance in Murine CCDs
RhoGDIα is a ubiquitous protein expressed in many cell types (24). Initial experiments tested whether RhoGDIα is expressed in mouse and rat CCDs, a nephron segment with high activity of ENaC (40). To study RhoGDIα abundance we used kidneys isolated from 8-week-old C57BL/6J mice. Shown in Fig. 1A are images taken from two consecutively cut slices of a kidney immunohistochemically stained for RhoGDIα (shown in brown) at 20× and 40× magnifications. Negative control (control; stained with secondary antibodies in the absence of primary antibodies) is also shown. Additional negative control experiments (stained without primary or secondary antibodies) also did not reveal any staining (data not shown). As seen from Fig. 1A, RhoGDIα is highly expressed in the CCDs of C57BL/6J mice compared with other nephron segments. Fig. 1B represents additional negative control stained with non-immune IgG and secondary antibodies and shows the absence of any significant signal in the cortex. To ensure that the RhoGDIα signal observed in Fig. 1A was localized in CCD, we performed double staining of RhoGDIα and aquaporin-2, a marker for collecting duct principal cells. As shown in Fig. 2C both proteins are co-localized in CCDs.
FIGURE 1.

RhoGDIα abundance in the kidney of C57BL/6J mouse shown at ×20 and ×40 magnificence. A, consecutive slices were used to demonstrate control immunohistochemical staining without primary antibodies (left image) and staining with anti-RhoGDIα antibodies (central and right images). High expression of RhoGDIα was observed in the CCDs. B, negative control image of a mouse kidney stained with non-immune IgG and secondary antibodies. C, double staining of RhoGDIα (brown) and aquaporin-2 (red) demonstrating their colocalization in the CCDs. Scale bars are shown.
FIGURE 2.
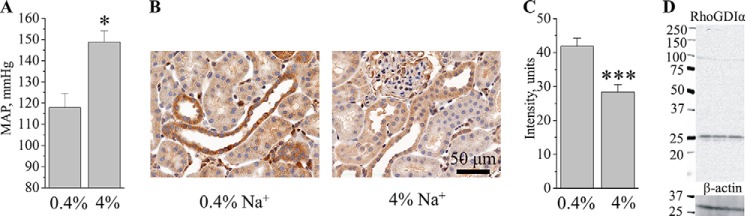
RhoGDIα abundance in the kidney of Dahl SS rats on normal (0.4%) and high (4%) salt diets. A, mean arterial pressure (MAP) measured with telemetry in SS rats fed a 0.4 or 4.0% Na+ diet for 3 weeks. B, representative immunohistochemical images of the kidneys from SS rats fed low and high salt diets as shown in A. A scale bar is shown. Magnification was 40×. C, summary graph of RhoGDIα signal intensity in the SS rat cortical collecting ducts. Number of animals in each group = 3. D, a representative Western blot of RhoGDIα in SS rat renal cortex demonstrates specificity of the used antibodies. *, p < 0.05; ***, p < 0.001 versus SS rats fed a low salt diet.
Further experiments tested RhoGDIα expression in Dahl SS rats. SS rats develop severe hypertension and renal damage when fed a high salt diet (41–43), and Rac1 hyperactivation was found to be critical in these processes (28). Similar to previously published data (34, 44, 45), changing of the diet from 0.4% to 4% resulted in the development of hypertension. As assessed by telemetry, the mean arterial pressure after 3 weeks on diets was 148.8 ± 5.3 and 117.9 ± 6.7 mm Hg in rats fed high and low salt diets, respectively (Fig. 2A). Immunohistochemistry analysis revealed that SS rats after 3 weeks on a high salt diet (4%) exhibit lower RhoGDIα abundance than on a normal salt diet (Figs. 2, B and C). Fig. 2D represents a Western blot analysis of RhoGDIα signal in the cortical lysate collected from three SS rats fed a 0.4% diet. The image demonstrates specificity of the used antibodies, which detect only one significant band at ∼25 kDa.
Effect of RhoGDIα Deficiency on ENaC-dependent Current Formation
To investigate the physiological significance of RhoGDIα, we generated a stable knockdown of this protein in mCCDcl1 cells. These cells were derived from mouse CCD principal cells and retain its characteristics, including morphology and CCD antigens (35, 36, 46, 47). We used short hairpin RNA (shRNA)-mediated silencing of RhoGDIα mRNA in the cells, and one successful clone was used for further experiments. Western blot analysis confirmed that utilization of control shRNA did not affect RhoGDIα level, whereas anti-RhoGDIα significantly reduced the abundance of the protein to ∼13% compared with the control cells (Figs. 3, A and B).
FIGURE 3.
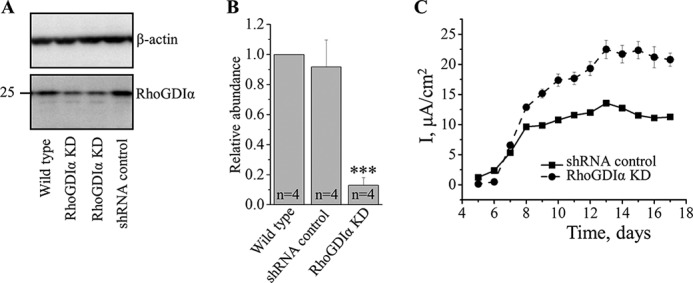
RhoGDIα knocked down mCCDcl1 cells develop increased ENaC-dependent current. Western blot analysis (A) and summary graphs (B) of RhoGDIα expression in wild type, RhoGDIα knockdown (KD), and control shRNA-expressing cells. Protein signal intensities were normalized to β-actin level. C, time line of transepithelial current development in RhoGDIα knockdown and control mCCDcl1 cells grown on permeable membranes (n = 6 in each group). ***, p < 0.001 versus wild type and cells expressing scrambled shRNA.
Control cells, when seeded onto permeable membranes, form an epithelium-like monolayer and develop amiloride-sensitive sodium current of ∼10 μA/cm2 within 12–14 days after seeding. These basal values were similar to those reported previously (10, 48, 49). RhoGDIα-deficient cells generated a significantly higher current of ∼20 μA/cm2 (Fig. 3C). These data demonstrate that RhoGDIα exhibits a suppressive role in formation of ENaC-mediated transepithelial flux.
Effect of RhoGDIα Deficiency on ENaC Subunits Abundance in M-1 Cells
M-1 is a commercially available cell culture line retaining many characteristics of cortical collecting duct cells including morphology and CCD antigens (50). The line was also used to generate RhoGDIα-knocked down immortalized cells for independent study on ENaC subunit abundance. Fig. 4A illustrates Western blot analysis of protein levels in wild type, RhoGDIα-deficient, and control shRNA-expressing cells. As summarized in Fig. 4B, RhoGDIα knockdown causes significant increases in expression of α-, β-, and γ-ENaC subunits. These data are in accord with the assessment of transepithelial currents shown in mCCDcl1 cells (Fig. 3).
FIGURE 4.
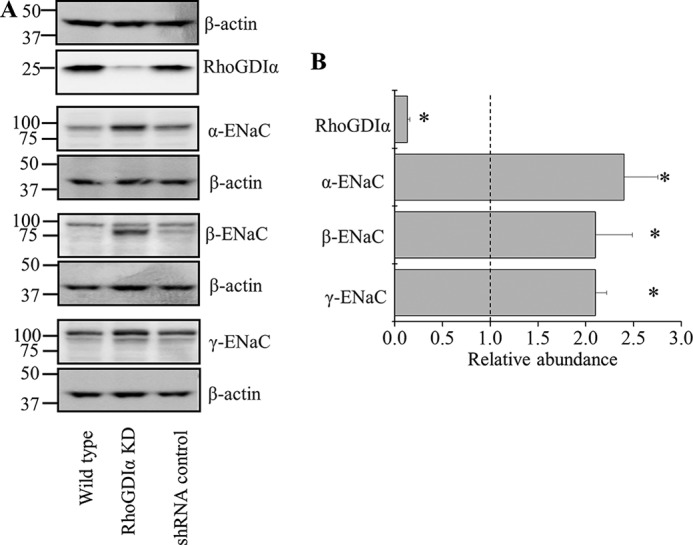
RhoGDIα knocked down M-1 cells exhibit increased ENaC subunits abundance. A, Western blot analysis of RhoGDIα and ENaC subunits expression in wild type, RhoGDIα knockdown (KD), and control scrambled shRNA-expressing cells. B, summary graph of RhoGDIα and α-, β-, and γ-ENaC subunit expression in RhoGDIα knockdown cells relatively to control cells with scrambled shRNA. Protein signal intensities were initially normalized to the β-actin level. Experiments were repeated three times. *, p < 0.05 versus control cells.
Effect of EGF Application on ENaC Activity in the RhoGDIα-deficient Cells
Our previous studies show that basolateral application of EGF on polarized mouse-collecting duct cells up-regulates ENaC activity (10), and this effect was blunted in Rac1 knockdown cells (32). We tested the effect of EGF in the mCCDcl1 cells lacking RhoGDIα (cell line described above). Application of EGF (10 ng/ml) to the basolateral side increased amiloride-sensitive flux in the control cells. However, in the RhoGDIα-deficient cells response to EGF application was significantly enhanced compared with control cells (Fig. 5A). These findings reveal that RhoGDIα participates in the regulation of ENaC activity by EGF. However, a 2-h treatment with EGF did not change the abundance of RhoGDIα proteins in control mCCDcl1 cells (Fig. 5B).
FIGURE 5.
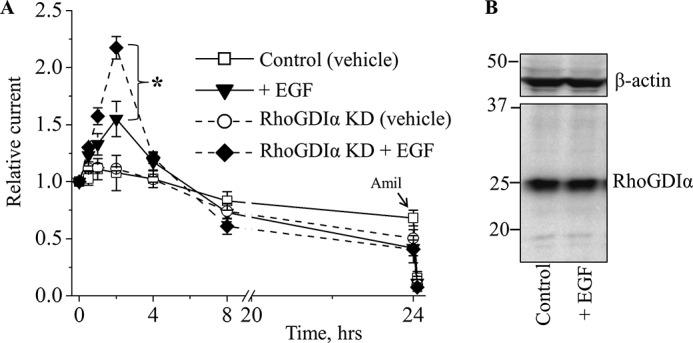
RhoGDIα-deficient cells exhibit enhanced response to EGF treatment. A, summary graph of relative transepithelial current in control and RhoGDIα knockdown (KD) mCCDcl1 principal cells in response to EGF (10 ng/ml). mCCDcl1 cells were serum-starved overnight. EGF and vehicle (control) were added basolaterally at time 0, and current was normalized to the starting level. Amiloride (10 μm; arrow) was added to the apical membrane at the end of experiment. n = between 6 and 12 in each group; *, p < 0.05. B, a representative Western blot (n = 4) of RhoGDIα in mCCDcl1 principal cells not treated and treated with EGF (10 ng/ml; 2 h).
Effect of EGF Application on ENaC Activity in the RhoGDIα-overexpressing Cells
As the transepithelial current measurements indicate that RhoGDIα deficiency produces an enhanced response to EGF in mCCDcl1 cells, we tested the effect of EGF on RhoGDIα-overexpressing cells. For this aim we transfected mCCDcl1 cells with RhoGDIα- and GFP-encoding cDNA plasmids. Because the transfection was performed on the formed epithelial monolayer, its efficiency was low for assessment of transepithelial current but allowed the use of individual transfected cells for patch clamp analysis. Fig. 6A illustrates representative ENaC activity traces recorded in cell attached configuration from apical membranes of the cells transfected with GFP only or with RhoGDIα and treated with vehicle or EGF (50 ng/ml; 2 h). As summarized on Fig. 6B, cotransfection of mCCDcl1 cells with RhoGDIα significantly decreased ENaC activity, which represents direct evidence of the critical role of this protein in the control of ENaC activity. Treatment with EGF increased the activity of ENaC in both control and RhoGDIα-overexpressing cells.
FIGURE 6.
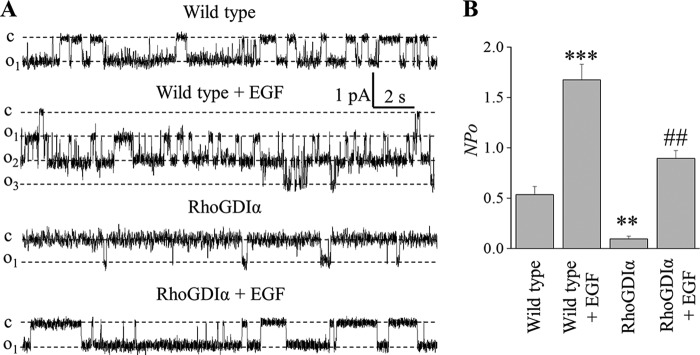
EGF stimulates ENaC activity in wild type and RhoGDIα-overexpressing cells. A, representative current traces from cell-attached patches that were made on the apical membrane of wild type and RhoGDIα-overexpressing mCCDcl1 cells treated with vehicle or 50 ng/ml EGF (2 h). These patches were held at a −40-mV test potential during the course of the experiment. B, summary graph of ENaC activity (NPo), n = 5 in each group. ** and *** p < 0.01 and 0.001 versus wild type; ##, p < 0.01 versus RhoGDIα-overexpressing cells.
Effect of RhoGDIα on RhoA- and Rac1-dependent Up-regulation of ENaC Activity
RhoGDIα controls the Rho family proteins by keeping them in an inactive state. Our previous studies in the CHO expression system revealed that neither wild type nor constitutively active Cdc42 overexpression affects ENaC activity (29). Wild type and constitutively active forms of RhoA and Rac1 were demonstrated to increase ENaC current density (29, 30).
Here we used CHO cells for transfection with RhoGDIα to investigate its effect on ENaC activity. Overexpression of RhoGDIα with all three mouse α-, β-, and γ-ENaC subunits significantly decreased amiloride-sensitive ENaC current density (Fig. 7). Moreover, co-transfection of RhoGDIα with RhoA or Rac1 precluded ENaC up-regulation by these small G proteins. We assume that the high concentration of RhoGDIα in the cytosol keeps Rho proteins in inactive state and prevents augmentation of ENaC activity.
FIGURE 7.
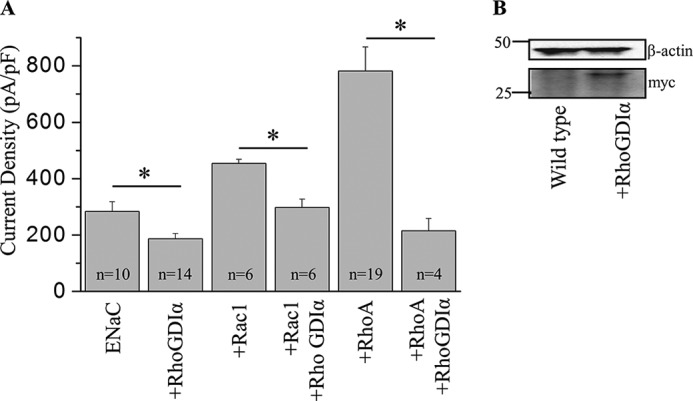
RhoGDIα inhibits Rac1 and Rho-dependent increase of ENaC activity. A, summary graph of amiloride-sensitive current density at −80 mV for CHO cells expressing mouse ENaC alone or co-expressed with RhoGDIα in the presence or absence of Rac1or RhoA. Whole-cell currents were evoked with a voltage ramp (60 to −100 mV from a holding potential of 40 mV). The number of observations for each group is shown. *, p < 0.05 versus the corresponding group without RhoGDIα. pF, picofarads. B, Western blot demonstrates expression of myc-tagged RhoGDIα in CHO cells transiently expressed with corresponding plasmid.
Rac1 Involvement in the Effects of EGF on ENaC Sodium Reabsorption in Native CCD Cell Monolayer
RhoGDIα has inhibitory effects on the small G proteins of the Rho family: RhoA, Rac1, and Cdc42. We have shown in the heterologous expression system that Cdc42 does not affect ENaC activity, whereas RhoA and Rac1 up-regulate it. We further tested the role of RhoA and Rac1 in the mCCDcl1 principal cell line. To determine if RhoA participated in the EGF-dependent regulation of ENaC activity, we used Rho kinase inhibitor Y27632 (51), which is a downstream target of RhoA (30, 52). These experiments were designed similarly to data shown on Fig. 5, but control and RhoGDIα-deficient cells were preincubated for 1 h with 2 μm Y27632 (applied bilaterally). However, Y27632 had no effect on either EGF-treated control or RhoGDIα-deficient cells (Fig. 8).
FIGURE 8.
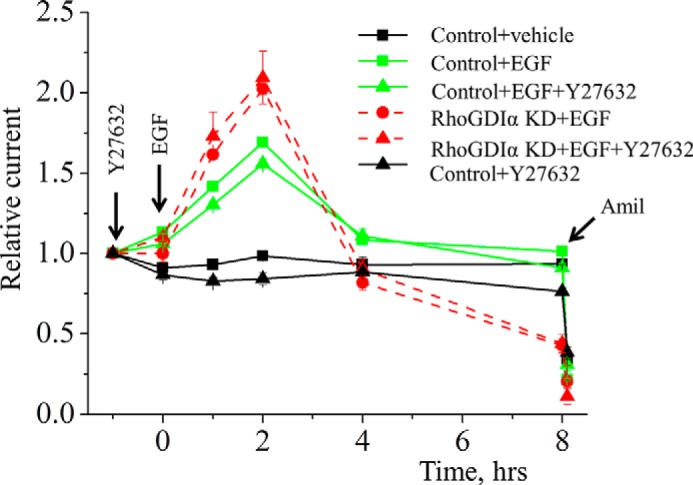
RhoA is not involved in EGF-dependent regulation of ENaC in mCCDcl1 cells. RhoA inhibition with Y27632 (1 h of pretreatment with 2 μm Y27632) does not affect ENaC response to EGF application as measured in mCCDc11 monolayer. EGF (10 ng/ml) and vehicle (control) were added basolaterally at time 0, and current was normalized to the starting level (1 h before EGF). Amiloride (Amil; 10 μm; arrow) was added to the apical membrane at the end of experiment. n = 6 in each group.
We further tested the levels of active Rac1 in mCCDcl1 cells before and after EGF treatment (10 ng/ml; 2 h). Detection of the GTP-bound form of Rac1 revealed a significantly increased active Rac1/total Rac1 ratio after EGF application compared with vehicle-treated control cells. Moreover, cells lacking RhoGDIα demonstrate an increased active Rac1 level over control cells with similar levels in EGF-treated and -untreated cells (Fig. 9). Control experiments with the application of GDP and GTPγs confirmed reduction and increase of Rac1 activity, respectively. Thus, these data demonstrate that in CCD cultured cells Rac1 activity is mediated by EGF, and RhoGDIα might be involved in this mechanism.
FIGURE 9.
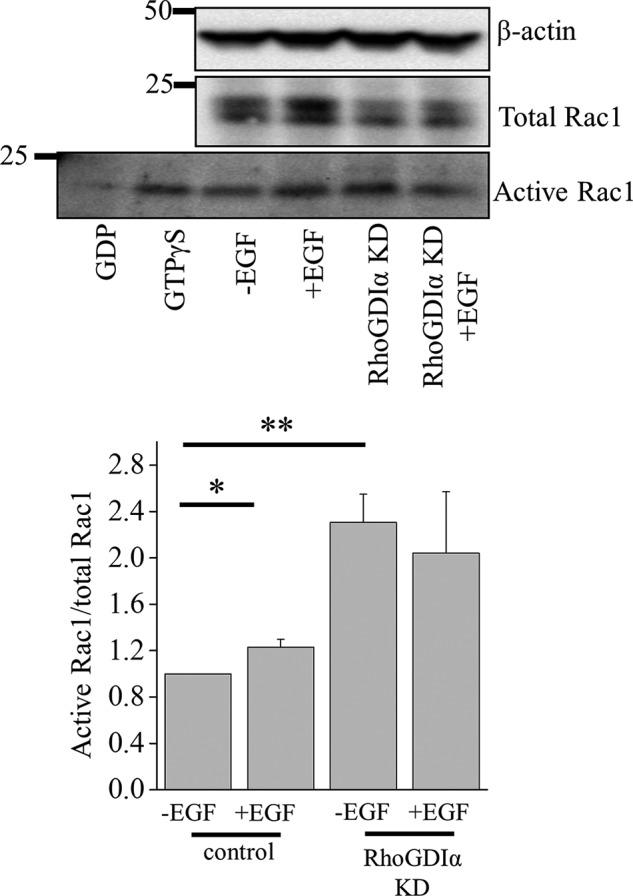
EGF up-regulates Rac1 activity in cortical collecting duct principal cells. Western blot analysis of active and total Rac1 abundance in mCCDcl1 cell lysate. Treatments with GDP and GTPγS were used as negative and positive controls, respectively. Below is a summary graph of the active Rac1/total Rac1 ratio in wild type and RhoGDIα knockdown (KD) cells after a 2-h treatment with EGF (10 ng/ml). *, p < 0.05; **, p < 0.05 versus control cells without EGF treatment.
DISCUSSION
We demonstrate that RhoGDIα modulates Rho family protein Rac1 in the CCD. This interaction seems to play a permissive role in EGF-dependent regulation of Rac1 and ENaC. Fig. 10 provides schematic illustration of this regulation. It appears from our data that EGF affects Rac1 activity, whereas RhoGDIα modulates bioavailability of Rac1 for activation. Regulation of Rac1 seems to be critical for physiological regulation of ENaC. Rac1 might mediate its effects on ENaC either through the MAPK pathway (11, 53) or WAVE (the WASP (Wiskott-Aldrich syndrome protein) family of verprolin homologous) proteins (14) and corresponding changes in the actin cytoskeleton (54–58). In addition to its role in various intracellular pathways, Rac1 is one of the key subunits of the NADPH oxidase complex and is involved in the production of reactive oxygen species (32). At least the acute effects of EGF correlate with reactive oxygen species production as pretreatment with the nonselective NADPH oxidase activity inhibitor apocynin blunted both generation of reactive oxygen species and an increase in ENaC-mediates current in response to EGF (32). An imbalance of NO, O2⨪, and H2O2 within the kidney has important consequences on Na+ homeostasis and blood pressure (59–61). Most likely, the increase in hydrogen peroxide levels mediates this effect as it was shown that H2O2 is critical for ENaC activity (62–65).
FIGURE 10.
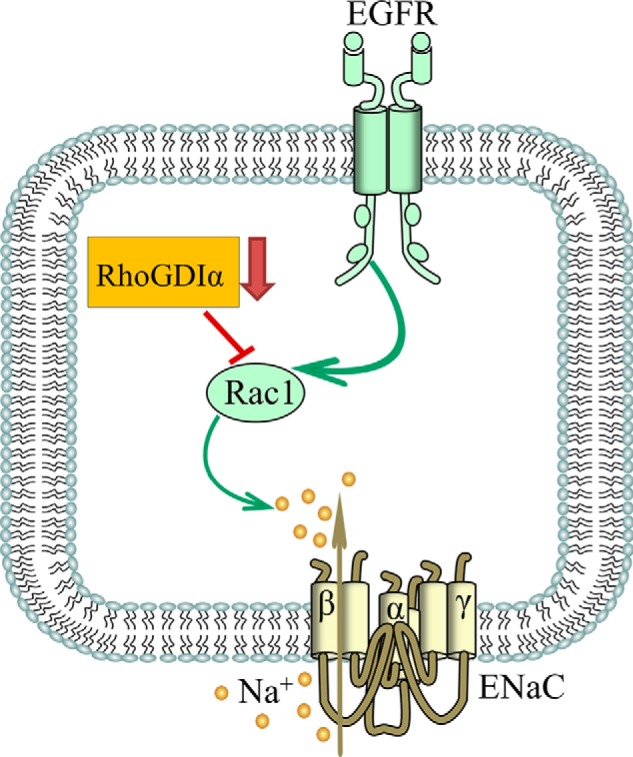
Schematic illustration of the proposed role for RhoGDIα and Rac1 in the effect of EGF on ENaC.
The mechanism described in this manuscript is specific to Rac1. Our data show that two other members of Rho family identified in CCD, RhoA and Cdc42, are not involved in EGF-mediated regulation of ENaC activity. We previously reported that Cdc42 does not alter ENaC activity when overexpressed in the CHO expression system (29). Interestingly, although RhoA was able to increase ENaC current density in CHO cells, the protein does not affect flow-mediated activation of ENaC (52). Similar results were observed in whole animals under pathological conditions (27). RhoGDIα deficiency in Arhgdia−/− mice led to renal abnormalities that were accompanied with increased levels of active Rac1 but not RhoA. To investigate the physiological role of Rac1 and Rho kinase in the development of abnormalities in these mice, small GTPase activity was inhibited with NSC23766 and fasudil, respectively. Administration of NSC23766 (but not fasudil) significantly reduced mean blood pressure, albuminuria, and kidney damage concomitantly with repression of renal Rac1 activity (27). Studies by Fujita and co-workers (28) showed that high salt loading activates Rac1 in the kidneys in rodent models of salt-sensitive hypertension, leading to blood pressure elevation and renal injury via a mineralocorticoid receptor (MR)-dependent pathway. A high salt diet caused renal Rac1 up-regulation in SS rats and down-regulation in salt-insensitive Dahl rats. Despite a reduction of serum aldosterone levels, salt-loaded SS rats showed increased MR signaling in the kidneys, and Rac1 inhibition prevented hypertension and renal damage with MR repression. Shibata et al. (28) further demonstrated in aldosterone-infused rats as well as adrenalectomized SS rats with aldosterone supplementation that salt-induced Rac1 and aldosterone acted interdependently to cause MR over-activity and hypertension. Recent review articles by Nagase and Fujita (66, 67), who pioneered this area of research, nicely summarized the role of RhoGDIα and Rac1-MR signaling in cardiorenal diseases and, specifically, mechanisms implicated in the pathogenesis of salt-sensitive hypertension and kidney injury.
Recent studies also identified that mutations in ARHGDIA cause nephrotic syndrome in humans and this effect is mediated via Rac1 and Cdc42 but not RhoA (68). Another study using whole exome sequencing revealed that RhoGDIα is implicated in congenital nephrotic syndrome (69).
In addition to identifying a critical role of RhoGDIα in control of ENaC activity, our data also demonstrate that EGF at least partially mediates these effects. As followed from our experiments, RhoGDIα does not directly mediate the effect of EGF on ENaC but has a permissive influence. In addition there is a possibility that other GDIs controlling Rac1 activity might be involved in effects of EGF. Besides, the critical role of GEFs in the control of kidney function and specifically ENaC activity was demonstrated. For instance we demonstrated that Rac1 GEF βPix is involved in the control of ENaC (29, 70). Thus, precise balance between small GTPases in the Rho family and GDIs and GEFs controlling their activity might also GEF critical for EGF-mediated effects on ENaC.
The decreased RhoGDIα abundance discovered on a high salt diet (Figs. 2) could play a permissive role for Rac1 activation described is SS rats (28). As high salt consumption in SS rats leads to cortical EGF deficiency (12), this impairs the chronic suppression of ENaC-mediated reabsorption in CCD (10) and results in increased fluid retention in the body and blood pressure. In summary, these findings provide a mechanistic insight on intracellular machinery underlying the contribution of RhoGDIα to the regulation of ENaC and the development of salt-sensitive hypertension.
Acknowledgments
We recognize Christine B. Duris and Glenn R. Slocum (both of the Medical College of Wisconsin) for excellent technical assistance with immunohistochemistry experiments. Mohamad M. Hejazi (Alfaisal University, Saudi Arabia) is also appreciated for help with transepithelial current measurements.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL108880 (to A. S.) and K99 HL116603 (NHLBI). This work was also supported by National Kidney Foundation Grant IG1724 and a Medical College of Wisconsin Research Affairs Committee pilot grant (to T. S. P.).
- ENaC
- epithelial Na+ channel
- CCD
- cortical collecting duct
- GEF
- guanine nucleotide exchange factor
- GDI
- guanine dissociation inhibitor
- RhoGDIα
- MR, mineralocorticoid receptor
- SS
- salt-sensitive
- GTPγS
- guanosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Staruschenko A., Palygin O., Ilatovskaya D. V., Pavlov T. S. (2013) Epidermal growth factors in the kidney and relationship to hypertension. Am. J. Physiol. Renal Physiol. 305, F12–F20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muto S., Furuya H., Tabei K., Asano Y. (1991) Site and mechanism of action of epidermal growth factor in rabbit cortical collecting duct. Am. J. Physiol. 260, F163–F169 [DOI] [PubMed] [Google Scholar]
- 3. Vehaskari V. M., Hering-Smith K. S., Moskowitz D. W., Weiner I. D., Hamm L. L. (1989) Effect of epidermal growth factor on sodium transport in the cortical collecting tubule. Am. J. Physiol. 256, F803–F809 [DOI] [PubMed] [Google Scholar]
- 4. Vehaskari V. M., Herndon J., Hamm L. L. (1991) Mechanism of sodium transport inhibition by epidermal growth factor in cortical collecting ducts. Am. J. Physiol. 261, F896–F903 [DOI] [PubMed] [Google Scholar]
- 5. Warden D. H., Stokes J. B. (1993) EGF and PGE2 inhibit rabbit CCD Na+ transport by different mechanisms: PGE2 inhibits Na+-K+ pump. Am. J. Physiol. 264, F670–F677 [DOI] [PubMed] [Google Scholar]
- 6. Falin R., Veizis I. E., Cotton C. U. (2005) A role for ERK1/2 in EGF- and ATP-dependent regulation of amiloride-sensitive sodium absorption. Am. J. Physiol. Cell Physiol. 288, C1003–C1011 [DOI] [PubMed] [Google Scholar]
- 7. Falin R. A., Cotton C. U. (2007) Acute down-regulation of ENaC by EGF involves the PY motif and putative ERK phosphorylation site. J. Gen. Physiol. 130, 313–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grossmann C., Freudinger R., Mildenberger S., Krug A. W., Gekle M. (2004) Evidence for epidermal growth factor receptor as negative-feedback control in aldosterone-induced Na+ reabsorption. Am. J. Physiol. Renal Physiol. 286, F1226–F1231 [DOI] [PubMed] [Google Scholar]
- 9. Shen J. P., Cotton C. U. (2003) Epidermal growth factor inhibits amiloride-sensitive sodium absorption in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 284, F57–F64 [DOI] [PubMed] [Google Scholar]
- 10. Levchenko V., Zheleznova N. N., Pavlov T. S., Vandewalle A., Wilson P. D., Staruschenko A. (2010) EGF and its related growth factors mediate sodium transport in mpkCCDc14 cells via ErbB2 (neu/HER-2) receptor. J. Cell. Physiol. 223, 252–259 [DOI] [PubMed] [Google Scholar]
- 11. Liu L., Duke B. J., Malik B., Yue Q., Eaton D. C. (2009) Biphasic regulation of ENaC by TGF-α and EGF in renal epithelial cells. Am. J. Physiol. Renal Physiol. 296, F1417–F1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pavlov T. S., Levchenko V., O'Connor P. M., Ilatovskaya D. V., Palygin O., Mori T., Mattson D. L., Sorokin A., Lombard J. H., Cowley A. W., Jr., Staruschenko A. (2013) Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J. Am. Soc. Nephrol. 24, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pochynyuk O., Stockand J. D., Staruschenko A. (2007) Ion channel regulation by Ras, Rho, and Rab small GTPases. Exp. Biol. Med. 232, 1258–1265 [DOI] [PubMed] [Google Scholar]
- 14. Karpushev A. V., Levchenko V., Ilatovskaya D. V., Pavlov T. S., Staruschenko A. (2011) Novel role of Rac1/WAVE signaling mechanism in regulation of the epithelial Na+ channel. Hypertension 57, 996–1002 [DOI] [PubMed] [Google Scholar]
- 15. Karpushev A. V., Levchenko V., Pavlov T. S., Lam V. Y., Vinnakota K. C., Vandewalle A., Wakatsuki T., Staruschenko A. (2008) Regulation of ENaC expression at the cell surface by Rab11. Biochem. Biophys. Res. Commun. 377, 521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pochynyuk O., Staruschenko A., Bugaj V., Lagrange L., Stockand J. D. (2007) Quantifying RhoA facilitated trafficking of the epithelial Na+ channel toward the plasma membrane with total internal reflection fluorescence-fluorescence recovery after photobleaching. J. Biol. Chem. 282, 14576–14585 [DOI] [PubMed] [Google Scholar]
- 17. Staruschenko A., Pochynyuk O. M., Tong Q., Stockand J. D. (2005) Ras couples phosphoinositide 3-OH kinase to the epithelial Na+ channel. Biochim. Biophys. Acta 1669, 108–115 [DOI] [PubMed] [Google Scholar]
- 18. Staruschenko A., Patel P., Tong Q., Medina J. L., Stockand J. D. (2004) Ras activates the epithelial Na+ channel through phosphoinositide 3-OH kinase signaling. J. Biol. Chem. 279, 37771–37778 [DOI] [PubMed] [Google Scholar]
- 19. Butterworth M. B., Edinger R. S., Silvis M. R., Gallo L. I., Liang X., Apodaca G., Frizzell R. A., Fizzell R. A., Johnson J. P. (2012) Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC). Am. J. Physiol. Renal Physiol. 302, F581–F590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takemura Y., Goodson P., Bao H. F., Jain L., Helms M. N. (2010) Rac1-mediated NADPH oxidase release of O2- regulates epithelial sodium channel activity in the alveolar epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L509–L520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saxena S. K., Kaur S. (2006) Regulation of epithelial ion channels by Rab GTPases. Biochem. Biophys. Res. Commun. 351, 582–587 [DOI] [PubMed] [Google Scholar]
- 22. Bos J. L., Rehmann H., Wittinghofer A. (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 23. Cherfils J., Zeghouf M. (2013) Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 24. DerMardirossian C., Bokoch G. M. (2005) GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 15, 356–363 [DOI] [PubMed] [Google Scholar]
- 25. Fukumoto Y., Kaibuchi K., Hori Y., Fujioka H., Araki S., Ueda T., Kikuchi A., Takai Y. (1990) Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 5, 1321–1328 [PubMed] [Google Scholar]
- 26. Togawa A., Miyoshi J., Ishizaki H., Tanaka M., Takakura A., Nishioka H., Yoshida H., Doi T., Mizoguchi A., Matsuura N., Niho Y., Nishimune Y., Nishikawa S. i., Takai Y. (1999) Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIα. Oncogene 18, 5373–5380 [DOI] [PubMed] [Google Scholar]
- 27. Shibata S., Nagase M., Yoshida S., Kawarazaki W., Kurihara H., Tanaka H., Miyoshi J., Takai Y., Fujita T. (2008) Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat. Med. 14, 1370–1376 [DOI] [PubMed] [Google Scholar]
- 28. Shibata S., Mu S., Kawarazaki H., Muraoka K., Ishizawa K., Yoshida S., Kawarazaki W., Takeuchi M., Ayuzawa N., Miyoshi J., Takai Y., Ishikawa A., Shimosawa T., Ando K., Nagase M., Fujita T. (2011) Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J. Clin. Invest. 121, 3233–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pavlov T. S., Chahdi A., Ilatovskaya D. V., Levchenko V., Vandewalle A., Pochynyuk O., Sorokin A., Staruschenko A. (2010) Endothelin-1 inhibits the epithelial Na+ channel through βPix/14-3-3/Nedd4–2. J. Am. Soc. Nephrol. 21, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Staruschenko A., Nichols A., Medina J. L., Camacho P., Zheleznova N. N., Stockand J. D. (2004) Rho small GTPases activate the epithelial Na+ channel. J. Biol. Chem. 279, 49989–49994 [DOI] [PubMed] [Google Scholar]
- 31. Pochynyuk O., Medina J., Gamper N., Genth H., Stockand J. D., Staruschenko A. (2006) Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J. Biol. Chem. 281, 26520–26527 [DOI] [PubMed] [Google Scholar]
- 32. Ilatovskaya D. V., Pavlov T. S., Levchenko V., Staruschenko A. (2013) ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am. J. Physiol. Cell Physiol. 304, C102–C111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawarazaki W., Nagase M., Yoshida S., Takeuchi M., Ishizawa K., Ayuzawa N., Ueda K., Fujita T. (2012) Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J. Am. Soc. Nephrol. 23, 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cowley A. W., Jr., Ryan R. P., Kurth T., Skelton M. M., Schock-Kusch D., Gretz N. (2013) Progression of glomerular filtration rate reduction determined in conscious Dahl salt-sensitive hypertensive rats. Hypertension 62, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaeggeler H.-P., Gonzalez-Rodriguez E., Jaeger N. F., Loffing-Cueni D., Norregaard R., Loffing J., Horisberger J.-D., Rossier B. C. (2005) Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J. Am. Soc. Nephrol. 16, 878–891 [DOI] [PubMed] [Google Scholar]
- 36. Butterworth M. B., Zhang L., Heidrich E. M., Myerburg M. M., Thibodeau P. H. (2012) Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J. Biol. Chem. 287, 32556–32565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., Vandewalle A. (1999) Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 38. Staruschenko A., Booth R. E., Pochynyuk O., Stockand J. D., Tong Q. (2006) Functional reconstitution of the human epithelial Na+ channel in a mammalian expression system. Methods Mol. Biol. 337, 3–13 [DOI] [PubMed] [Google Scholar]
- 39. Pavlov T. S., Ilatovskaya D. V., Levchenko V., Mattson D. L., Roman R. J., Staruschenko A. (2011) Effects of cytochrome P450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC). Am. J. Physiol. Renal Physiol. 301, F672–F681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Staruschenko A. (2012) Regulation of transport in the connecting tubule and cortical collecting duct. Compr. Physiol. 2, 1541–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cowley A. W., Jr. (1992) Long-term control of arterial blood pressure. Physiol. Rev. 72, 231–300 [DOI] [PubMed] [Google Scholar]
- 42. Cowley A. W., Jr. (2006) The genetic dissection of essential hypertension. Nat. Rev. Genet. 7, 829–840 [DOI] [PubMed] [Google Scholar]
- 43. Sanders P. W. (1996) Salt-sensitive hypertension: lessons from animal models. Am. J. Kidney Dis. 28, 775–782 [DOI] [PubMed] [Google Scholar]
- 44. Mattson D. L., Dwinell M. R., Greene A. S., Kwitek A. E., Roman R. J., Jacob H. J., Cowley A. W., Jr. (2008) Chromosome substitution reveals the genetic basis of Dahl salt-sensitive hypertension and renal disease. Am. J. Physiol. Renal Physiol. 295, F837–F842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Miguel C., Das S., Lund H., Mattson D. L. (2010) T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1136–R1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gonzalez-Rodriguez E., Gaeggeler H. P., Rossier B. C. (2007) IGF-1 vs insulin: respective roles in modulating sodium transport via the PI-3 kinase/Sgk1 pathway in a cortical collecting duct cell line. Kidney Int. 71, 116–125 [DOI] [PubMed] [Google Scholar]
- 47. Gaeggeler H. P., Guillod Y., Loffing-Cueni D., Loffing J., Rossier B. C. (2011) Vasopressin-dependent coupling between sodium transport and water flow in a mouse cortical collecting duct cell line. Kidney Int. 79, 843–852 [DOI] [PubMed] [Google Scholar]
- 48. Nofziger C., Chen L., Shane M. A., Smith C. D., Brown K. K., Blazer-Yost B. L. (2005) PPARγ agonists do not directly enhance basal or insulin-stimulated Na+ transport via the epithelial Na+ channel. Pflugers Arch. 451, 445–453 [DOI] [PubMed] [Google Scholar]
- 49. Pochynyuk O., Bugaj V., Vandewalle A., Stockand J. D. (2008) Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am. J. Physiol. Renal Physiol. 294, F38–F46 [DOI] [PubMed] [Google Scholar]
- 50. Stoos B. A., Náray-Fejes-Tóth A., Carretero O. A., Ito S., Fejes-Tóth G. (1991) Characterization of a mouse cortical collecting duct cell line. Kidney Int. 39, 1168–1175 [DOI] [PubMed] [Google Scholar]
- 51. Ishizaki T., Uehata M., Tamechika I., Keel J., Nonomura K., Maekawa M., Narumiya S. (2000) Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 57, 976–983 [PubMed] [Google Scholar]
- 52. Karpushev A. V., Ilatovskaya D. V., Staruschenko A. (2010) The actin cytoskeleton and small G protein RhoA are not involved in flow-dependent activation of ENaC. BMC Res. Notes 3, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Soundararajan R., Melters D., Shih I. C., Wang J., Pearce D. (2009) Epithelial sodium channel regulated by differential composition of a signaling complex. Proc. Natl. Acad. Sci. U.S.A. 106, 7804–7809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karpushev A. V., Ilatovskaya D. V., Pavlov T. S., Negulyaev Y. A., Staruschenko A. (2010) Intact cytoskeleton is required for small G protein dependent activation of the epithelial Na+ channel. PLoS ONE 5, e8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ilatovskaya D. V., Pavlov T. S., Levchenko V., Negulyaev Y. A., Staruschenko A. (2011) Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. FASEB J. 25, 2688–2699 [DOI] [PubMed] [Google Scholar]
- 56. Mazzochi C., Benos D. J., Smith P. R. (2006) Interaction of epithelial ion channels with the actin-based cytoskeleton. Am. J. Physiol. Renal Physiol. 291, F1113–F1122 [DOI] [PubMed] [Google Scholar]
- 57. Cantiello H. F., Stow J. L., Prat A. G., Ausiello D. A. (1991) Actin filaments regulate epithelial Na+ channel activity. Am. J. Physiol. 261, C882–C888 [DOI] [PubMed] [Google Scholar]
- 58. Gonzalez-Perrett S., Batelli M., Kim K., Essafi M., Timpanaro G., Moltabetti N., Reisin I. L., Arnaout M. A., Cantiello H. F. (2002) Voltage dependence and pH regulation of human polycystin-2-mediated cation channel activity. J. Biol. Chem. 277, 24959–24966 [DOI] [PubMed] [Google Scholar]
- 59. Lassègue B., San Martín A., Griendling K. K. (2012) Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 110, 1364–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cai H., Griendling K. K., Harrison D. G. (2003) The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 24, 471–478 [DOI] [PubMed] [Google Scholar]
- 61. Cowley A. W., Jr. (2008) Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension 52, 777–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Downs C. A., Kumar A., Kreiner L. H., Johnson N. M., Helms M. N. (2013) H2O2 regulates lung ENaC via ubiquitin-like protein Nedd8. J. Biol. Chem. 288, 8136–8145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ma H. P. (2011) Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. J. Biol. Chem. 286, 32444–32453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mamenko M., Zaika O., Ilatovskaya D. V., Staruschenko A., Pochynyuk O. (2012) Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in the distal nephron additively to aldosterone. J. Biol. Chem. 287, 660–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun P., Yue P., Wang W. H. (2012) Angiotensin II stimulates epithelial sodium channels (ENaC) in the cortical collecting duct of the rat kidney. Am. J. Physiol. Renal Physiol. 302, F679–F687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nagase M., Fujita T. (2013) Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat. Rev. Nephrol. 9, 86–98 [DOI] [PubMed] [Google Scholar]
- 67. Nagase M. (2013) Role of Rac1 GTPase in salt-sensitive hypertension. Curr. Opin. Nephrol. Hypertens. 22, 148–155 [DOI] [PubMed] [Google Scholar]
- 68. Gee H. Y., Saisawat P., Ashraf S., Hurd T. W., Vega-Warner V., Fang H., Beck B. B., Gribouval O., Zhou W., Diaz K. A., Natarajan S., Wiggins R. C., Lovric S., Chernin G., Schoeb D. S., Ovunc B., Frishberg Y., Soliman N. A., Fathy H. M., Goebel H., Hoefele J., Weber L. T., Innis J. W., Faul C., Han Z., Washburn J., Antignac C., Levy S., Otto E. A., Hildebrandt F. (2013) ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Invest. 123, 3243–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gupta I. R., Baldwin C., Auguste D., Ha K. C., El Andalousi J., Fahiminiya S., Bitzan M., Bernard C., Akbari M. R., Narod S. A., Rosenblatt D. S., Majewski J., Takano T. (2013) ARHGDIA: a novel gene implicated in nephrotic syndrome. J. Med. Genet. 50, 330–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Staruschenko A., Sorokin A. (2012) Role of βPix in the Kidney. Front. Physiol. 3, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]