

Background: α-Dicarbonyls are central intermediates in the formation of advanced glycation end products (AGEs).
Results: A quantitation method for the complete spectrum relevant in vivo was established.
Conclusion: Non-enzymatic chemistry of glucose and l-ascorbic acid as precursors and α-dicarbonyl intermediates play an important role in vivo.
Significance: Knowledge of plasma levels of α-dicarbonyls is crucial to understand the complex pathways of AGE formation in vivo.
Keywords: Ascorbic Acid, Carbohydrate Chemistry, Glycation, High Performance Liquid Chromatography (HPLC), Mass Spectrometry (MS), Plasma, beta-Cleavage, Dicarbonyls, Maillard Reaction, Quinoxalines
Abstract
Maillard α-dicarbonyl compounds are known as central intermediates in advanced glycation end product (AGE) formation. Glucose is the primary source of energy for the human body, whereas l-threo-ascorbic acid (vitamin C) is an essential nutrient, involved in a variety of enzymatic reactions. Thus, the Maillard degradation of glucose and ascorbic acid is of major importance in vivo. To understand the complex mechanistic pathways of AGE formation, it is crucial to extend the knowledge on plasma concentrations of reactive key α-dicarbonyl compounds (e.g. 1-deoxyglucosone). With the present work, we introduce a highly sensitive LC-MS/MS multimethod for human blood plasma based on derivatization with o-phenylenediamine under acidic conditions. The impact of workup and reaction conditions, particularly of pH, was thoroughly evaluated. A comprehensive validation provided the limit of detection, limit of quantitation, coefficients of variation, and recovery rates. The method includes the α-dicarbonyls 1-deoxyglucosone, 3-deoxyglucosone, glucosone, Lederer's glucosone, dehydroascorbic acid, 2,3-diketogulonic acid, 1-deoxypentosone, 3-deoxypentosone, 3,4-dideoxypentosone, pentosone, 1-deoxythreosone, 3-deoxythreosone, threosone, methylglyoxal, glyoxal; the α-keto-carboxylic acids pyruvic acid and glyoxylic acid; and the dicarboxylic acid oxalic acid. The method was then applied to the analyses of 15 healthy subjects and 24 uremic patients undergoing hemodialysis. The comparison of the results revealed a clear shift in the product spectrum. In most cases, the plasma levels of target analytes were significantly higher. Thus, this is the first time that a complete spectrum of α-dicarbonyl compounds relevant in vivo has been established. The results provide further insights into the chemistry of AGE formation and will be helpful to find specific markers to differentiate between the various precursors of glycation.
Introduction
Advanced glycation of proteins has been investigated in aging, diabetes, and nutrition (1–3). The Maillard reaction, a non-enzymatic process, is initiated when proteins are exposed to glucose or other carbohydrates. Through a series of reactions, it eventually yields the irreversible advanced glycation end products (AGEs).2 There are multiple sources and mechanisms of AGE formation in vivo, involving oxidative and non-oxidative chemistry of reducing sugars, Schiff bases, Amadori adducts, ascorbic acid and metabolic intermediates (4, 5). Because they are correlated to the severity of diabetic and uremic complications their clinical relevance was established (6–8).
AGE concentrations in vivo are markedly amplified in diabetes but also in non-diabetic uremia with a significant loss of renal clearance (9). Thus, besides substrate concentration, carbonyl stress, as described by Baynes et al. (10), is an alternative explanation for the increase of chemical protein modifications in various diseases. Carbonyl stress is caused by a generalized increase in the concentration of reactive carbonyl precursors of AGEs. Among those, α-dicarbonyl compounds (α-DCs) play a very prominent role. Their relevance as key intermediates in AGE formation was extensively investigated in in vitro model systems (11–15).
Endogenous formation is supposed to be the predominant source of α-DCs in human circulation (16). However, information on the physiological importance of exogenous dicarbonyl intake is scarce and subject to recent investigations (17–19). Regarding this topic, foods and peritoneal dialysis fluids are considered to release considerable amounts of reactive α-DCs (20–24).
For a comprehensive understanding of the complex formation pathways of AGEs in vivo, the analytical assessment of the complete α-DC spectrum is crucial. Nemet and Monnier (25) examined the l-threo-ascorbic acid (ASA, vitamin C) degradation products dehydroascorbic acid (DHA), 2,3-diketogulonic acid (2,3-DKG), threosone, 3-deoxythresosone (3-DT), and xylosone in human lens. In human blood plasma, however, only glyoxal (GO), methylglyoxal (MGO), 3-deoxyglucosone (3-DG), and DHA have been described in detail so far (26–33). The implemented analytical methods vary not only in the inevitable derivatization procedure but also in the chromatographic technique used. The common alternative approach for the quantitation of DHA is the measurement of the difference of ASA before and after a reduction step (34, 35). Consequently, concentrations of published plasma levels of healthy subjects differ in a wide range and are thus not comparable (e.g. for GO from 220 to 1150 pmol/ml, for MGO from 120 to 650 pmol/ml, and for DHA from 550 to 6800 pmol/ml (mean values)).
Glomb and Tschirnich compared common derivatization approaches (36). According to them, the use of aromatic o-diamines (e.g. o-phenylenediamine (OPD)) is prerequisite for the analysis of highly reactive α-DCs, such as 1-deoxyglucosone (1-DG). The detection of these short lived intermediates is limited by the rate of condensation of the reagent with the carbonyl moiety. However, these trapping reagents impose high oxidative stress on the system investigated and could lead to artifact formation, especially of α-DCs that originate from oxidative pathways (e.g. d-arabino-hexos-2-ulose (glucosone)).
The work group of Thornalley (31) developed a reliable method for the detection of MGO as 6,7-dimethoxy-2-methylquinoxaline in human plasma. They stressed the importance of pH control to avoid the degradation of dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P) to MGO, both central intermediates in several metabolic pathways in living organisms.
Although many methods for the detection of a few selected α-DCs were described already, a comprehensive method including all relevant compounds in vivo has been missing up to now. The particular analytical challenge in this regard is the very diverging polarity of the target substances but also the baseline separation of isomeric pairs with identical molecular masses (e.g. 1-/3-deoxypentosone (1-/3-DP)).
We developed and validated a highly sensitive LC-MS/MS multimethod suitable for routine analysis based on the derivatization with OPD, including 15 α-dicarbonyl compounds and two α-keto-carboxylic acids. Both substance categories are hereafter referred to as α-DCs for reasons of simplicity. In detail, the method covers 1-DG, 3-DG, glucosone, N6-(3,6-dideoxyhexos-2-ulos-6-yl)-l-lysine (Lederer's glucosone), DHA, 2,3-DKG, 4,5-dihydroxy-2,3-pentanedione (1-DP), 4,5-dihydroxy-2-oxopentanal (3-DP), 3,4-dideoxypentosone (3,4-DDP), pentos-2-ulose (pentosone), 1-deoxythreosone (1-DT), 3-DT, threosone, MGO, ethanedial (glyoxal, GO), 2-oxopropanoic acid (pyruvic acid), oxoethanoic acid (glyoxylic acid), and ethanedioic acid (oxalic acid) and was used to screen an initial set of plasma samples from 15 healthy human subjects and 24 uremic patients undergoing hemodialysis (HD patients). The results are discussed with respect to the sources and mechanistic relationships of the α-DCs detected.
EXPERIMENTAL PROCEDURES
Materials and Plasma Samples
Chemicals of the highest grade available were obtained from Sigma-Aldrich and Fisher unless otherwise indicated. NMR solvents were purchased from ARMAR Chemicals (Leipzig/Doettingen, Germany). The quinoxaline derivatives of the α-DCs will be marked hereafter with the suffix “-Q”. 1-DG-Q, 3-DG-Q, glucosone-Q, Lederer's glucosone-Q, 1-DP-Q, 3-DP-Q, 3,4-DDP-Q, pentosone-Q, 1-DT-Q, 3-DT-Q, threosone-Q, pyruvic acid-Q, glyoxylic acid-Q, oxalic acid-Q, and 3-DG were synthesized according to our previous work (37–39). The identities of target compounds were verified by nuclear magnetic resonance (NMR) experiments.
Written informed consent was obtained from all patients. The study was approved by the Ethics Committee of the Medical Faculty of the Martin Luther University Halle-Wittenberg. Blood samples were obtained from 15 healthy subjects with normal renal function and 24 non-diabetic patients undergoing hemodialysis using EDTA as an anticoagulant (2 mg/ml whole blood). In HD patients, samples were obtained predialysis before the midweek treatment session. Hemodialysis was performed three times weekly for 4–5 h using polysulfone dialyzers. All patients were treated with bicarbonate hemodialysis (acid concentrate type 257, 8.4% sodium bicarbonate type 200, MTN Neubrandenburg GmbH, Neubrandenburg, Germany) with ultrapure water quality (by reverse osmosis and sterile filters). Plasma was derived by centrifugation (3000 × g, 10 min, 4 °C) within 20 min of collection and immediately subjected to the assay procedure described below. HbA1c, creatinine, and C-reactive protein were measured by routine methods at the central laboratory of Martin-Luther-University Clinical Center, Halle (Saale, Germany).
2-(2′(R),3′(R),4′-Trihydroxybutyl)quinoxaline (3-DGal-Q)
3-Deoxy-d-threo-hexos-2-ulose (3-deoxygalactosone; 3-DGal) was synthesized according to the literature (40) with the exception of the 3-DGal bis(benzoyl hydrazone) cleavage. Here, instead of benzaldehyde, sodium nitrite was used, following the procedure of Henseke and Bauer (41). Purification of crude 3-DGal-Q was achieved by preparative high performance liquid chromatography with ultraviolet detection (HPLC-UV). NMR results were in line with those of Hellwig et al. (40).
3-(d-erythro-Glycerol-1-yl)-quinoxaline-2-carboxylic Acid o-Aminoanilide (DHA Precursor-Q)
DHA (250 mg; 1.44 mmol) was suspended in methanol (25 ml), and OPD was added (308.5 mg; 2.85 mmol). The reaction mixture was heated for 2 h at 40 °C and allowed to cool. The precipitated solid was isolated by filtration; washed with water, ethanol, and ether; and dried (yield: 282.6 mg, 76%). Recrystallization from ethanol gave yellow needles.
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 3.44–3.55 (m, 2H), 4.03 (m, 1H), 5.42 (m, 1H), 6.63 (m, 1H), 6.8 (dd, J = 8.0, 1.4 Hz, 1H), 7.0 (m, 1H), 7.36 (dd, J = 8.0, 1.5 Hz, 1H), 7.96 (m, 2H), 8.17 (m, 1H), 8.23 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 63.3, 72.0, 74.5, 116.4, 116.6, 122.9, 126.4, 127.2, 128.9, 129.5, 131.0, 132.0, 139.6, 141.2, 143.2, 147.4, 156.0, 165.1. HR-MS: m/z 393.09645 (found); m/z 393.09596 (calculated for C18H18O4N4K [M + K]+).
3-(d-erythro-Glycerol-1-yl)-quinoxaline-2-carboxylic γ-Lactone (DHA-Q)
A suspension of precursor DHA-Q (500 mg) in water (10 ml) was treated with 0.1 m hydrochloric acid (25 ml) and stirred at room temperature. The reaction process was followed by thin layer chromatography (TLC; EtOAc, UV detection). The reaction mixture was extracted with EtOAc (3 × 35 ml), the organic layers were combined, and the solvent was evaporated. The residue was purified by column chromatography (silica gel 60, 63–200 μm (Merck), EtOAc). Fractions including the compound with Rf 0.24 (TLC: EtOAc, UV detection) were collectively concentrated in vacuo to afford 180 mg (66%) of light yellow needles. NMR results were in line with those of Nemet and Monnier (25). HR-MS: m/z 269.05301 (found); m/z 269.05328 (calculated for C12H10O4N2Na [M + Na]+).
2,3-DKG
2,3-DKG as sodium salt was synthesized according to the method of Otsuka et al. (42). The product was obtained as a white hygroscopic precipitate. It was characterized by the LC-MS/MS method mentioned below after derivatization with OPD and directly utilized for quinoxaline synthesis.
3-((1S,2R)-1,2,3-Trihydroxypropyl)-quinoxaline-2-carboxylic Acid (2,3-DKG-Q)
The corresponding quinoxaline of 2,3-DKG was obtained according to Nemet and Monnier (25). Purification of the quinoxaline solution was done by preparative HPLC-UV (tR = 62 min). Fractions including the pure compound were collectively evaporated under reduced pressure, dissolved in water, and lyophilized to afford 2,3-DKG-Q as a light yellow powder in quantitative yield. NMR data were in line with the literature (25). HR-MS: m/z 263.06749 (found); m/z 263.06735 (calculated for C12H11O5N2 [M − H]+).
Isotopically Labeled 2-Hydroxy-3-methyl-2,3-13C2-quinoxaline (Pyruvic Acid-13C2-Q)
Pyruvic acid-13C2-Q was synthesized according to the method of Arun et al. (43). Briefly, a solution of OPD in distilled water (306.9 mg in 8 ml of ultrapure water, 2.84 mm) was added to 1,2-13C2-pyruvic acid (250.0 mg in 8 ml ultrapure water, 2.84 mm) dropwise with constant stirring. The precipitated pale yellow colored compound was filtered, washed with water, and lyophilized. The crude sample was recrystallized from 50% ethanol absolute (404.7 mg, 88%). NMR data were in line with the literature. HR-MS: m/z 163.0779 (found); m/z 163.0776 (calculated for C713C2H8ON2 [M + H]+).
Time Course of the OPD Reaction of 3-DG, MGO, GO, Glyoxylic Acid, Pyruvic Acid, Oxalic Acid, DHA Precursor-Q, and 2,3-DKG-Q under Assay Conditions
All compounds (20 μm) were incubated under assay conditions in amber glass vials under argon atmosphere at 22 °C in 0.4 m formate buffer (pH 3.0) containing 0.55 mm OPD and 3.4 mm EDTA. Vials were tightly sealed by a screw cap with integrated polytetrafluoroethylene/silicone septum. At various time points, aliquots of the reaction mixtures were subjected to LC-UV analysis as described below. After 24 h, trifluoroacetic acid (TFA; 0.4 m) was added, and the incubation was continued for 1 h. Then pH 3.0 was adjusted again with 4 m ammonium hydroxide. The injection volume was adjusted to account for the dilution of the reaction mixture. The yields were determined against the authentic reference α-DC-Q standards.
Time Course of the OPD Reaction of ASA, DHA, 2,3-DKG, and 3-DG-Q under Assay Conditions
All compounds (20 μm) were incubated as described above over a prolonged time period. No TFA was added after 24 h. At various time points, aliquots of the reaction mixtures were subjected to LC-UV analysis as described below. The yields were determined against the authentic reference α-DC-Q standards. Due to the lack of authentic reference for the quinoxaline of 3,4-dideoxyglucosone-3-ene (3,4-DGE-Q), quantitation was done with 3-DGal-Q under the assumption of an equal extinction coefficient.
De Novo Formation of MGO-Q from G3P and DHAP with OPD under Various Assay Conditions
G3P and DHAP (50 and 100 μm, which corresponds to a plasma concentration of 100 and 200 μm, respectively) were incubated under assay conditions in amber glass vials under argon atmosphere at 22 °C in various buffer solutions containing 0.55 mm OPD and 3.4 mm EDTA. The buffers used were potassium phosphate buffer (0.4 m, pH 7.0), sodium formate buffer (0.4 m, pH 3.0), and perchloric acid (0.4 m, pH <0). Vials were tightly sealed by a screw cap with integrated polytetrafluoroethylene/silicone septum. Reaction mixtures were subjected to LC-UV analysis as described below not only after 24 h but immediately after the addition of OPD and after a 1-h incubation time to account for possible MGO impurities of the starting materials. The yield of MGO was determined against the authentic reference α-DC-Q standards.
Assay of α-DCs in Plasma
Sodium formate buffer (2 m, pH 3.0, 200 μl), the internal standard pyruvic acid-13C2-Q (1.25 μm, 100 μl) and the derivatizing agent OPD (2.75 mm, 200 μl) were added to 500 μl of blood plasma. The sample was incubated for 24 h in the dark at room temperature under argon atmosphere. TFA (2 m, 250 μl) was added, and the incubation continued for 1 h under the same conditions. The pH of the sample was then adjusted to pH 3.0 with ammonium hydroxide (4 m, 415 μl). Water was added (85 μl) to give a total sample volume of 1750 μl. The protein precipitate was separated by centrifugation (16,000 × g and 20 °C). The supernatant (storage at −80 °C) was administered to LC-MS/MS analysis.
HPLC-UV
A Besta HD 2-200 pump (Wilhelmsfeld, Germany) was used at a flow rate of 15 ml/min. Elution of material was monitored by a UV detector (Jasco UV-2075 with a preparative flow cell (Gross-Umstadt, Germany)). The detection wavelength was 320 nm. Chromatographic separations were performed on a stainless steel column (KNAUER, 250 × 20 mm, Eurospher-100 C18, 10 μm (Berlin, Germany)). The mobile phase used consisted of water (solvent A) and methanol/water (7:3 (v/v), solvent B). To both solvents (A and B) 0.8 ml/liter formic acid was added. Samples were injected at 5% B for 2,3-DKG-Q and at 35% B for 3-DGal-Q and purified under isocratic conditions.
NMR Spectroscopy
NMR spectra were recorded on a Varian VXR 400 spectrometer operating at 400 MHz for 1H and 100 MHz for 13C or on a Varian Unity Inova 500 instrument operating at 500 MHz for 1H and 125 MHz for 13C, respectively. Chemical shifts are given relative to external SiMe4.
Accurate Mass Determination (High Resolution MS)
The high resolution positive and negative ion electrospray ionization mass spectra (electrospray ionization-high resolution MS) were taken on a Bruker Apex III Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonics, Billerica, MA) equipped with an Infinity cell, a 7.0-tesla superconducting magnet, a radio frequency-only hexapole ion guide, and an external off-axis electrospray source (Apollo; Agilent, Santa Clara, CA). Nitrogen was used as a drying gas at 150 °C. The samples were dissolved in methanol, and the solutions were introduced continuously via a syringe pump at a flow rate of 120 μl/h. The data were acquired with 256,000 data points and zero-filled to 1,024,000 by averaging 32 scans.
High Performance Liquid Chromatography with Coupled Ultraviolet-Mass Spectrometry Detection (LC-UV-MS/MS)
The HPLC apparatus (Jasco, Gross-Umstadt, Germany) consisted of a pump (PU-2080 Plus) with degasser (LG-2080-02) and quaternary gradient mixer (LG-2080-04), a column oven (Jasco Jetstream II), an autosampler (AS-2057 Plus), and a UV detector (UV-2075). Mass spectrometric detection was conducted on an API 4000 QTrap LC-MS/MS system (Applied Biosystems/MDS Sciex, Concord, Canada) equipped with a turbo ion spray source using electrospray ionization in positive mode: sprayer capillary voltage, 4.5 kV; nebulizing gas flow, 50 ml/min; heating gas, 60 ml/min at 550 °C; and curtain gas, 40 ml/min.
Chromatographic separation was performed on a stainless steel column packed with RP-18 material (KNAUER, 250 × 4.6 mm, Eurospher-100 C18, 5 μm (Berlin, Germany)) using a flow rate of 1.0 ml/min. The mobile phase used was water (solvent A) and methanol/water (7:3 (v/v), solvent B). To both solvents (A and B), 0.6 ml/liter heptafluorobutyric acid was added. Analysis was performed at 25 °C column temperature using gradient elution: 5% B (0–0.1 min) to 35% B (0.5 min) to 47% B (12 min) to 100% B (17–25 min). For mass spectrometric detection, the scheduled multiple-reaction monitoring mode was used, utilizing collision-induced dissociation (CID) of the protonated molecules with compound-specific orifice potentials and fragment-specific collision energies (Table 1). Quantitation was performed using the standard addition method. More precisely, increasing concentrations of authentic α-DC-Q reference compounds at factors of 0.5, 1, 2, and 3 times the concentration of the analyte in the sample were added to separate aliquots of the sample after the workup procedure. The aliquots were analyzed, and a regression of response versus concentration is used to determine the concentration of the analyte in the sample. Spikes were run for one of approximately every 30 samples. Calibration with this method resolves potential matrix interferences. Potential losses during the workup procedure and intrabatch changes of instrument sensitivity were corrected with pyruvic acid-13C2 as an internal standard.
TABLE 1.
LC-MS/MS parameters for quinoxaline quantitation
| Quinoxaline and letter assignment (see Fig. 2) | Retention time | Precursor ion |
Product ion 1a |
Product ion 2b |
Product ion 3b |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m/z | DPc | m/z | CEd | CXPe | m/z | CE | CXP | m/z | CE | CXP | ||
| min | amuf | V | amu | eV | V | amu | eV | V | amu | eV | V | |
| 2,3-DKG (a) | 9.5 | 265.1 | 40 | 185.1 | 23.0 | 11.0 | 221.1 | 17.0 | 17.0 | 161.0 | 27.0 | 11.0 |
| Glucosone (b) | 9.9 | 251.1 | 55 | 173.1 | 24.0 | 16.0 | 161.1 | 30.0 | 13.0 | 233.2 | 19.0 | 16.0 |
| Oxalic acid (c) | 10.5 | 162.9 | 70 | 90.1 | 42.0 | 15.0 | 145.1 | 25.0 | 10.0 | 117.1 | 31.0 | 7.0 |
| Pentosone (d) | 11.2 | 221.1 | 50 | 173.1 | 22.0 | 14.0 | 161.0 | 27.0 | 13.0 | 132.1 | 45.0 | 10.0 |
| 1-DG (e) | 11.9 | 235.2 | 55 | 175.1 | 28.0 | 12.0 | 187.1 | 27.0 | 11.0 | 217.2 | 18.0 | 16.0 |
| DHA (f) | 12.0 | 247.1 | 55 | 229.0 | 18.0 | 20.0 | 185.3 | 20.0 | 15.0 | 171.1 | 33.0 | 10.0 |
| 3-DG (g) | 13.9 | 235.2 | 55 | 199.1 | 24.0 | 17.0 | 145.0 | 31.0 | 9.0 | 157.1 | 12.0 | 30.0 |
| Threosone (h) | 14.0 | 191.2 | 45 | 173.1 | 17.5 | 14.0 | 144.1 | 37.0 | 11.0 | 161.2 | 24.0 | 12.0 |
| 1-DP (i) | 16.8 | 205.1 | 50 | 187.1 | 21.0 | 14.0 | 145.1 | 27.0 | 12.0 | 169.2 | 30.0 | 11.0 |
| Glyoxylic acid (j) | 17.1 | 147.1 | 65 | 129.0 | 26.5 | 9.0 | 119.1 | 30.0 | 10.0 | 102.1 | 39.0 | 8.5 |
| 3-DP (k) | 17.1 | 205.1 | 50 | 187.2 | 19.5 | 10.0 | 158.1 | 34.0 | 14.0 | 174.1 | 32.5 | 13.0 |
| Lederer's glucosone | 18.4 | 363.3 | 65 | 187.1 | 27.0 | 16.0 | 159.1 | 29.0 | 10.0 | 219.1 | 24.0 | 12.0 |
| 3-DT (l) | 19.2 | 175.1 | 55 | 157.2 | 24.0 | 10.0 | 129.2 | 35.0 | 9.0 | 145.0 | 25.5 | 10.0 |
| DHA precursor (m) | 19.5 | 355.3 | 52 | 109.2 | 19.0 | 8.0 | 247.2 | 15.0 | 16.0 | 337.2 | 15.0 | 12.0 |
| Pyruvic acid (n) | 19.5 | 160.9 | 60 | 133.1 | 28.0 | 10.0 | 91.9 | 40.0 | 7.0 | 64.9 | 54.0 | 11.0 |
| Pyruvic acid-13C2 | 19.5 | 163.1 | 60 | 134.0 | 28.0 | 10.0 | 92.1 | 40.0 | 7.0 | 64.9 | 54.0 | 11.0 |
| 1-DT (o) | 19.6 | 175.1 | 45 | 157.1 | 22.0 | 7.0 | 156.1 | 35.0 | 15.0 | 89.1 | 55.0 | 5.0 |
| 3,4-DDP (p) | 20.5 | 189.1 | 55 | 171.2 | 25.0 | 15.0 | 145.1 | 25.0 | 11.0 | 102.1 | 52.0 | 16.0 |
| GO (q) | 20.9 | 131.1 | 32 | 77.0 | 40.0 | 6.0 | 104.0 | 30.0 | 8.0 | |||
| MGO (r) | 22.3 | 145.0 | 50 | 77.0 | 41.0 | 5.0 | 118.1 | 30.5 | 7.0 | 65.0 | 45.0 | 5.0 |
a MRM transition used for quantitation (quantifier).
b MRM transition used for confirmation (qualifier).
c DP, declustering potential.
d CE, collision energy.
e CXP, collision cell exit potential.
f amu, atomic mass units.
To obtain fragmentation spectra of α-DCs-Q in plasma workup solutions, target material was first enriched by repeated collection from the above HPLC system. After solvent evaporation in a vacuum concentrator (Savant SpeedVac Plus SC 110 A combined with a Vapor Trap RVT 400, Thermo Fisher Scientific GmbH (Dreieich, Germany)), the residue was dissolved in water and reinjected, using a CID experiment. The fragmentation spectra of the authentic references were obtained with the same parameters. For 1-DG-Q, the following parameters were used: declustering potential, 50 V; collision energy, 30 eV; collision cell exit potential, 10 V; scan range, m/z 130–250 (4 s). All plasma workup samples and incubations were analyzed in single batches to exclude interassay variations.
Validation of the Quantitation Method
Intraassay coefficients of variation were determined by repeated workup and analysis of a plasma sample (n = 6). In addition, the limit of detection and limit of quantitation with all steps of the analysis included were estimated according to DIN 32645 (n = 6, confidence level p = 0.95, k = 3) (44). To determine recovery rates, the respective alpha-DCs-Q were added at four different concentrations to three parallel sets of blood samples of one subject. The native and the spiked plasma sample were subjected to the assay of α-DCs in plasma as described above. The recovery rate was estimated as the quotient of (spiked α-DC-Q amount − amount α-DC-Q in native plasma sample)/amount of added α-DC-Q × 100%.
RESULTS
α-Dicarbonyl Compounds in Blood Plasma of Healthy Human Subjects and Hemodialysis Patients
Plasma was obtained from 15 healthy subjects with normal renal function and no proteinuria and 24 uremic patients undergoing hemodialysis (HD patients). Details are given in Table 2. Normal renal function was defined as a serum creatinine level below 102 μmol/liter. To ensure the absence of inflammatory reactions, C-reactive protein had to be 5 μmol/liter or below for all healthy subjects.
TABLE 2.
Profile of subjects examined in this study
| Healthy subjects | HD patients | |
|---|---|---|
| No. of participants | 15 | 24 |
| Sex, female/male | 6/9 | 6/18 |
| Age (years) | 24–34 | 30–85 |
| HbA1c (mmol/mol) | 37 ± 2 | 40 ± 17 |
| Serum creatinine (μmol/liter) | 82 ± 13 | 861 ± 322 |
| C-reactive protein (mg/liter) | <5 | 11 ± 21 |
After derivatization of target α-DCs to the respective quinoxalines and separation from the protein residue, the plasma samples were subjected to the described LC-MS/MS method. All plasma workup samples of controls and HD patients were each analyzed in single batches to exclude interassay variations. Intraassay coefficients of variation were determined by independent analyses of three different blood samples for each subject and were <10% in all cases. The results are presented in Table 3.
TABLE 3.
Levels of all relevant α-dicarbonyl compounds in human blood plasma
Values shown are the mean ± S.D. of 15 healthy subjects and 24 patients undergoing hemodialysis (HD patients), replicate analyses, n = 3. Student's t test was used for statistical evaluation of significant differences between both groups: *, p < 0.01 versus healthy subjects; **, p < 0.001 versus healthy subjects.
| Quinoxaline | Healthy subjects |
HD patients |
||
|---|---|---|---|---|
| Mean ± S.D. | Range | Mean ± S.D. | Range | |
| pmol/ml | pmol/ml | |||
| Glucosone | 46 ± 11 | 28–67 | 96 ± 49** | 57–276 |
| 1-DG | 22 ± 3 | 16–28 | 30 ± 16 | 12–82 |
| 3-DG | 43 ± 5 | 35–56 | 65 ± 20** | 36–125 |
| Lederer's glucosone | <LOD | 7.0 ± 2.5a | <LOD–13 | |
| Pentosone | 15 ± 10a | <LOD–32 | 11 ± 5a | <LOD–23 |
| 1-DP | 3.6 ± 1.2a | 2.6–7.8 | 6.2 ± 2.6a** | 3.2–15 |
| 3-DP | 11 ± 2 | 9–17 | 33 ± 9** | 20–53 |
| Threosone | 5.4 ± 0.7a | 4.2–6.6 | 9.0 ± 3.5a** | 4.2–19 |
| 1-DT | 3.1 ± 0.3 | 2.6–3.8 | 3.8 ± 1.0* | 2.0–6.6 |
| 3-DT | 10 ± 2 | 8–14 | 45 ± 11** | 28–75 |
| MGO | 61 ± 7 | 51–76 | 219 ± 129** | 42–617 |
| GO | 491 ± 47 | 405–564 | 1273 ± 980** | 400–4914 |
| Pyruvic acid | 7250 ± 2549 | 2818–12,038 | 35,874 ± 19,080** | 8399–97,935 |
| Glyoxylic acid | 1264 ± 353 | 783–1942 | 2031 ± 608** | 1026–3514 |
| Oxalic acid | 40 ± 21 | 18–82 | 36 ± 12 | 16–70 |
| 3,4-DDP | 6.0 ± 1.0 | 4.4–7.7 | 19 ± 8** | 6–42 |
| DHA precursor | 687 ± 351b | 337–1303 | 1925 ± 1106b** | 308–4829 |
| DHA | 15,400 ± 4019b | 6809–23,967 | 12,538 ± 7183b | 2737–26,680 |
| 2,3-DKG | 1741 ± 674c | 567–2669 | 411 ± 238c** | 145–937 |
a LOD < × < LOQ.
b The sum of DHA-Q and DHA precursor-Q account for approximately 38% of the true DHA content.
c Values adjusted as described under “Disussion.”
In selected cases, the identity of the detected compounds in plasma was confirmed by comparison of the native fragmentation spectrum with authentic reference standards. The results were in full compliance and are exemplarily shown for 1-DG-Q in Fig. 1. The protonated molecular ion [M + H]+ at m/z 235.2 is expected to undergo dehydration to the ion at m/z 217.2 and 199.2. Subsequent decay leads to signals at m/z 191.0, 187.1, 175.1, 171.2, and 158.1.
FIGURE 1.

Verification of 1-DG by CID of m/z 235.2 [M + H]+. A, authentic reference; B, plasma workup.
Development of an LC-MS/MS Method for 17 Relevant Dicarbonyl Compounds as Their Corresponding Quinoxalines
The complex matrix and the low concentration of most target analytes hamper the use of UV detectors for qualitative and quantitative analysis. Tandem mass spectrometric detection in the multiple-reaction monitoring mode was prerequisite for the detection of the 17 relevant α-DCs in blood plasma. To achieve highly sensitive detection utilizing collision-induced dissociation of the protonated molecules, the compound-specific orifice potentials and fragment-specific collision energies had to be determined by authentic reference material (Table 1).
HPLC separation was based on a RP-18 phase with methanol as the organic eluent. Optimization of gradient, column temperature, and ion pair reagent led to almost baseline separation of all critical quinoxalines possessing the same molecular weight, which therefore could not be distinguished via MS detection (1-DG-Q versus 3-DG-Q versus 4-DG-Q versus 3-DGal-Q, 1-DP-Q versus 3-DP-Q, 1-DT-Q versus 3-DT-Q). Fig. 2 shows the typical chromatogram of a plasma sample after derivatization with OPD.
FIGURE 2.
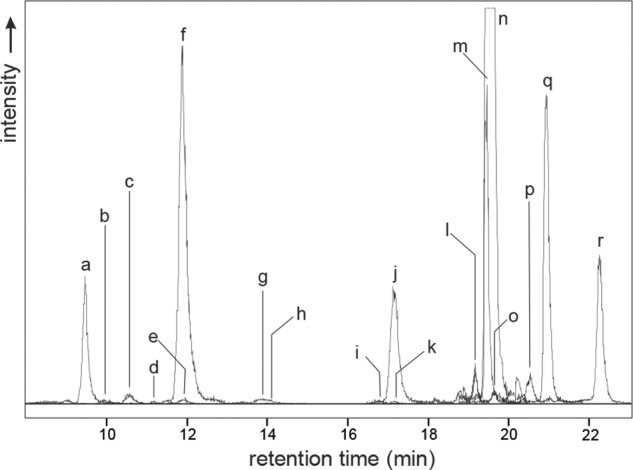
LC-MS/MS chromatogram of a plasma sample after standard workup procedure. For clarity, only the quantifier mass transition for each analyte of the scheduled multiple-reaction monitoring is shown. For letter assignments (a–r), see Table 1.
Verification of the Derivatization Procedure
The reactivity of α-DCs required conversion into stable quinoxaline derivatives. After the blood sample is drawn from the living subject, the complex system blood is prone to significant alterations regarding its biological activity and chemical composition, especially under exposure to oxygen. Immediate plasma generation and instant start of the derivatization procedure under well defined conditions is therefore prerequisite for reproducible quantitative results.
To prevent underestimation of rather slow reacting compounds, OPD was chosen as the derivatization agent. It has a comparatively high conversion rate of α-DCs to the respective quinoxaline derivatives. To ensure complete derivatization, selected native α-DCs (GO, MGO, glyoxylic acid, pyruvic acid, oxalic acid, and 3-DG) were incubated with OPD under standard assay conditions, and the formation of respective quinoxalines was monitored between 0.5 and 48 h. The results presented in Fig. 3 revealed complete reaction (±5%) within 11 h, with the short-chained substances reacting much more rapidly, which is in accordance with our previous findings (36). The exception was oxalic acid, which showed no reaction with OPD. Furthermore, the stability of all quinoxaline compounds included in our method was monitored with authentic references under the derivatization conditions for 24 h. No degradation was observed.
FIGURE 3.
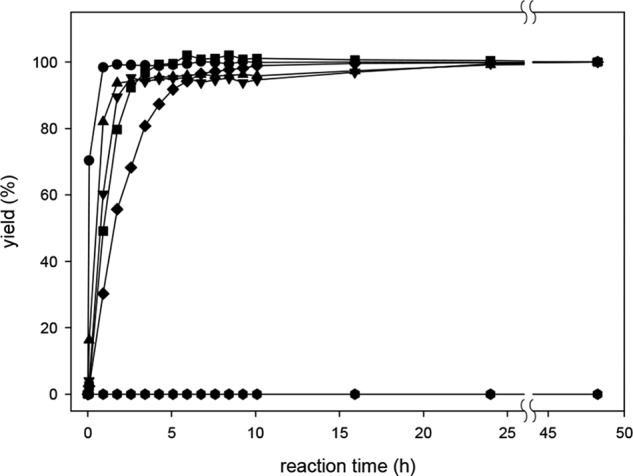
Reactions of selected dicarbonyl compounds with OPD in formate buffer (0.4 m, pH 3.0) at room temperature: 3-DG (♦), MGO (●), GO (■), pyruvic acid (▾), glyoxylic acid (▴), and oxalic acid (hexagons).
In addition, the derivatization of a plasma sample was monitored by repeated injection of aliquots and LC-MS/MS analyses of the complete α-DC spectrum. Besides the risk of underestimation of α-DCs in trace amounts, avoiding overestimation of certain dicarbonyl quinoxalines is an even bigger challenge. For example, de novo formation of glucosone-Q and GO-Q during derivatization with OPD are described in the literature (22, 36, 45). The main reasons are the strong oxidative conditions imposed on the system by the addition of OPD in combination with oxygen and trace amounts of metal ions. However, with our derivatization procedure, a stable plateau for all analytes was reached within the relevant incubation time, indicating no or negligible de novo formation in the plasma matrix. In this regard, it is important to note that the incubation took place under argon atmosphere to minimize the impact of oxygen. Furthermore, EDTA as a metal ion-chelating agent was present in the assay.
The plasma samples were diluted in formate buffer (pH 3.0). McLellan and Thornalley (31) applied different derivatization procedures to biological systems and discovered significant interferences from G3P and DHAP by spontaneous elimination of phosphate to form MGO during sample processing, depending on the chosen pH value. The rate of reaction increased with increasing pH. To avoid the degradation of G3P and DHAP, we conducted the derivatization step under strong acidic conditions with perchloric acid. However, under these harsh conditions, we discovered significant degradation of 3-DG-Q, 1-DP-Q, 3-DT-Q, and threosone-Q (data not shown). Especially 3-DG-Q with a half-life of 80 h was rapidly degraded to form the diastereomer 3-DGal-Q (Fig. 4). Mittelmaier et al. (46) already investigated the formation of 3-DGal in peritoneal dialysis fluids under sterilization conditions. They postulated a reaction mechanism that includes a reversible dehydration of free 3-DG leading to 3,4-dideoxyglucosone-3-ene (3,4-DGE). Subsequently, 3,4-DGE undergoes hydration to form 3-DGal. Obviously, this type of reaction also takes place with the corresponding quinoxalines under harsh acidic assay conditions. Whereas with the native α-DC, the carbonyl moiety is the driving force, with the quinoxaline, it is the extension of conjugated unsaturation.
FIGURE 4.
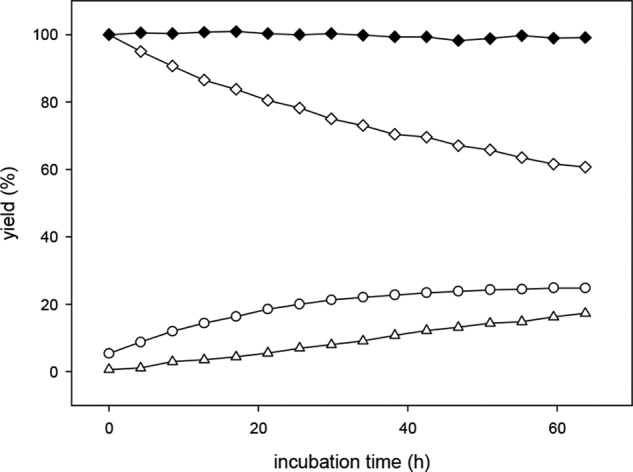
Degradation of 3-DG-Q at pH 0.6 (open symbols) and pH 3.0 (closed symbols): 3-DG (♦), 3-DGal (●), and 3,4-DGE (▴).
We monitored the degradation of 3-DG-Q in sodium formate solution at different pH values (data not shown). Based on our findings, we chose more gentle conditions and employed a formate buffer (0.4 m, pH 3.0). Here, no degradation of 3-DG-Q at 21 °C for 48 h was observed. In addition, formation of MGO caused by oxidative degradation of nucleic acids and related compounds during the derivatization process is accelerated by strong acids like perchloric acid but negligible with milder acidic treatment (47, 48).
To ensure reliable MGO quantitation with the method described herein, we conducted control experiments at different pH conditions with 100 and 200 μm concentrations of G3P and DHAP, respectively (Table 4). The chosen concentrations were based on the experiments conducted by McLellan and Thornalley (31). The conversion rate at pH 3.0 after 24 h in mol % was below 2% for G3P and below 8% for DHAP, independent from the starting concentration of both. The minor interferences found were inevitable, and the results were comparable with the well established method of McLellan. To ensure the absence of contaminants in water or chemicals used in the assay possibly leading to overestimation, a reactant blank was incubated along with the plasma samples.
TABLE 4.
Conversion rate in mol % of G3P and DHAP, respectively, to MGO over an incubation period of 24 h in the indicated medium in presence of 0.55 mm OPD and 3.4 mm EDTA
| G3P |
DHAP |
|||
|---|---|---|---|---|
| 100 μm | 200 μm | 100 μm | 200 μm | |
| mol % | mol % | |||
| pH <0; 0.4 m perchloric acid | 21.3 | 21.2 | 8.5 | 8.3 |
| pH 3.0; 0.4 m sodium formate buffer | 1.4 | 1.5 | 7.7 | 7.6 |
| pH 7.0; 0.4 m potassium phosphate buffer | 18.3 | 19.8 | 96.6 | 98.6 |
Mechanistic Relationship of DHA, 2,3-DKG, and Their Corresponding Quinoxalines under Assay Conditions
Investigation of the derivatization reaction of DHA in sodium formate buffer (0.4 m, pH 3.0) with OPD (0.55 mm) gave rather surprising results shown in Fig. 5 and in the scheme in Fig. 6. Initially, a precursor compound (DHA precursor-Q) with two molecules of OPD was formed. The precursor then converted to DHA-Q. However, after 24 h, no plateau was reached, and the precursor compound was still detectable in significant amounts. A quantitative reaction of DHA with OPD was only achieved after more than 6 days, which is not feasible for the present analytical assessment. In addition, oxidative conditions imposed by OPD lead to formation of DHA from ASA. When 20 μm ASA was incubated under assay conditions, 50 mol % of the ASA was converted to DHA-Q after 6 days, whereas after 24 h, de novo formation of DHA-Q from ASA was negligible (<2%).
FIGURE 5.
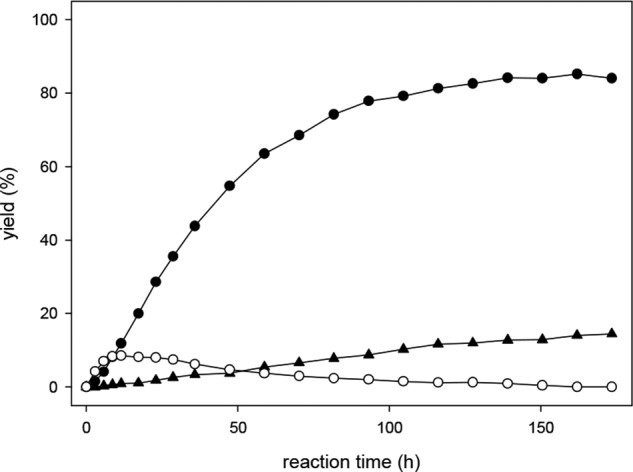
Reaction of DHA with OPD in formate buffer (0.4 m, pH 3.0) at room temperature yields DHA precursor-Q (○), DHA-Q (●), and 2,3-DKG-Q (▴).
FIGURE 6.
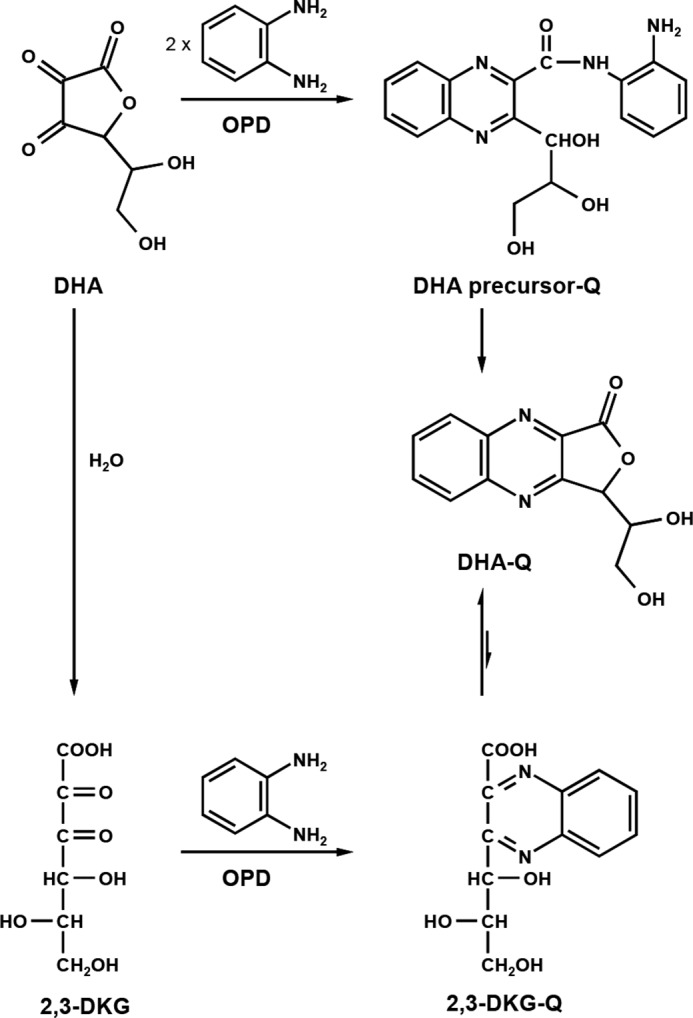
Mechanistic relationship of DHA, 2,3-DKG, and their corresponding derivatization products.
In contrast, conversion of 2,3-DKG to its corresponding quinoxaline is completed after 8 h (data not shown). Obviously, DHA is the least reactive of all α-DCs assessed (with the exception of oxalic acid). Consequently, it is important to note that the plasma levels of DHA-Q and DHA precursor-Q given in Table 3 have to be added and account for ∼38% of the true DHA plasma content.
To monitor the conversion of DHA precursor-Q to DHA-Q, the precursor was synthesized independently. The authentic reference DHA precursor-Q was then incubated under assay conditions (Fig. 7). A half-life of 5 h was observed. After 24 h, 81% of DHA precursor-Q was converted to DHA-Q. Residual amounts of DHA precursor-Q (10% based on initial concentration) remained and had to be considered for the calculation of DHA concentration. Interestingly, also small amounts of 2,3-DKG-Q were detected (7%). A reverse reaction of the quinoxalines to form free DHA with subsequent hydrolytic opening of the lactone ring system to yield 2,3-DKG and its corresponding quinoxaline 2,3-DKG-Q can be excluded. Thus, DHA-Q has to be hydrolyzed slowly to form 2,3-DKG-Q directly. To further investigate the relationship of the two quinoxalines, 2,3-DKG-Q was incubated under assay conditions, including the protein precipitation step with TFA after 24 h. The results in Fig. 8 show conversion of 2,3-DKG-Q to DHA-Q, which was significantly triggered at the lower pH during the 1-h precipitation step. 2,3-DKG-Q yields ∼25% DHA-Q after the complete workup procedure. Obviously, there is a steady state between DHA-Q and 2,3-DKG-Q with preferred DHA-Q formation as the more stable product under acidic conditions.
FIGURE 7.
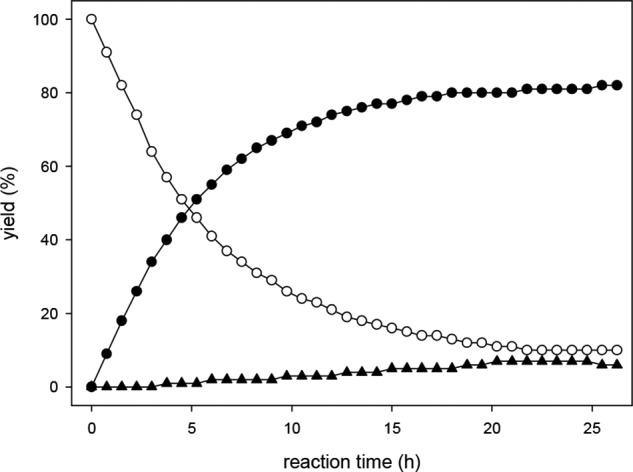
Formation of DHA-Q (●) and 2,3-DKG-Q (▴) starting from DHA precursor-Q (○) under standard workup conditions.
FIGURE 8.
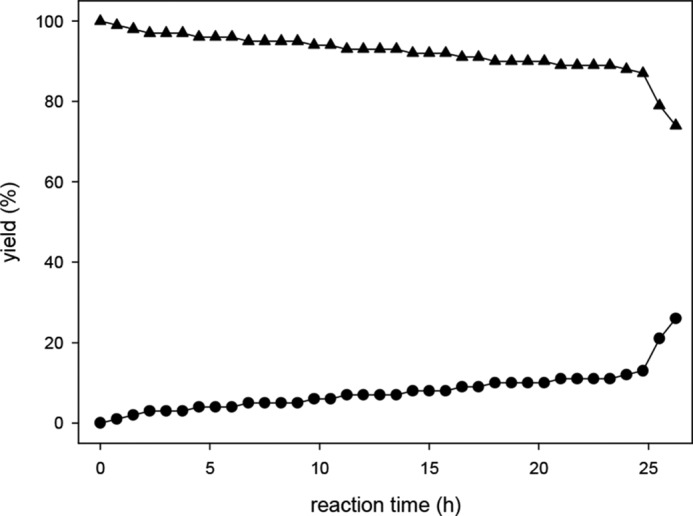
Degradation of 2,3-DKG-Q (▴) under standard workup conditions yields DHA-Q (●).
Validation of the Proposed Method
The limit of detection and limit of quantitation with all steps of the analytical method included were calculated as described under “Experimental Procedures.” The limit of detection differs from 0.5 to 42.2 pmol/liter. The accuracy of the method was determined as the recovery rate at three different concentration levels and ranged between 82 and 120%. Repeatability was expressed as the coefficient of variation and was 10% or better. The complete validation data are shown in Table 5.
TABLE 5.
Coefficient of variation (CV), recovery rates, limit of detection (LOD), and limit of quantitation (LOQ) (all steps of the analysis included) of plasma samples
| Quinoxaline | CVa | Recoveryb | LODb | LOQb |
|---|---|---|---|---|
| % | % | pmol/ml | pmol/ml | |
| Glucosone | 6 | 109 | 6.1 | 18.3 |
| 1-DG | 4 | 112 | 2.2 | 6.6 |
| 3-DG | 5 | 97 | 5.4 | 16.2 |
| Lederer's glucosone | —c | 114 | 3.5 | 10.5 |
| Pentosone | 7 | 98 | 8.3 | 24.9 |
| 1-DP | 9 | 82 | 2.5 | 7.5 |
| 3-DP | 8 | 93 | 3.5 | 10.5 |
| Threosone | 3 | 90 | 4.2 | 12.6 |
| 1-DT | 3 | 91 | 0.5 | 1.5 |
| 3-DT | 3 | 98 | 2.0 | 6.0 |
| MGO | 8 | 106 | 3.0 | 9.0 |
| GO | 9 | 98 | 6.9 | 20.7 |
| Pyruvic acid | 2 | 99 | 14.1 | 42.3 |
| Glyoxylic acid | 4 | 92 | 42.2 | 126.6 |
| Oxalic acid | 7 | 89 | 6.8 | 20.4 |
| 3,4-DDP | 3 | 110 | 0.8 | 2.4 |
| DHA precursor | —d | —d | 2.9 | 8.7 |
| DHA | 10 | 91 | 22.9 | 68.7 |
| 2,3-DKG | 6 | 119 | 6.1 | 18.3 |
a Repeatability conditions, n = 6.
b Replicate analyses, n = 3.
c Estimation not possible because analyte was below the limit of detection.
d Estimation not possible because compound is the intermediate in DHA-Q formation.
DISCUSSION
Up to now, only GO, MGO, 3-DG, and DHA were quantitated in human plasma using established methods, although the spectrum of α-DCs in vivo is supposed to be far more complex. Major endogenous sources are the degradation of blood glucose and ascorbic acid. As a result, the corresponding degradation products already identified in model experiments should also be present in vivo. Beyond that, lipid peroxidation as well as enzymatic and non-enzymatic pathways irrespective of Maillard reactions contribute to the content of α-DCs. It is speculated that even exogenous sources like foods add to the plasma level of α-DCs.
Because derivatization with OPD is prone to both underestimation of α-DC levels and de novo formation during the derivatization procedure, recovery levels for each compound were thoroughly evaluated, and conversion conditions were kept strictly constant. A blank was prepared for each derivatization set to monitor the quality of all incorporated reagents. The derivatization period was adjusted to provide quantitative conversion of all α-DCs except DHA and oxalic acid, which will be discussed below. The sample deproteinization step by the addition of TFA was kept as short as possible. Immediately after protein separation, the pH was adjusted to pH 3.0 again. Under these conditions, even critical quinoxaline structures like 3-DG-Q proved to be stable, and oxidative degradation of nucleic acids and related compounds as well as degradation of G3P and DHAP, which will lead to overestimation of MGO, were avoided.
α-DCs are known to exhibit a high reactivity toward N-terminal amino acids and amino acid residues of proteins, especially to the sulfhydryl group of cysteine, the guanidino group of arginine, and the ϵ-amino group of lysine. Because these amino functionalities are readily available in excess in human blood plasma, we reason that nearly all α-DCs are bound reversibly or irreversibly to these residues. In the case of MGO, in vitro experiments of Lo et al. (49) showed that only 1% exists in a free form. The reversibly bound MGO is in a dynamic equilibrium with its free form and can be measured by trapping with OPD (50, 51). Therefore, our approach included the separation of the high molecular plasma fraction only after the derivatization procedure to cover the total amount of free and reversibly bound α-DCs in a reliable way. Although, the definition of “reversibly bound α-DCs” remains the object of critical discussion (48) because some reversible reactions under assay conditions might be instead irreversible under physiological conditions, the good validation results proved the reliability of the newly developed workup and derivatization procedure.
The formation of MGO in physiological systems was studied extensively in the literature, reviewed recently by Rabbani and Thornalley (16). The enzymatic and spontaneous degradation of the triose phosphates G3P and DHAP are known sources for MGO. Oxidation of acetone in the catabolism of ketone bodies, oxidation of aminoacetone in the catabolism of threonine, degradation of proteins glycated by glucose, and degradation of ASA also contribute to the total plasma content of MGO. GO is formed by DNA oxidation, lipid peroxidation (52), sugar autoxidation (53), and oxidative degradation of glycated proteins (11). GO and MGO are both detoxified mainly by the glyoxalase system with glutathione as a cofactor to give glycolate and lactate, respectively (55). Because C2- and C3-fragments originate by various non-enzymatic and enzymatic pathways besides the breakdown of glucose and ASA, they must be evaluated as very vague parameters from the mechanistic point of view. The same applies for pyruvic acid as a central intermediate in several metabolic processes, which explains its high concentration in plasma.
The C6-dicarbonyl compound glucosone and its analogues 1-DG and 3-DG do not arise from ascorbate degradation and therefore are markers for glucose-derived α-DCs in the context of Maillard chemistry (see scheme in Fig. 9). 3-DG is formed non-oxidatively from the Amadori product of glucose via 1,2-enolization and dehydration, whereas 2,3-enolization yields 1-DG (56). Oxidation of the Amadori compound leads to glucosone (37). Besides from the Maillard reaction, an important endogenous route leading to 3-DG formation from glucose is the enzymatic polyol pathway (57). Although the bioconversion of glucose into glucosone by pyranose oxidase for synthetic purposes is described in the literature (58), no such enzymatic pathway has been identified in vivo so far.
FIGURE 9.

Formation pathways of α-DCs from glucose and ascorbic acid.
3-DG is reviewed in the literature as the most abundant C6-dicarbonyl in vivo (59) but possesses only a very limited glycating reactivity (37). Thus, the chemistry of 3-DG has to be considered as of minor relevance regarding Maillard processes under physiological conditions. This is supported by the fact that 3-DG-derived AGEs (e.g. Nδ-[5-hydro-5-(2,3,4-trihydroxybutyl)-4-imidazolon-2-yl]ornithine (3-DG-H1), 6-(2-formyl-5-hydroxymethyl-1-pyrrolyl)-l-norleucine (pyrraline), and Nϵ-{2-{[(4S)-4-ammonio-5-oxido-5-oxopentyl]amino}-5-[(2S,3R)-2,3,4-trihydroxybutyl]-3,5-dihydro-4H-imidazol-4-ylidene}-l-lysinate (DODIC or DOGDIC)) are only of minor quantitative importance in blood plasma. 1-DG and glucosone were identified as the central intermediates leading to C4- and C5-fragments, respectively (60). Their reductone structure with an α-oxo-enediol moiety boasts significantly higher reactivity. More generally, this applies to all analog C4- and C5-dicarbonyls. As reported before, the half-life of 1-DG is about 0.5 h under physiological conditions (61) versus 8 h for glucosone (37) and 40 h for 3-DG (36). Hence, 1-DG is by far the most reactive and thus important α-DC intermediate regarding glucose-derived AGEs. In particular, amine-induced β-cleavage in the presence of lysine leads directly to carboxylic acid amides, a novel class of amide-AGEs (14, 62) that are of quantitative importance in vivo (9).
Lederer's glucosone was not detected in the plasma of healthy subjects, although it is a relative stable non-reductone structure like 3-DG. However, unlike 3-DG, the required enolization along the entire carbon backbone makes this α-DC susceptible to multiple degradation processes. The existence of Lederer's glucosone in vivo is evident from the detection of its AGE follow-up structure glucosepane at low levels in human blood and extracellular matrix (63, 64).
DHA is a C6-dicarbonyl structure exclusively assigned to the degradation of ASA, formed by oxidation. As mentioned above, the DHA level in plasma can only be roughly estimated by the analytical assay described herein, mainly because of the incomplete conversion to its corresponding quinoxaline in the given time period. However, DHA in aqueous solution hydrolyzes irreversibly to 2,3-DKG, which is the central intermediate of ASA degradation (65). Therefore, it is important to differentiate between DHA as part of the redox equilibrium ASA-DHA, mediated in vivo by enzymatic pathways, and 2,3-DKG as the direct precursor of fragmentation products with a carbon backbone smaller than C6.
The yields of 2,3-DKG-Q from DHA in Fig. 5 and from DHA precursor-Q in Fig. 7 after 24 h were almost equal, which certainly excludes de novo formation of 2,3-DKG via hydrolyzation of DHA. In addition, we confirmed that ASA and DHA did not yield any C4- or C5-dicarbonyl degradation compounds during the incubation period. Indeed, the plasma level of 2,3-DKG can be estimated fairly accurately, taking into account the comprehensively investigated relationship of DHA-Q and 2,3-DKG-Q under assay conditions. Based on our results, 6.5% of DHA-Q was converted to 2,3-DKG-Q during the derivatization time of 24 h (Figs. 7 and 8). On the other hand, 25% of the formed 2,3-DKG-Q was converted to DHA-Q. The values given in Table 3 for 2,3-DKG are already adjusted accordingly. The adjustments described above are only valid if DHA-Q is in significant excess, which is given by the situation in blood plasma. Only in this case, the change in DHA-Q level due to the conversion is negligible compared with that of 2,3-DKG-Q.
α-DCs with a carbon skeleton smaller than C6 arise from the degradation of glucose as well as of ASA. As established in previous papers of our group (39, 66), the C4-dicarbonyls threosone, 1-DT, and 3-DT are formed from both 1-DG and 2,3-DKG via β-dicarbonyl cleavage with an C4-enediol as the reactive intermediate. Oxidation of the latter leads to threosone, whereas dehydration at C3 results in 3-DT. The enediol may also undergo isomerization to give 1-DT in an equivalent reaction. As a consequence under deaeration, which represents the situation in vivo, 3-DT was the prominent structure because the reductone 1-DT has to be considered a much more reactive and, thus, short lived intermediate.
It has to be noted that differentiation between l-threo-pentos-2-ulose (arising from ASA and often referred to as xylosone) and the C4-stereoisomer d-erythro-pentos-2-ulose (arising from glucose) was not possible. For the applied LC-MS/MS method, MRM parameters were determined with l-threo-pentos-2-ulose. In model incubations of glucose and lysine, a signal with retention time and mass transitions identical to those of l-threo-pentos-2-ulose was detected (data not shown). By definition, with glucose as precursor, the detected compound has to be d-erythro-pentos-2-ulose. Thus, both structures coelute and are therefore summarized under the term pentosone regardless of its origin. Yet, the plasma levels of pentosone were comparatively high and did not fit into the picture. Decarboxylation is a well established mechanism of ASA degradation and leads to C5-compounds, including pentosone (65, 67), but under physiological conditions, the formation of C4-dicarbonyls from 2,3-DKG is favored (25, 39). Glucose-derived pentosone stems from glucosone by the same mechanism of hydrolytic β-cleavage as threosone from 1-DG as the precursor. After isomerization and hydration, formic acid is cleaved off and gives an C5-1,2-enediol as the reactive intermediate. Oxidation leads to pentosone, whereas water elimination yields 3-DP or, after 2,3-enolization, 1-DP (60). Considering the need for an oxidation step in order to obtain pentosone from both 2,3-DKG or glucosone, formation of 1-DP and 3-DP should be favored. The results therefore strongly suggest an additional alternative source for pentosone formation in vivo.
In Maillard chemistry, glyoxylic acid is assigned to disaccharide chemistry (38) but can also arise from oxidation of glyoxal (68) and degradation of DHA (65, 69). However, this cannot account solely for the plasma levels, which were 2.5-fold higher than GO and in the same range as 2,3-DKG. An alternative source is the degradation of hydroxyproline with the subsequent glyoxylate metabolism in the human organism, which is rather complex, involving several enzymatic and non-enzymatic reactions, and has been subject to recent investigation (54).
Oxalic acid can originate via β-dicarbonyl fragmentation as well as via oxidative α-DC cleavage from 2,3-DKG and is the main degradation product of the latter (39). In addition, oxalate is also part of the glyoxylate metabolism mentioned above. However, under assay conditions, the dicarboxylic acid oxalic acid is not converted to its corresponding quinoxaline. This is expected, because at the chosen workup pH, the carboxylic acid groups do not show sufficient carbonyl activity to react with OPD. Consequently, there must be alternative precursors for oxalic acid-Q formation to oxalic acid itself. 3,4-DDP is a known intermediate of maltose degradation but was found in neither the glucose nor ASA reaction systems (38). Hence, the origin of the detected 3,4-DDP-Q remains unknown.
The presented LC-MS/MS method provides for the first time the opportunity to identify and quantitate the complete spectrum of relevant α-DCs in plasma of healthy human subjects in a single chromatographic run. For 14 compounds, the plasma levels were determined. Three compounds were below the limit of quantitation but were unequivocally identified. 10 substances have not been reported for human plasma samples before. To evaluate the clinical relevance of the assay described herein, an initial set of 24 uremic patients undergoing hemodialysis was analyzed. Uremia is related to an increase in oxidative and carbonyl stress and thus should lead to a clear shift in the dicarbonyl spectrum. Indeed, most α-DCs were considerably higher in HD patients. Glucose-derived glucosone exhibited a 2-fold increase, which is expected under conditions of elevated oxidative stress. In contrast, 1-DG does not require an oxidation step for its formation and remained nearly at the level of healthy subjects. Interestingly, plasma levels of 2,3-DKG were considerably decreased in HD patients. This must be explained by a significantly accelerated degradation via oxidative pathways. GO and MGO are further compounds of published interest in regard to certain chronic diseases like uremia. A 3-fold elevation of GO and a 4-fold increase of MGO were observed in uremic plasma. This is in line with the literature (27, 29, 32), although the absolute values differ significantly, depending on the respective study. The assessment of α-DC plasma levels depends strongly on the analytical approach, specifically on workup conditions, derivatization procedure, and chromatographic method. Most importantly, the derivatization was conducted in the presence of protein to assess both free and reversibly bound α-DCs. Therefore, a direct comparison of the plasma levels of the present study and those reported previously is not possible.
Nevertheless, validation of the present method for the detection of α-DC compounds in plasma has been carried out extensively regarding the formation of artifacts and mechanistic relationships to exclude false quantitative data. In general, the elevated levels of α-DCs found explain the elevated levels of AGEs in uremia. The newly developed method has now to be extended to follow-up studies with patients with various complications. The results for a wide range of highly reactive carbonyl intermediates will help us to understand the complex mechanisms and factors that influence α-DC formation and consequently open new perspectives regarding the formation and relevance of AGE chemistry in vivo.
Acknowledgment
We thank Dr. J. Schmidt (Leibnitz Institute of Plant Biochemistry, Halle, Germany) for performing accurate mass determination.
This work was supported by German Federal Ministry of Education and Research (BMBF) Project 13N11798.
- AGE
- advanced glycation end products
- α-DC
- α-dicarbonyl compound
- ASA
- l-threo-ascorbic acid
- DHA
- dehydroascorbic acid
- 2,3-DKG
- 2,3-diketogulonic acid (l-threo-2,3-hexodiulosonic acid)
- 3-DT
- 3-deoxythresosone (4-hydroxy-2-oxobutanal)
- GO
- glyoxal
- MGO
- methylglyoxal (2-oxopropanal)
- 3-DG
- 3-deoxyglucosone (3-deoxy-d-erythro-hexos-2-ulose)
- OPD
- o-phenylenediamine
- threosone
- 3,4-dihydroxy-2-oxobutanal
- 1-DG
- 1-deoxyglucosone (1-deoxy-d-erythro-hexo-2,3-diulose)
- DHA
- dehydroascorbic acid
- DHAP
- dihydroxyacetone phosphate
- G3P
- glyceraldehyde 3-phosphate
- 1-DP
- 1-deoxypentosone
- 3-DP
- 3-deoxypentosone
- 3,4-DDP
- 3,4-dideoxypentosone (3,4-dideoxypentos-2-ulose)
- 1-DT
- 1-deoxythresosone (1-hydroxy-2,3-butanedione)
- Q
- quinoxaline
- HD patients
- patients undergoing hemodialysis
- 3-DGal
- 3-deoxygalactosone
- EtOAc
- ethyl acetate
- 3,4-DGE
- 3,4-dideoxyglucosone-3-ene
- MeOH
- methanol
- CID
- collision-induced dissociation
- glucosone
- d-arabino-hexos-2-ulose
- Lederer's glucosone
- N6-(3,6-dideoxyhexos-2-ulos-6-yl)-l-lysine
- pentosone
- pentos-2-ulose.
REFERENCES
- 1. Monnier V. M., Mustata G. T., Biemel K. L., Reihl O., Lederer M. O., Zhenyu D., Sell D. R. (2005) Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on “a puzzle nearing resolution”. Ann. N.Y. Acad. Sci. 1043, 533–544 [DOI] [PubMed] [Google Scholar]
- 2. Baynes J. W. (2001) The role of AGEs in aging: causation or correlation. Exp. Gerontol. 36, 1527–1537 [DOI] [PubMed] [Google Scholar]
- 3. Tessier F. J., Birlouez-Aragon I. (2012) Health effects of dietary Maillard reaction products: the results of ICARE and other studies. Amino Acids 42, 1119–1131 [DOI] [PubMed] [Google Scholar]
- 4. Degenhardt T. P., Brinkmann-Frye E., Thorpe S. R., Baynes J. W. (1998) Role of carbonyl stress in aging and age-related diseases. in The Maillard Reaction in Foods and Medicine (O'Brien J., ed) pp. 3–10, Royal Society of Chemistry, London [Google Scholar]
- 5. Hamada Y., Araki N., Koh N., Nakamura J., Horiuchi S., Hotta N. (1996) Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem. Biophys. Res. Commun. 228, 539–543 [DOI] [PubMed] [Google Scholar]
- 6. Sell D. R., Lapolla A., Odetti P., Fogarty J., Monnier V. M. (1992) Pentosidine formation in skin correlates with severity of complications in individuals with long-standing IDDM. Diabetes 41, 1286–1292 [DOI] [PubMed] [Google Scholar]
- 7. McCance D. R., Dyer D. G., Dunn J. A., Bailie K. E., Thorpe S. R., Baynes J. W., Lyons T. J. (1993) Maillard reaction products and their relation to complications in insulin-dependent diabetes mellitus. J. Clin. Invest. 91, 2470–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sugiyama S., Miyata T., Ueda Y., Tanaka H., Maeda K., Kawashima S., Van Ypersele de Strihou C., Kurokawa K. (1998) Plasma levels of pentosidine in diabetic patients: an advanced glycation end product. J. Am. Soc. Nephrol. 9, 1681–1688 [DOI] [PubMed] [Google Scholar]
- 9. Henning C., Smuda M., Girndt M., Ulrich C., Glomb M. A. (2011) Molecular basis of maillard amide-advanced glycation end product (AGE) formation in vivo. J. Biol. Chem. 286, 44350–44356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baynes J. W., Thorpe S. R. (1999) Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48, 1–9 [DOI] [PubMed] [Google Scholar]
- 11. Glomb M. A., Monnier V. M. (1995) Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J. Biol. Chem. 270, 10017–10026 [DOI] [PubMed] [Google Scholar]
- 12. Glomb M. A., Lang G. (2001) Isolation and characterization of glyoxal-arginine modifications. J. Agric. Food Chem. 49, 1493–1501 [DOI] [PubMed] [Google Scholar]
- 13. Smuda M., Glomb M. A. (2013) Fragmentation pathways during Maillard-induced carbohydrate degradation. J. Agric. Food Chem. 61, 10198–10208 [DOI] [PubMed] [Google Scholar]
- 14. Smuda M., Voigt M., Glomb M. A. (2010) Degradation of 1-deoxy-d-erythro-hexo-2,3-diulose in the presence of lysine leads to formation of carboxylic acid amides. J. Agric. Food Chem. 58, 6458–6464 [DOI] [PubMed] [Google Scholar]
- 15. Ahmed N., Thornalley P. J. (2002) Chromatographic assay of glycation adducts in human serum albumin glycated in vitro by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and intrinsic fluorescence. Biochem. J. 364, 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rabbani N., Thornalley P. J. (2012) Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 42, 1133–1142 [DOI] [PubMed] [Google Scholar]
- 17. Linden T., Musi B., Järkelid L., Forsbäck G., Kjellstrand P., Deppisch R., Wieslander A. (2001) Glucose degradation products in peritoneal dialysis fluids may have both local and systemic effects: a study of residual fluid and mesothelial cells. Perit. Dial. Int. 21, 607–610 [PubMed] [Google Scholar]
- 18. Tauer A., Bender T. O., Fleischmann E. H., Niwa T., Jörres A., Pischetsrieder M. (2005) Fate of the glucose degradation products 3-deoxyglucosone and glyoxal during peritoneal dialysis. Mol. Nutr. Food Res. 49, 710–715 [DOI] [PubMed] [Google Scholar]
- 19. Ankrah N. A., Appiah-Opong R. (1999) Toxicity of low levels of methylglyoxal: depletion of blood glutathione and adverse effect on glucose tolerance in mice. Toxicol. Lett. 109, 61–67 [DOI] [PubMed] [Google Scholar]
- 20. Degen J., Hellwig M., Henle T. (2012) 1,2-Dicarbonyl compounds in commonly consumed foods. J. Agric. Food Chem. 60, 7071–7079 [DOI] [PubMed] [Google Scholar]
- 21. Arribas-Lorenzo G., Morales F. J. (2010) Analysis, distribution, and dietary exposure of glyoxal and methylglyoxal in cookies and their relationship with other heat-induced contaminants. J. Agric. Food Chem. 58, 2966–2972 [DOI] [PubMed] [Google Scholar]
- 22. Gensberger S., Mittelmaier S., Glomb M. A., Pischetsrieder M. (2012) Identification and quantification of six major α-dicarbonyl process contaminants in high-fructose corn syrup. Anal. Bioanal. Chem. 403, 2923–2931 [DOI] [PubMed] [Google Scholar]
- 23. Schalkwijk C. G., Posthuma N., ten Brink H. J., ter Wee P. M., Teerlink T. (1999) Induction of 1,2-dicarbonyl compounds, intermediates in the formation of advanced glycation end-products, during heat-sterilization of glucose-based peritoneal dialysis fluids. Perit. Dial. Int. 19, 325–333 [PubMed] [Google Scholar]
- 24. Mittelmaier S., Fünfrocken M., Fenn D., Berlich R., Pischetsrieder M. (2011) Quantification of the six major α-dicarbonyl contaminants in peritoneal dialysis fluids by UHPLC/DAD/MSMS. Anal. Bioanal. Chem. 401, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 25. Nemet I., Monnier V. M. (2011) Vitamin C degradation products and pathways in the human lens. J. Biol. Chem. 286, 37128–37136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mirza M. A., Kandhro A. J., Memon S. Q., Khuhawar M. Y., Arain R. (2007) Determination of glyoxal and methylglyoxal in the serum of diabetic patients by MEKC using stilbenediamine as derivatizing reagent. Electrophoresis 28, 3940–3947 [DOI] [PubMed] [Google Scholar]
- 27. Odani H., Shinzato T., Matsumoto Y., Usami J., Maeda K. (1999) Increase in three α,β-dicarbonyl compound levels in human uremic plasma: specific in vivo determination of intermediates in advanced Maillard reaction. Biochem. Biophys. Res. Commun. 256, 89–93 [DOI] [PubMed] [Google Scholar]
- 28. Lapolla A., Reitano R., Seraglia R., Sartore G., Ragazzi E., Traldi P. (2005) Evaluation of advanced glycation end products and carbonyl compounds in patients with different conditions of oxidative stress. Mol. Nutr. Food Res. 49, 685–690 [DOI] [PubMed] [Google Scholar]
- 29. Lapolla A., Flamini R., Lupo A., Aricò N. C., Rugiu C., Reitano R., Tubaro M., Ragazzi E., Seraglia R., Traldi P. (2005) Evaluation of glyoxal and methylglyoxal levels in uremic patients under peritoneal dialysis. Ann. N.Y. Acad. Sci. 1043, 217–224 [DOI] [PubMed] [Google Scholar]
- 30. Knecht K. J., Feather M. S., Baynes J. W. (1992) Detection of 3-deoxyfructose and 3-deoxyglucosone in human urine and plasma: evidence for intermediate stages of the Maillard reaction in vivo. Arch. Biochem. Biophys. 294, 130–137 [DOI] [PubMed] [Google Scholar]
- 31. McLellan A. C., Phillips S. A., Thornalley P. J. (1992) The assay of methylglyoxal in biological systems by derivatization with 1,2-diamino-4,5-dimethoxybenzene. Anal. Biochem. 206, 17–23 [DOI] [PubMed] [Google Scholar]
- 32. Nakayama K., Nakayama M., Iwabuchi M., Terawaki H., Sato T., Kohno M., Ito S. (2008) Plasma α-oxoaldehyde levels in diabetic and nondiabetic chronic kidney disease patients. Am. J. Nephrol. 28, 871–878 [DOI] [PubMed] [Google Scholar]
- 33. Lopez-Anaya A., Mayersohn M. (1987) Ascorbic and dehydroascorbic acids simultaneously quantified in biological fluids by liquid chromatography with fluorescence detection, and comparison with a colorimetric assay. Clin. Chem. 33, 1874–1878 [PubMed] [Google Scholar]
- 34. Margolis S. A., Ziegler R. G., Helzlsouer K. J. (1991) Ascorbic and dehydroascorbic acid measurement in human serum and plasma. Am. J. Clin. Nutr. 54, 1315S–1318S [DOI] [PubMed] [Google Scholar]
- 35. Lykkesfeldt J., Loft S., Nielsen J. B., Poulsen H. E. (1997) Ascorbic acid and dehydroascorbic acid as biomarkers of oxidative stress caused by smoking. Am. J. Clin. Nutr. 65, 959–963 [DOI] [PubMed] [Google Scholar]
- 36. Glomb M. A., Tschirnich R. (2001) Detection of α-dicarbonyl compounds in Maillard reaction systems and in vivo. J. Agric. Food Chem. 49, 5543–5550 [DOI] [PubMed] [Google Scholar]
- 37. Gobert J., Glomb M. A. (2009) Degradation of glucose: reinvestigation of reactive α-dicarbonyl compounds. J. Agric. Food Chem. 57, 8591–8597 [DOI] [PubMed] [Google Scholar]
- 38. Smuda M., Glomb M. A. (2011) Novel insights into the Maillard catalyzed degradation of maltose. J. Agric. Food Chem. 59, 13254–13264 [DOI] [PubMed] [Google Scholar]
- 39. Smuda M., Glomb M. A. (2013) Maillard degradation pathways of vitamin C. Angew. Chem. Int. Ed. Engl. 52, 4887–4891 [DOI] [PubMed] [Google Scholar]
- 40. Hellwig M., Degen J., Henle T. (2010) 3-deoxygalactosone, a “new” 1,2-dicarbonyl compound in milk products. J. Agric. Food Chem. 58, 10752–10760 [DOI] [PubMed] [Google Scholar]
- 41. Henseke G., Bauer C. (1959) Heterocyclische Verbindungen, V. Chinoxalinsynthesen mit Osonhydrazonen. Chem. Ber. 10.1002/cber.19590920236 [DOI] [Google Scholar]
- 42. Otsuka M., Kurata T., Arakawa N. (1986) Isolation and characterization of an intermediate product in the degradation of 2,3-diketo-l-gulonic acid. Agric. Biol. Chem. 50, 531–533 [Google Scholar]
- 43. Arun V., Robinson P. P., Manju S., Leeju P., Varsha G., Digna V., Yusuff K. K. M. (2009) A novel fluorescent bisazomethine dye derived from 3-hydroxyquinoxaline-2-carboxaldehyde and 2,3-diaminomaleonitrile. Dyes Pigments 82, 268–275 [Google Scholar]
- 44. Materials Testing Standards Committee NA 062 of the Deutsches Institut fuer Normung (German Institute for Standardization) (1994) DIN 32645; Chemical Analysis: Decision Limit, Detection Limit and Determination Limit; Estimation in Case of Repeatability; Terms, Methods, Evaluation, Beuth Verlag, Berlin [Google Scholar]
- 45. Mittelmaier S., Fünfrocken M., Fenn D., Fichert T., Pischetsrieder M. (2010) Identification and quantification of the glucose degradation product glucosone in peritoneal dialysis fluids by HPLC/DAD/MSMS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 877–882 [DOI] [PubMed] [Google Scholar]
- 46. Mittelmaier S., Fünfrocken M., Fenn D., Pischetsrieder M. (2011) 3-Deoxygalactosone, a new glucose degradation product in peritoneal dialysis fluids: identification, quantification by HPLC/DAD/MSMS and its pathway of formation. Anal. Bioanal. Chem. 399, 1689–1697 [DOI] [PubMed] [Google Scholar]
- 47. Chaplen F. W., Fahl W. E., Cameron D. C. (1996) Detection of methylglyoxal as a degradation product of DNA and nucleic acid components treated with strong acid. Anal. Biochem. 236, 262–269 [DOI] [PubMed] [Google Scholar]
- 48. Chaplen F. W., Fahl W. E., Cameron D. C. (1998) Evidence of high levels of methylglyoxal in cultured Chinese hamster ovary cells. Proc. Natl. Acad. Sci. U.S.A. 95, 5533–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lo T. W., Westwood M. E., McLellan A. C., Selwood T., Thornalley P. J. (1994) Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N-α-acetylarginine, N-α-acetylcysteine, and N-α-acetyllysine, and bovine serum albumin. J. Biol. Chem. 269, 32299–32305 [PubMed] [Google Scholar]
- 50. Dhar A., Desai K., Liu J., Wu L. (2009) Methylglyoxal, protein binding and biological samples: are we getting the true measure? J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877, 1093–1100 [DOI] [PubMed] [Google Scholar]
- 51. Chaplen F. W., Fahl W. E., Cameron D. C. (1996) Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal. Biochem. 238, 171–178 [DOI] [PubMed] [Google Scholar]
- 52. Fu M. X., Requena J. R., Jenkins A. J., Lyons T. J., Baynes J. W., Thorpe S. R. (1996) The advanced glycation end product, N-ϵ-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 271, 9982–9986 [DOI] [PubMed] [Google Scholar]
- 53. Wells-Knecht K. J., Zyzak D. V., Litchfield J. E., Thorpe S. R., Baynes J. W. (1995) Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry 34, 3702–3709 [DOI] [PubMed] [Google Scholar]
- 54. Duarte N. C., Becker S. A., Jamshidi N., Thiele I., Mo M. L., Vo T. D., Srivas R., Palsson B. Ø. (2007) Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. U.S.A. 104, 1777–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thornalley P. J. (1998) Glutathione-dependent detoxification of α-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem. Biol. Interact. 111, 137–151 [DOI] [PubMed] [Google Scholar]
- 56. Niwa T. (1999) 3-Deoxyglucosone: metabolism, analysis, biological activity, and clinical implication. J. Chromatogr. B Biomed. Sci. Appl. 731, 23–36 [DOI] [PubMed] [Google Scholar]
- 57. Tsukushi S., Katsuzaki T., Aoyama I., Takayama F., Miyazaki T., Shimokata K., Niwa T. (1999) Increased erythrocyte 3-DG and AGEs in diabetic hemodialysis patients: role of the polyol pathway. Kidney Int. 55, 1970–1976 [DOI] [PubMed] [Google Scholar]
- 58. Karmali A., Coelho J. (2011) Bioconversion of d-glucose into d-glucosone by glucose 2-oxidase from Coriolus versicolor at moderate pressures. Appl. Biochem. Biotechnol. 163, 906–917 [DOI] [PubMed] [Google Scholar]
- 59. Vistoli G., De Maddis D., Cipak A., Zarkovic N., Carini M., Aldini G. (2013) Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic. Res. 47, 3–27 [DOI] [PubMed] [Google Scholar]
- 60. Glomb M. A., Gobert J., Voigt M. (2010) Dicarbonyls from Maillard degradation of glucose and maltose. ACS Symp. Ser. 1042, 35–44 [Google Scholar]
- 61. Glomb M. A., Pfahler C. (2000) Synthesis of 1-deoxy-d-erythro-hexo-2,3-diulose, a major hexose Maillard intermediate. Carbohydr. Res. 329, 515–523 [DOI] [PubMed] [Google Scholar]
- 62. Glomb M. A., Pfahler C. (2001) Amides are novel protein modifications formed by physiological sugars. J. Biol. Chem. 276, 41638–41647 [DOI] [PubMed] [Google Scholar]
- 63. Biemel K. M., Friedl D. A., Lederer M. O. (2002) Identification and quantification of major maillard cross-links in human serum albumin and lens protein. Evidence for glucosepane as the dominant compound. J. Biol. Chem. 277, 24907–24915 [DOI] [PubMed] [Google Scholar]
- 64. Sell D. R., Biemel K. M., Reihl O., Lederer M. O., Strauch C. M., Monnier V. M. (2005) Glucosepane is a major protein cross-link of the senescent human extracellular matrix. Relationship with diabetes. J. Biol. Chem. 280, 12310–12315 [DOI] [PubMed] [Google Scholar]
- 65. Shin D. B., Feather M. S. (1990) The degradation of l-ascorbic acid in neutral solutions containing oxygen. J. Carbohydr. Chem. 9, 461–469 [Google Scholar]
- 66. Voigt M., Glomb M. A. (2009) Reactivity of 1-deoxy-d-erythro-hexo-2,3-diulose: a key intermediate in the Maillard chemistry of hexoses. J. Agric. Food Chem. 57, 4765–4770 [DOI] [PubMed] [Google Scholar]
- 67. Reihl O., Lederer M. O., Schwack W. (2004) Characterization and detection of lysine-arginine cross-links derived from dehydroascorbic acid. Carbohydr. Res. 339, 483–491 [DOI] [PubMed] [Google Scholar]
- 68. Rossner J., Velisek J., Pudil F., Davidek J. (2001) Strecker degradation products of aspartic and glutamic acids and their amides. Czech J. Food Sci. 19, 41–45 [Google Scholar]
- 69. Takagi M., Morita N. (1987) Active oxygens and the peroxidation of linoleic acid catalysed by degraded species of ascorbic acid. Bioelectrochem. Bioenerg. 18, 171–178 [Google Scholar]