

Abstract
PI3Kα, a heterodimeric lipid kinase, catalyzes the conversion of phosphoinositide-4,5-bisphosphate (PIP2) to phosphoinositide-3,4,5-trisphosphate (PIP3), a lipid that recruits to the plasma membrane proteins that regulate signaling cascades that control key cellular processes such as cell proliferation, carbohydrate metabolism, cell motility, and apoptosis. PI3Kα is composed of two subunits, p110α and p85, that are activated by binding to phosphorylated receptor tyrosine kinases (RTKs) or their substrates. The gene coding for p110α, PIK3CA, has been found to be mutated in a large number of tumors; these mutations result in increased PI3Kα kinase activity. The structure of the complex of p110α with a fragment of p85 containing the nSH2 and the iSH2 domains has provided valuable information about the mechanisms underlying the physiological activation of PI3Kα and its pathological activation by oncogenic mutations. This review discusses information derived from x-ray diffraction and theoretical calculations regarding the structural and dynamic effects of mutations in four highly mutated regions of PI3K p110α, as well as the proposed mechanisms by which these mutations increase kinase activity. During the physiological activation of PI3Kα, the phosphorylated tyrosine of RTKs binds to the nSH2 domain of p85, dislodging an inhibitory interaction between the p85 nSH2 and a loop of the helical domain of p110α. Several of the oncogenic mutations in p110α activate the enzyme by weakening this autoinhibitory interaction. These effects involve structural changes as well as changes in the dynamics of the enzyme. One of the most common p110α mutations, H1047R, activates PI3Kα by a different mechanism: it increases the interaction of the enzyme with the membrane, maximizing the access of the PI3Kα to its substrate PIP2, a membrane lipid.
Keywords: PIK3R1, p85, PIK3CA, PI3K, Somatic mutation, PIP2, PIP3
Introduction
Class I phosphatidylinositide-3-kinases (PI3Ks) are lipid kinases that catalyze the phosphorylation of phosphoinositide-4,5-bisphosphate (PIP2) at the 3-position of the inositol to generate phosphoinositide-3,4,5-trisphosphate (PIP3). PIP3 acts as a second messenger by recruiting to the membrane proteins that contain a pleckstrin homology (PH) domain (Vanhaesebroeck and Alessi 2000). This results in activation of several important signaling proteins including AKT (also called protein kinase B, PKB), a serine/threonine protein kinase that regulates cell proliferation, carbohydrate metabolism, cell motility and apoptosis, and other cellular processes (Vanhaesebroeck and Waterfield 1999; Katso et al. 2001).
Class I PI3Ks are further divided into subclasses IA and IB. PI3Kα, PI3Kβ, and PI3Kδ belong to subclass IA and PI3Kγ to subclass IB. Subclass IA kinases are heterodimers composed of two subunits, a catalytic p110 subunit and a regulatory p85 subunit. The three PI3K isoforms (PI3Kα, PI3Kβ, and PI3Kδ) contain different p110 sequences (p110α, p110β, and p110δ) (Gabelli et al. 2010).
The action of PI3K is reversed by PTEN (phosphatase and tensin homolog), an enzyme that hydrolyzes the phosphate at the 3−position of PIP3, regenerating PIP2. PTEN is a well–characterized tumor suppressor, suggesting that genes encoding PI3Ks might be oncogenes. Indeed, PIK3CA, the gene encoding p110α, has been found to be mutated with high frequency in a wide variety of tumors (Samuels et al. 2004; Rudd et al. 2011). To date, over 5,500 mutations have been identified in p110α (Forbes et al. 2011). Furthermore, the mutant proteins that have been characterized show an increased PI3Kα activity (Kang et al. 2004; Carson et al. 2008; Zhao and Vogt 2008a, b).
Due to its high clinical relevance, many efforts are underway to understand the mechanism of PI3K activation by its physiological activators and by oncogenic mutations. Advances in this area are crucial to the design PI3K-targeted therapies. This review discusses recent advances in understanding the mechanism of the physiological activation of PI3K as well as the ways by which oncogenic mutations increase PI3K activity.
Structure and activation of PI3Kα
The p110α subunit of PI3Kα is composed of five domains: an adaptor binding domain (ABD; residues 1–108), a Ras binding domain (RBD; 191–291), a C2 domain (residues 330–480), a helical domain (residues 525–696), and a kinase domain (residues 697–1068) (Walke et al. 1999). The p85α subunit is also composed of five domains: an SH3 domain, a GAP domain, two SH2 domains (nSH2 and cSH2) separated by a coiled-coil, the inter-SH2 or iSH2 domain (Fig. 1a) (Dhand et al. 1994).
Fig. 1.
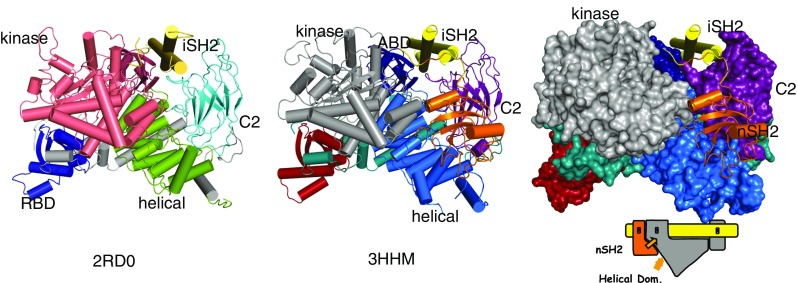
Structure of PI3Kα. a Structure of wild-type p110α/ni p85α heterodimer (pdb id 2RD0); the nSH2 domain is not shown because the experimental electron density for this domain was weak and diffuse. b Structure of the p110α/ni p85α heterodimer of the hot spot mutant H1047R showing the nSH2 domain. c Surface representation of the H1047R p110α/ni p85α with the kinase, C2 domains and helical in contact with nSH2
The multidomain architecture of p110 and p85 contribute to the mechanisms through which activation of PI3Kα is controlled (Geering et al. 2007). Although p85 is necessary for the cellular stability of PI3Kα, the baseline kinase activity of p110 is partially inhibited by binding p85. The nSH2 domain of p85 is responsible for this inhibition (Wu et al. 2007; Carson et al. 2008). PI3Kα is activated by binding, through its nSH2 (and cSH2) domain (Shepherd et al. 1997), to receptor tyrosine kinases (RTKs) or their substrates (such as insulin receptor substrate 1, IRS−1) once activated by phosphorylation. Binding to phosphorylated activators relieves the partial inhibition of PI3Kα by the nSH2 domain (Shepherd et al. 1997). In this sense, activation of the enzyme represents a reversal of the autoinhibition by the nSH2 domain.
The structure of a human PI3Kα heterodimer containing the complete p110α in complex with the nSH2 and the iSH2 domains of p85 (Mandelker et al. 2009) (pdb id 3HMM) provided a rationale for the importance of the nSH2 domain (Fig. 1b, c). In the structure, all p110 domains have interfaces with one another as well as with domains of p85. The iSH2 domain of p85 is an extended coiled-coil that traverses the complex and interacts directly with the ABD and the C2 domain of p110 (Huang et al. 2007; Miled et al. 2007; Hon et al. 2012) (Fig. 1). The nSH2 domain has a key central position, contacting the helical, the kinase, and the C2 domains of p110, and is connected by chain continuity to the iSH2 coiled-coil (Mandelker et al. 2009). These interactions, in particular those with the kinase domain, are responsible for the inhibition of the PI3K activity in the absence of phosphorylated substrates. A key interface involves the interaction of an exposed loop of the helical domain with a groove of the nSH2 domain (Fig. 2a). In this interface, the most noticeable contacts are formed between the positively charged groove of the nSH2 and two negatively charged residues of the loop of the p110 helical domain: Glu542 and Glu545. Although the structure of the p110α/niSH2 p85 with a bound phosphotyrosine peptide (pY-peptide) is not available, the structures of the isolated nSH2 domain of human p85 in complex with several pY-peptides containing p-Tyr-Met/Val-Xaa-Met sequences have been determined (PDB id 2IUI, 2IUH) (Holt et al. 1994; Nolte et al. 1996). Using these structures, the position of the pY-peptides in the p110α/niSH2 p85structure can be accurately modeled. Interestingly, the pY-peptides bind in the same groove that in the heterodimer is occupied by the exposed loop of the helical domain (Mandelker et al. 2009). This observation provides a simple explanation for the activation of PI3Kα by phosphorylated tyrosines of activated RTKs or their activated substrates: binding of the phosphorylated tyrosine (pY) is incompatible with the binding of nSH2 to the same position, thereby releasing its inhibition of the enzyme mediated by nSH2. It is important to realize that, if the exposed loop of the helical domain were continuously occupying the nSH2 groove, it would not be possible for the pY to bind. For relief of inhibition to be possible, the equilibrium ensemble of the heterodimer must include conformations in which the nSH2 is not interacting with the helical domain (Burke et al. 2011). In other words, physiological activation by the pY of the RTKs relies on the dynamics of the protein. Support for this concept can be found by comparing the structures of the wild-type (WT) p110α/niSH2 p85 heterodimer and that of the H1047R mutant (alone and in complex with the inhibitor wortmannin) (Fig. 3). While the structure of the WT complex shows diffuse density for the nSH2 domain (PDB id 2RD0, 4A55), the H1047R structures have well-defined densities for nSH2 (PDB id 3HHM, 3HIZ), suggesting that the WT structure contains members of the ensemble in which the nSH2 is not in contact with the helical domain (Huang et al. 2007; Mandelker et al. 2009; Hon et al. 2012). Additional support for the dynamic nature of the interaction of the nSH2 domain of p85 with the helical domain of p110 was found in hydrogen/deuterium exchange mass spectrometry experiments (Burke et al. 2012). Also, normal mode analysis using either all-atom modes or elastic network models showed that the nSH2 experiences large high-amplitude, low-frequency movements in the WT protein (Fig. 4, see below). It should be noted that it is possible that the mechanism of activation may be isoform specific.
Fig. 2.
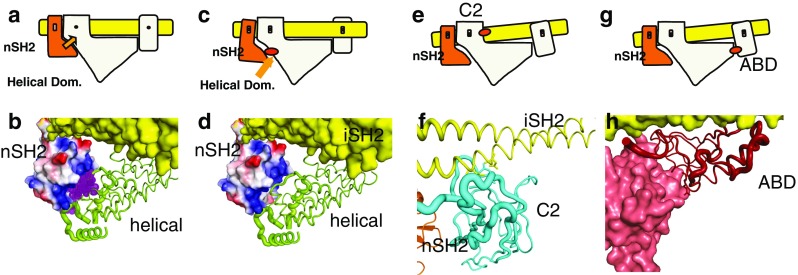
Schematic representation of the mechanism of activation by phosphorylated RTK peptides and by oncogenic mutations at domain interfaces. a Wild-type resting PI3K. Orange arrow indicates inhibition by nSH2. b Binding of phosphorylated peptide (purple) disrupts the inhibitory interaction as shown diagramatically in (c). d Mutation in the helical domain have the same effect as phosphorylated binding shown in (c and e). e Mutations in the C2 domain of p110α change the C2/iSH2 interaction resulting in a weakening of the inhibitory interaction of the nSH2 with the helical domain. g, h ABD mutations in the ABD/Kinase domain affect the ABD/iSH2 interaction at one end, which in turn lessens the nSH2/ helical domain interaction
Fig. 3.

Position of the H1047R mutation in the p110α/ni p85αheterodimer structure. The top of the figure corresponds to the position of the membrane surface. The position of ATP is derived from the structure 1e8x (Walker et al. 1999). The kinase domain is colored gray and the other domains have been omitted
Fig. 4.

Fluctuations of the nSH2 and iSH2 p85α domains. Normal modes were computed by the Anisotropic Network Model (Atilgan et al. 2001). The profiles calculated by generating random linear combinations of the amplitudes of the lowest first six (left) or first 12 (right) vibrational modes. Fluctuations of the wild-type PI3Kα (p110α/ni SH2 p85α) are in orange, fluctuations of the E542R/E545R mutant in the p110α/niSH2 p85α are in gray, and those of the E542K/E545K mutant in the p110α/nip85α complex are in green
Oncogenic mutations
Mutations in PIK3CA, the gene that codes for the p110α subunit of the PI3Kα, have been found in diverse tumors including those of the breast, squamous cell lung carcinoma, brain, colon, head and neck, uterus, ovary, cervical, and stomach (Bachman et al. 2004; Broderick et al. 2004; Campbell et al. 2004; Samuels et al. 2004; Levine et al. 2005). Many of these mutations are present in four “highly mutated regions (HMRs)” in the ABD, the C2, the helical, and the kinase domains (Gymnopoulos et al. 2007; Vogt et al. 2007), including two “hot spots” (in the helical and kinase domains). The structures of the p110α/niSH2 p85 provide insight into the mechanisms by which these mutations may result in higher enzymatic activity (Huang et al. 2007; Carson et al. 2008; Zhao and Vogt 2008a, b; Mandelker et al. 2009; Hon et al. 2012). Interestingly, three of the HMRs affect residues that are located at interfaces between pairs of PI3K domains: the helical and the nSH2 domain, the C2 and the iSH2 domains, and the ABD and the kinase domain.
Two glutamate residues in the helical domain, Glu542 and Glu545 are frequently mutated to positively charged residues in tumors (Bachman et al. 2004; Broderick et al. 2004; Campbell et al. 2004; Lee et al. 2005; Levine et al. 2005; Engelman et al. 2006). As mentioned above, the structures of p110α/niSH2 complexes show that these residues are directly involved in the interaction of the helical domain and the nSH2 domain of p85. Mutations at these positions weaken the inhibitory interaction of the nSH2 domain in a manner similar to that of binding pY. That is, mutations at this HMR activate the enzyme by the same mechanism employed by the physiological activation. If this mechanism of activation is operational, these mutants should not show further activation by binding pY-peptides. This is indeed the case: addition of pY-peptides at concentrations that significantly increase the activity of the WT do not increase the activity of the mutants (Carson et al. 2008). These observations suggest that the effect of these mutations is to increase the fraction of the time that the nSH2 domains are not in an inhibitory position; i.e. the amplitude of the excursions of the nSH2 away from the helical and the kinase domains should be larger. One way to look at these motions is to use of normal mode analysis (Eyal et al. 2011; Gur et al. 2013). Normal mode analysis of the helical domain double mutants E542K/E545K and E542R/E545R shows that, in both cases, the nSH2 domain of p85 experiences a much larger amplitude of movement (expressed as the average fluctuations) than in the WT protein (Fig. 4; unpublished results). The regions with increased mobility in the mutants are concentrated in three regions of the nSH2: residues 380–410, around residue 340, and around residue 360 (Fig. 2). In a comparable region of the iSH2, shown as a control, although there are regions with high mobility, the amplitude of the fluctuations is the same for the WT and the mutants (Figs. 2, 4).
Another HMR is present in the C2 domain, where Asn345 is frequently mutated to lysine. This residue is within hydrogen bonding distance (2.8 and 3.0 Å) of Asn564 and Asp560 of iSH2, respectively. Replacing Asn345 will disrupt one of the two major interactions between the p110 and the p85 subunits. This weakening of the p110–p85 interaction will be transmitted to the nSH2 domain and reduce the autoinhibitory interaction between the nSH2 domain of p85 and the p110 subunit (Fig. 2). At the time this mechanism was proposed, no mutations had been identified in the p85 subunit. This situation changed after the discovery of Asn560 and Asn564 mutations in p85 (CGARN 2008). As these residues are those in H-bonding contact with Asn345 of the C2 domain, the p85 mutations should have a similar effect to those of the mutations in the C2 domain of p110.
The third HMR involves two residues of the ABD: Arg38 and Arg88. These two arginines are located at a contact between the ABD and the kinase domains, within hydrogen bonding distance of Gln738, Asp743, and Asp746, three residues in consecutive turns of a helix of the kinase domain. A deletion mutant of the ABD domain has higher enzymatic activity than the WT (Zhao and Vogt 2008a, b). Disruption of these interactions will weaken the interaction between the ABD and the kinase domain (Fig. 2). As a result, the main attachment of the end of the p85 iSH2 coiled-coil farthest from the nSH2 is hindered. Given the intrinsic rigidity of the coiled-coil, the disruption can be transmitted to the opposite end of the coil, where it connects to the nSH2, which then loses its strong inhibitory interaction with the helical domain. This mechanism provides an answer for the most puzzling question raised by this mutation: how can a mutation so far from the nSH2 domain relieve the inhibition by nSH2?
The fourth HMR, the most common location of p110α mutations in tumors (a hot-spot), includes the H1047R mutation (Fig. 2). Understanding the mechanism of activation by this mutant required the determination of its structure. As mentioned above, the structure of this mutant was determined alone and in the covalent complex with the pan-PI3K inhibitor wortmannin (Mandelker et al. 2009). Although the overall structure of the mutant is highly similar to that of the WT protein, the two differ in one important way involving the residues that follow the mutation, 1047–1062 (Fig. 3). In the WT, the main chain reverses direction at residue H1047, continues for one additional residue, and becomes disordered thereafter. In contrast, in the mutant, the direction of the main chain is changed in such a way that it continues straight from the position of the arginine, changes direction at position 1053, and forms a complex loop that extends from Thr1053 to the end of the chain at residue Ile1062. In this new conformation, residues following residue 1053 are in a position where they can interact with the cell membrane. In addition, ordering of residues 1050–1062 requires a change in the conformation of another loop of the kinase domain (residues 864–874) that had been previously proposed to interact with the membrane. These two changes strongly suggest that the H1047R mutation alters the interaction of PI3K with the cell membrane. If this is the case, the elevated catalytic activity could be the result of an increased interaction of the enzyme with the membrane, which, in turn, would increase the accessibility of the enzyme to its substrate PIP2, a membrane component. Partial support for this mechanism was provided by experiments utilizing lipid vesicles of different compositions that included PIP2 as the substrate (Mandelker, Gabelli et al. 2009). In these experiments, the sensitivity of the kinase activity to the lipid composition of the vesicles was quite different between the WT and the H1047R mutant, suggesting that the two proteins interact differently with membranes. A study using surface plasmon resonance aimed at examining lipid binding by PI3Kα in immobilized liposomes provides independent support for this hypothesis (Hon et al. 2012).
This mechanism may also explain why the activity of the H1047R mutant is independent of Ras binding (Zhao and Vogt 2008a, b). Ras is attached to the cell membrane through a farnesyl group and binds p110α through the Ras-binding domain (RBD), suggesting that Ras may activate the WT form of p110α by increasing tethering of PI3Kα to the cell membrane. The H1047R mutation may circumvent the need for Ras by directly increasing the interaction of p110α with the membrane by virtue of the altered conformations of loops 864–874 and 1050–1062. Furthermore, when a myristoylation site is engineered into an otherwise WT p110α, the expressed protein, now constitutively attached to the membrane, activates AKT and other downstream targets in a manner similar to that of the His1047Arg mutant.
Summary
PI3Kα is a signaling heterodimer composed of two subunits, p110α and p85α, that play crucial roles in cell growth, differentiation, apoptosis, and metabolism. The gene coding for the p110α subunit, PIK3CA, is frequently mutated in tumors of diverse origins. PI3Kα expressing these mutations exhibits increased kinase activity resulting in activation of signaling pathways that may contribute to tumor aggressiveness. The structures of PI3K complexes consisting of both the catalytic subunit p110α and portions of the regulatory subunit p85 provided invaluable information about the mechanism of the physiological activation of PI3Kα as well as about the mechanism by which oncogenic mutations increase PI3Kα activity. The most common mutations are proposed to activate the enzyme by two mechanisms: release of the autoinhibition by the nSH2 domain of p85 and increase of the interaction of the protein with the membrane that results in a greater accessibility of the enzyme to its substrate, a membrane component. Insights provided by these structures are crucial for the development of novel strategies aimed at creating more effective chemotherapeutic interventions against tumors harboring these mutations. Design of isoform-specific and mutation-specific inhibitors will be helped by the availability of inhibitor complexes of PI3Kα as well of those of other PI3K isoforms (Mandelker et al. 2009; Berndt et al. 2010; Certal et al. 2012; Hon et al. 2012; Murray et al. 2012).
Acknowledgments
This work was supported by the Virginia and D.K. Ludwig Fund for Cancer Research and by NIH grant CA43460. SBG is a Stewart Trust fellow.
Conflict of interest
Authors Sandra B. Gabelli, Ignacia Echeverria, Megan Alexander, Krisna C. Duong-Ly, Daniele Chaves-Moreira, Evan T. Brower, B. Vogelstein, and L. Mario Amzel declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Footnotes
Special Issue Advances in Biophysics in Latin America
Contributor Information
Sandra B. Gabelli, Email: gabelli@jhmi.edu
L. Mario Amzel, Email: mamzel@jhmi.edu.
References
- Atilgan A, Durell S, et al. Anisotropy of fluctuation dynamics of proteins iwth an elastic network model. Biophys J. 2001;80(1):505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman KE, Argani P, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3(8):772–775. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- Berndt A, Miller S, et al. The p110delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat Chem Biol. 2010;6(3):244. doi: 10.1038/nchembio0310-244b. [DOI] [PubMed] [Google Scholar]
- Broderick DK, Di C, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64(15):5048–5050. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- Burke JE, Perisic O, et al. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA) Proc Natl Acad Sci USA. 2012;109(38):15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Vadas O, et al. Dynamics of the phosphoinositide 3-kinase p110delta interaction with p85alpha and membranes reveals aspects of regulation distinct from p110alpha. Structure. 2011;19(8):1127–1137. doi: 10.1016/j.str.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IG, Russell SE, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64(21):7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- Carson JD, Van Aller G, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409(2):519–524. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- Certal V, Halley F, et al. Discovery and optimization of new benzimidazole- and benzoxazole-pyrimidone selective PI3Kbeta inhibitors for the treatment of phosphatase and TENsin homologue (PTEN)-deficient cancers. J Med Chem. 2012;55(10):4788–4805. doi: 10.1021/jm300241b. [DOI] [PubMed] [Google Scholar]
- Cgarn CGARN. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhand R, Hara K, et al. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 1994;13(3):511–521. doi: 10.1002/j.1460-2075.1994.tb06289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Luo J, et al. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Eyal, E., A. Dutta, et al. (2011) Cooperative dynamics of proteins unraveled by network models. WIREs Computational Molecular Science 1 (May-June) [DOI] [PMC free article] [PubMed]
- Forbes SA, Bindal N, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli SB, Mandelker D, et al. Somatic mutations in PI3Kalpha: structural basis for enzyme activation and drug design. Biochim Biophys Acta. 2010;1804(3):533–540. doi: 10.1016/j.bbapap.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering B, Cutillas PR, et al. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci USA. 2007;104(19):7809–7814. doi: 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur M, Zomot E et al (2013) Global motions exhibited by proteins in micro- to milliseconds simulations concur with anisotropic network model predictions. J Chem Phys 139(121912) [DOI] [PMC free article] [PubMed]
- Gymnopoulos M, Elsliger MA, et al. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci USA. 2007;104(13):5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt KH, Olson L, et al. Phosphatidylinositol 3-kinase activation is mediated by high-affinity interactions between distinct domains within the p110 and p85 subunits. Mol Cell Biol. 1994;14(1):42–49. doi: 10.1128/mcb.14.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon WC, Berndt A, et al. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene. 2012;31(32):3655–3666. doi: 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Mandelker D, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318(5857):1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- Kang S, Bader AG, et al. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA. 2004;102(3):802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- Lee JW, Soung YH, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24(8):1477–1480. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- Levine DA, Bogomolniy F, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11(8):2875–2878. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- Mandelker D, Gabelli SB, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106(40):16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miled N, Yan Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317(5835):239–242. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- Murray JM, Sweeney ZK, et al. Potent and highly selective benzimidazole inhibitors of PI3-kinase delta. J Med Chem. 2012;55(17):7686–7695. doi: 10.1021/jm300717c. [DOI] [PubMed] [Google Scholar]
- Nolte RT, Eck MJ, et al. “Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes”. Nat Struct Biol. 1996;3(4):364–374. doi: 10.1038/nsb0496-364. [DOI] [PubMed] [Google Scholar]
- Rudd ML, Price JC, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17(6):1331–1340. doi: 10.1158/1078-0432.CCR-10-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Siddle K, et al. Is stimulation of class-1 phosphatidylinositol 3-kinase activity by insulin sufficient to activate pathways involved in glucose metabolism. Biochem Soc Trans. 1997;25(3):978–981. doi: 10.1042/bst0250978. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346(Pt 3):561–576. doi: 10.1042/0264-6021:3460561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253(1):239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Kang S, et al. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007;32(7):342–349. doi: 10.1016/j.tibs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, et al. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402(6759):313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Wu H, Yan Y, et al. Regulation of class IA PI3Ks. Biochem Soc Trans. 2007;35(Pt 2):242–244. doi: 10.1042/BST0350242. [DOI] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27(41):5486–5496. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA. 2008;105(7):2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]