

Abstract
Post-translational modification by the small ubiquitin-like modifier-1 (SUMO1) limits insulin secretion from β-cells by inhibiting insulin exocytosis and glucagon-like peptide-1 (GLP-1) receptor signalling. The secretion of glucagon from α-cells is regulated in a manner opposite to that of insulin; it is inhibited by elevated glucose and GLP-1, and increased by adrenergic signalling. We therefore sought to determine whether SUMO1 modulates mouse and human α-cell function. Action potentials (APs), ion channel function and exocytosis in single α-cells from mice and humans, identified by glucagon immunostaining, and glucagon secretion from intact islets were measured. The effects of SUMO1 on α-cell function and the respective inhibitory and stimulatory effects of exendin 4 and adrenaline were examined. Upregulation of SUMO1 increased α-cell AP duration, frequency and amplitude, in part as a result of increased Ca2+ channel activity that led to elevated exocytosis. The ability of SUMO1 to enhance α-cell exocytosis was cAMP-dependent and resulted from an increased L-type Ca2+ current and a shift away from exocytosis dependent on non-L-type channels, an effect that was mimicked by knockdown of the deSUMOylating enzyme sentrin/SUMO-specific protease-1 (SENP1). Finally, although SUMO1 prevented GLP-1 receptor-mediated inhibition of α-cell Na+ channels and single-cell exocytosis, it failed to prevent the exendin 4-mediated inhibition of glucagon secretion. Consistent with its cAMP dependence, however, SUMO1 enhanced α-cell exocytosis and glucagon secretion stimulated by adrenaline. Thus, by contrast with its inhibitory role in β-cell exocytosis, SUMO1 is a positive regulator of α-cell exocytosis and glucagon secretion under conditions of elevated cAMP.
Key points
SUMOylation is the reversible modification of proteins by the attachment of small ubiquitin-like modifier (SUMO) peptides, which in pancreatic β-cells inhibits insulin exocytosis and glucagon-like peptide-1 (GLP-1) receptor signalling.
We find in glucagon-secreting pancreatic α-cells that SUMOylation increases excitability and enhances exocytosis by increasing L-type Ca2+ currents.
The ability of SUMOylation to facilitate α-cell exocytosis is cAMP-dependent, leading to enhanced adrenaline-stimulated glucagon secretion.
SUMO1 prevents inhibition of α-cell Na+ current and exocytosis by a GLP-1 receptor agonist, but does not prevent GLP-1 receptor-dependent inhibition of glucagon secretion.
SUMOylation modifies α-cell responses to cAMP-dependent signalling and, by contrast with its inhibitory effects in β-cells, enhances α-cell exocytosis and glucagon secretion.
Introduction
Insulin and glucagon are the two major hormones regulating glucose storage and mobilization, respectively. Their secretion from the pancreatic islets of Langerhans is regulated in opposing manners by metabolic and hormonal inputs. Like β-cells, the function of α-cells depends on action potential (AP) firing, which is observed at low glucose concentrations and is coupled to the exocytosis of glucagon-containing secretory granules (Barg et al. 2000). Several ion channels are required for appropriate α-cell AP firing. Voltage-dependent Na+ (Nav) channels are active at physiological membrane potentials in α-cells, and contribute to the AP upstroke (Göpel et al. 2000; Vignali et al. 2006). Inhibition of these decreases AP amplitudes and blunts glucagon secretion (Göpel et al. 2000; Ernst et al. 2009). Voltage-gated K+ (Kv) channels contribute to α-cell AP repolarization; inhibition of these blunts glucagon secretion (Ramracheya et al. 2010; Spigelman et al. 2010). Voltage-dependent Ca2+ channels (VDCCs) also contribute to the initiation and peak of α-cell APs, and are required for glucagon secretion and α-cell exocytosis. Under low glucose conditions, α-cell exocytosis and glucagon secretion can be blocked by ω-conotoxin (Macdonald et al. 2007), suggesting a coupling to N-type or perhaps P/Q-type VDCCs (Rorsman et al. 2012).
The non-L-type Ca2+ channels contribute <20% of α-cell Ca2+ current, although adrenergic stimulation is suggested to shift α-cell exocytosis towards a greater dependence upon Ca2+ entry through L-type VDCCs (De Marinis et al. 2010; Rorsman et al. 2012). Conversely, the intestinal incretin hormone glucagon-like peptide-1 (GLP-1) inhibits glucagon release (Baggio & Drucker, 2007). GLP-1 analogues and inhibitors of native GLP-1 degradation are now important tools in the clinical management of diabetes (Lovshin & Drucker, 2009; Hare et al. 2010; Henry et al. 2011; Rauch et al. 2012). Although the effects of both incretin and adrenergic receptor signalling are thought be cAMP-dependent, the mechanism(s) that underlie disparate regulation of glucagon secretion by GLP-1 and adrenaline are incompletely understood. One report suggests that α-cell AP firing and exocytosis may be differentially regulated by the magnitude of the cAMP response conferred by differing GLP-1 and adrenergic receptor expression levels in α-cells (De Marinis et al. 2010).
Insulin secretion is regulated by post-translational SUMOylation, whereby a small ubiquitin-like modifier (SUMO) peptide is covalently attached to protein targets (Yeh, 2009). In β-cells, SUMOylation controls ion channel activity, exocytosis, metabolic sensing by glucokinase, and gene transcription (Manning Fox et al. 2012; Aukrust et al. 2013). Additionally, SUMOylation may modulate responsiveness to circulating hormones. GLP-1 receptor signalling in β-cells is downregulated by SUMOylation (Rajan et al. 2012). In addition, the stimulus-dependent re-internalization of β-adrenergic receptors may require SUMOylation of arrestins (Wyatt et al. 2011). The role of SUMOylation in α-cell function has not been investigated.
We therefore sought to examine the role of SUMOylation in α-cell function and glucagon secretion, and to establish whether this modifies responses to GLP-1 and adrenergic receptor activation. We demonstrate that SUMO1 increases α-cell AP firing, which is probably attributable to an upregulation of L-type Ca2+ channels, and enhanced α-cell exocytosis. Although over-expressed SUMO1 also prevented the inhibition of Na+ currents and exocytosis by the GLP-1 receptor agonist exendin 4, it did not prevent the exendin 4-dependent suppression of glucagon secretion. Rather, the ability of SUMO1 to enhance α-cell exocytosis depended on the presence of cAMP within the patch clamp pipette. Indeed, SUMO1 was able to enhance the stimulatory action of forskolin and adrenaline on α-cell exocytosis, and potentiated adrenaline-induced glucagon secretion from intact islets. Thus, SUMOylation exerts an important control of the distal machinery governing glucagon exocytosis in a cAMP-dependent manner. The ability of SUMOylation to enhance α-cell exocytosis and glucagon secretion contrasts with its direct inhibitory action on insulin secretion.
Methods
Cells and cell culture
Mouse islets from male C57BL6 mice (Charles River Laboratories, Montreal, QC, Canada) were isolated by standard collagenase digestion followed by hand-picking and then cultured overnight in Roswell Park Memorial Institute (RPMI) medium (Gibco 11875; Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) (v/v) and 100 U ml−1 penicillin/streptomycin at 37°C and 5% CO2. Human islets from the Clinical Islet Laboratory at the University of Alberta and the Alberta Diabetes Institute IsletCore were cultured for 1–3 days in Dulbecco's modified Eagle's medium (DMEM) (Gibco 11885; Life Technologies) with 10% FBS and 100 U ml−1 penicillin/streptomycin at 37°C and 5% CO2. Islets were dispersed into single cells by incubation for 10 min at 37°C in a Hanks’ solution-based enzyme-free cell dissociation buffer (Life Technologies), followed by mechanical dispersion with a flame-polished glass pipette. All studies were approved by the Animal Care and Use Committee and the Human Research Ethics Board at the University of Alberta.
Constructs, adenoviruses and recombinant peptides
Recombinant adenoviruses expressing green fluorescent protein (GFP) alone (Ad-GFP) or together with SUMO1 (Ad-SUMO1), and expressing an shRNA sequence targeted against SENP1 (Ad-shSENP1) or a scrambled control (Ad-Scrambled), have been characterized (Dai et al. 2009, 2011). Ad-SUMO1 was constructed by cloning the human SUMO1 sequence into the pAdtrackCMV plasmid for adenoviral generation. Sequences for shRNA constructs were as follows: Ad-shSENP1 (GATCCCGCCGAAAGACCTCAAGTGGATTTTCAAGAGAAATCCACT-TGAGGTCTTTCGGTTTTTTCCAAA), and Ad-Scrambled (GATCCCGATTGCGCCAGATTGAAGA-TTCAAGAGATCTTCAATCTGGCGCAATCTTTTTTCCAAA). All constructs co-expressed GFP to allow identification of infected cells prior to patch clamp recordings and to estimate infection efficiency. Recombinant human SUMO1 and glutathione S-transferase (GST) proteins were sourced from Sigma-Aldrich Canada (Oakville, ON, Canada) and infused into cells at 4 μg ml−1.
Glucagon secretion assay
Glucagon secretion was assessed by perifusion using a BioRep PERI4 system with batches of 85 mouse islets at 36–48 h following infection with either Ad-GFP or Ad-SUMO1. Islets were perifused at 50 μl min−1 with Krebs–Ringer bicarbonate Hepes (KRBH) solution containing 140 mm NaCl, 3.6 mm KCl, 2.6 mm CaCl2, 0.5 mm NaH2PO4, 0.5 mm MgSO4·7H2O 5 mm Hepes, 2 mm NaHCO3, 0.05% w/v BSA and glucose as indicated at a pH of 7.45 with NaOH. Islets were pre-incubated at 3 mm glucose for 30 minutes prior to fraction collection at 2 min intervals. Samples were assayed using the Mouse/Rat Glucagon MULTI-ARRAY Assay System from Meso Scale Discovery (Rockville, MD, USA).
Patch clamp electrophysiology
The standard whole-cell technique with the sine+DC lock-in function of an EPC10 amplifier and Patchmaster software (HEKA Electronics, Lambrecht/Pfalz, Germany) was used in experiments performed at 32–35°C. Patch pipettes, pulled from borosilicate glass and coated with Sylgard, had a resistance of 3–4 MΩ when filled with pipette solution; liquid junction potentials were corrected as appropriate. For depolarization-stimulated capacitance measurements and voltage-gated Na+ and Ca2+ current by standard whole-cell patch clamp, the pipette solution contained 125 mm Cs-glutamate, 10 mm CsCl, 10 mm NaCl, 1 mm MgCl2, 0.05 mm EGTA, 5 mm Hepes, 0.1 mm cAMP and 3 mm MgATP (pH 7.15 with CsOH). The extracellular bath contained 118 mm NaCl, 20 mm tetraethylammonium chloride, 5.6 mm KCl, 1.2 mm MgCl2, 2.6 mm CaCl2, 1 mm glucose and 5 mm Hepes (pH 7.4 with NaOH). Some experiments were performed with perforated patch pipettes pulled from thick-walled borosilicate glass tubes, with resistances of 8–10 MΩ when filled with 76 mm K2SO4, 10 mm KCl, 10 mm NaCl, 1 mm MgCl2 and 5 mm Hepes (pH 7.25 with KOH), and back-filled with 0.24 mg ml−1 amphotericin B. The extracellular solution for these experiments consisted of 140 mm NaCl, 3.6 mm KCl, 1.5 mm CaCl2, 0.5 mm MgSO4, 10 mm Hepes, 0.5 mm NaH2PO4, 5 mm NaHCO3, 1 mm glucose (pH 7.3 with NaOH). VDCC activity was also measured by substituting Ba2+ for Ca2+. The pipette solution for this consisted of 140 mm Cs-glutamate, 1 mm MgCl2, 20 mm tetraethylammonium chloride, 5 mm EGTA, 20 mm Hepes and 3 mm MgATP (pH 7.3 with CsOH). The extracellular bath contained 20 mm BaCl2, 100 mm NaCl, 5 mm CsCl, 1 mm MgCl2, 1 mm glucose, 10 mm Hepes and 0.5 μm tetrodotoxin (pH 7.35 with CsOH). For Kv currents, the intracellular solution contained 140 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 10 mm Hepes, 10 mm EGTA and 3 mm MgATP (pH 7.3 with KOH). The bath solution was composed of 135 mm NaCl, 5.4 mm KCl, 1 mm CaCl2, 1.2 mm MgCl2, 10 mm Hepes and 1 mm glucose (pH 7.3 with NaOH).
Blockers of L-type Ca2+ channels (isradipine, 5 μm) and N-type (and perhaps P/Q-type) Ca2+ channels (ω-conotoxin, 100 nm), the GLP-1 receptor agonist exendin 4 (10 nm; Sigma-Aldrich Canada), or adrenaline (10 μm; Sigma-Aldrich Canada) were added to the bath as indicated. Measurements were performed in separate groups of cells (from the same human or mouse islet isolation). Cells were infected 4–6 h after dispersion (as above), and then for an additional 36 h (for over-expression) or 72 h (for shRNA experiments) before patch clamp experiments. Measurements were normalized to initial cell size and expressed as femtofarad (fF) per picofarad (pF) and picoampere (pA) per pF. In all experiments, mouse and human α-cells were positively identified by immunostaining for glucagon (guinea pig anti-glucagon, 1 : 5000; Linco Research, St Charles, MO, USA), and appropriate secondary antibodies (Alexa Fluor 594 goat anti-guinea pig, 1 : 200; Invitrogen, Eugene, OR, USA).
Data analysis
Analysis was performed using Fit-Master software (HEKA Electronik) and SigmaPlot Version 11 (Systat Software, Point Richmond, CA, USA). Groups were compared by multiple-comparison ANOVA followed by Bonferroni or Fisher's least significant difference (LSD) [area under the curve (AUC)] post-tests, or Student's unpaired t test. Data are presented as the mean ± s.e.m. P-values of < 0.05 are considered to indicate statistical significance.
Results
SUMO1 regulates α-cell APs through distinct effects on K+ and Ca2+ currents
SUMO1 inhibits β-cell Kv currents, increasing the duration of APs and reducing firing frequency (Dai et al. 2009). We expressed SUMO1 in mouse α-cells, identified by positive glucagon immunostaining, and examined AP firing in the presence of a hyperpolarizing −2 pA current injection (Fig. 1A and B) because we found previously that this facilitates α-cell AP firing (Spigelman et al. 2010). Ad-SUMO1 (n = 11) increased mouse α-cell AP half-width (P < 0.05), firing frequency (P < 0.01), decreased the threshold for AP firing (P < 0.01), and increased AP amplitudes (P < 0.001) compared with cells infected with the control Ad-GFP (n = 11–15) (Fig. 1A–G). The AP peak potentials were not increased by Ad-SUMO1, suggesting that the elevated AP amplitudes are almost entirely attributable to a lower firing threshold.
Figure 1. SUMO1 increases α-cell excitability and upregulates voltage-dependent Ca2+ currents.
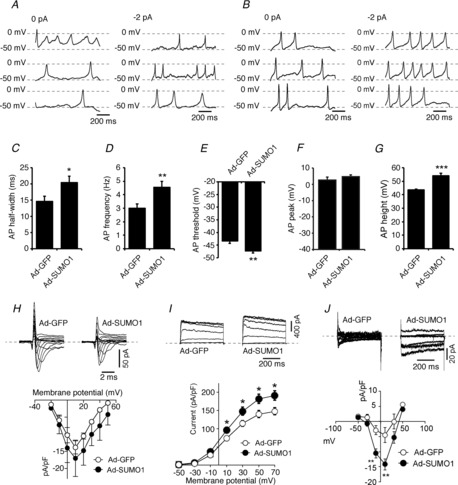
We measured action potentials (APs) from mouse α-cells in the perforated patch condition, positively identified by glucagon immunostaining, with 0 pA and −2 pA current injection. A, representative APs from three different α-cells transduced with green fluorescent protein (Ad-GFP). B, representative APs from three different α-cells transduced with SUMO1 (Ad-SUMO1). Upregulation of SUMO1 increased AP half-width (C) and firing frequency (D), decreased the firing threshold (E), and increased AP amplitudes without affecting peak potential (F, G). We then measured voltage-dependent Na+ (H), K+ (I) and Ba2+ (J) currents from mouse α-cells infected with Ad-GFP (open circles) or Ad-SUMO1 (black circles), positively identified by glucagon immunostaining, in the whole-cell configuration. *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons of Ad-SUMO1 with Ad-GFP.
We assessed the effect of SUMO1 on voltage-gated Na+, K+ and Ca2+ channels in mouse α-cells (Fig. 1H–J). Voltage-dependent Na+ currents, elicited by a series of short (10 ms) depolarizations from −70 mV to voltages between −60 mV and +60 mV, were unaffected by SUMO1 (n = 26). Kv currents were slightly, but significantly, increased in α-cells infected with Ad-SUMO1 (n = 20; P < 0.05) compared with Ad-GFP (n = 15). This may contribute to the observed increase in AP firing frequency. Conversely, voltage-dependent Ba2+ currents (a measure of Ca2+ channel activity) elicited by a 500 ms stepwise depolarization from −70 mV to 0 mV were significantly (n = 8; P < 0.01) increased when SUMO1 was upregulated. This occurred despite a hyperpolarizing shift in VDCC voltage-dependent inactivation (from V50 = −34.6 ± 3.9 mV to −48.5 ± 2.5 mV, n = 14 and n = 13; P < 0.01) that cannot explain the upregulated Ba2+ current. Thus, the increased AP amplitude and duration upon upregulation of SUMO1 are consistent with an increased Ca2+ current.
SUMOylation enhances α-cell exocytosis
Given that SUMO1 increases α-cell Ca2+ currents, we examined whether this translates into an elevated α-cell exocytotic response. Increasing SUMO1 in mouse α-cells enhanced exocytosis triggered by a series of membrane depolarizations (n = 43–50; P < 0.001) (Fig. 2A and B). The deSUMOylating enzyme SENP1 cleaves SUMO1 from its target proteins (Yeh, 2009). Expression of a SENP1-targeted shRNA adenoviral construct (Ad-shSENP1; n = 29) resulted in an increased exocytotic response in mouse α-cells compared with a scrambled control (Ad-Scrambled; n = 22; P < 0.01), similar to the effect of SUMO1. The SUMO1-dependent increase in exocytosis was also observed in human α-cells infected with Ad-SUMO1 (n = 32, n = 33; P < 0.01) (Fig. 2E and F). The direct intracellular infusion of recombinant SUMO1 peptide had no effect on α-cell exocytosis or Ca2+ currents (not shown). Finally, we confirmed that SUMO1 also increases Ca2+ current in human α-cells (n = 29; P < 0.05) (Fig. 2G and H). This increase in Ca2+ current could be prevented by the L-type channel blocker isradipine (5 μm, n = 11) (Fig. 2G and H).
Figure 2. SUMOylation increases the α-cell exocytotic response to membrane depolarization, and upregulates L-type Ca2+ currents.
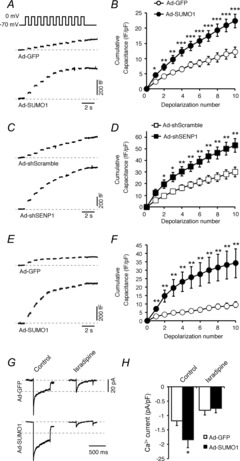
We measured exocytosis as increases in membrane capacitance in mouse and human α-cells, positively identified by glucagon immunostaining, during a series of 10 membrane depolarizations from −70 mV to 0 mV. Representative (A) and quantified (B) responses of mouse α-cells infected with green fluorescent protein (Ad-GFP; open circles) or SUMO1 (Ad-SUMO1; black circles)). Representative (C) and quantified (D) responses of mouse α-cells infected with a scrambled control (Ad-shScramble; open squares) or an shRNA sequence targeted against SENP1 (Ad-shSENP1; black squares)). Representative (E) and quantified (F) responses of human α-cells infected with Ad-GFP (open circles) or Ad-SUMO1 (black circles). Representative (G) and quantified (H) Ca2+ currents from human α-cells infected by Ad-GFP or Ad-SUMO1, elicited by a single depolarization from −70 mV to 0 mV, in the absence or presence of 5 μm isradipine. *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons with the control.
SUMO1-enhanced α-cell exocytosis is dependent on L-type Ca2+ channels
To determine the nature of α-cell exocytosis following upregulation of SUMO1, we examined the effects of the VDCC inhibitors isradipine and ω-conotixin. These block L- and N-type channels, respectively, although it has been suggested that ω-conotoxin may additionally block P/Q-type channels (Rorsman et al. 2012). The exocytotic response in mouse α-cells under control conditions (Ad-GFP) was suppressed by 100 nm ω-conotoxin (by 74%, n = 6) (Fig. 3A, left). We find, however, that ω-conotoxin is ineffective at suppressing exocytosis when SUMO1 is upregulated (n = 13) (Fig. 3A, right). Conversely, although isradipine (5 μm) had little effect on exocytosis in control α-cells (n = 9) (Fig. 3B and C), it prevented the upregulation of α-cell exocytosis by SUMO1 (n = 17; P < 0.01) (Fig. 3B and D). We also examined the effect of ω-conotoxin and isradipine on exocytosis in response to a 500 ms depolarization in mouse α-cells following SENP1 knockdown (Fig. 3E and F). Again, exocytosis was inhibited by ω-conotoxin only under the control condition (Ad-Scrambled; n = 15–24; P < 0.01), whereas isradipine was effective at inhibiting exocytosis in cells infected with Ad-shSENP1 (n = 12–15; P < 0.05) (Fig. 3E and F). Finally, to determine whether increased Ca2+ entry per se is responsible for the SUMO1-dependent increase in α-cell exocytosis, we monitored capacitance upon infusion of 200 nm free Ca2+ (Fig. 3G and H). Under these conditions, Ca2+-induced exocytosis was similar between α-cells infected with Ad-GFP (n = 8) and Ad-SUMO1 (n = 15).
Figure 3. SUMOylation shifts the dependence of exocytosis from non-L-type to L-type voltage-dependent Ca2+ channels (VDCCs).
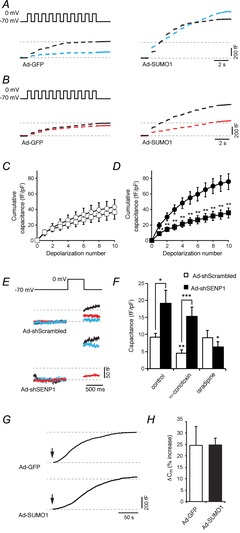
We measured exocytosis as increases in membrane capacitance in mouse α-cells, identified by glucagon immunostaining, in response to membrane depolarization. A, representative capacitance responses of α-cells infected with green fluorescent protein (Ad-GFP) or SUMO1 (Ad-SUMO1) under control conditions (black traces) or in the presence of 100 nm ω-conotoxin (blue traces). B, representative capacitance responses of α-cells infected with Ad-GFP or Ad-SUMO1 under control conditions (black traces) or in the presence of 5 μm isradipine (red traces). C, the cumulative capacitance response from α-cells expressing Ad-GFP in the absence (open circles) and presence (open squares) of 5 μm isradipine. D, the cumulative capacitance response from α-cells expressing Ad-SUMO1 in the absence (black circles) and presence (black squares) of 5 μm isradipine. E, representative capacitance traces in response to a single depolarization of α-cells infected with a scrambled control (Ad-Scrambled) or an shRNA sequence targeted against SENP1 (Ad-shSENP1) in the absence of a voltage-dependent Ca2+ channel (VDCC) inhibitor (black traces), or in the presence of 100 nm ω-conotoxin (blue traces) or 5 μm isradipine (red traces). F, average capacitance responses to the single depolarization in cells infected with Ad-Scrambled (open bars) or Ad-shSENP1 (black bars) in the absence or presence of 100 nm ω-conotoxin and 5 μm isradipine. G, continuous recording of membrane capacitance during infusion of 200 nm free Ca2+ following establishment of whole-cell access (arrows). H, the percentage increase in membrane capacitance (Cm) over 200 s. *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons with the respective control, or as indicated.
SUMO1 prevents the suppression of α-cell Na+ currents and exocytosis by exendin 4
As SUMO1 negatively regulates the trafficking and activity of the GLP-1 receptor in β-cells (Rajan et al. 2012), we examined whether SUMO1 could modulate the cellular response(s) to the GLP-1 receptor agonist exendin 4 in α-cells. In our hands, the effects of exendin 4 on α-cell APs are consistent with an inhibition of voltage-gated Na+ currents, leading to decreased AP height and peak potential (not shown). Indeed, the rapidly inactivating Na+ currents in mouse α-cells, elicited by short (10 ms) depolarizations from −70 mV to voltages between −60 mV and +60 mV, were significantly (n = 8; P < 0.01) blunted by exendin 4 (10 nm) (Fig. 4A and B). This results from a hyperpolarizing shift in voltage-dependent inactivation of the Na+ current from a half-inactivation (V50) of −64.6 ± 1.5 mV (n = 10) to −82.8 ± 1.6 mV (n = 10; P < 0.001) (Fig. 4C and D). Although SUMO1 did not affect α-cell Na+ current amplitude (n = 26) or inactivation time constant at 0 mV (τ = 4.3 ± 0.7 ms versus τ = 3.5 ± 0.7 ms, n = 14, n = 9), consistent with the findings in Fig. 1, infection of α-cells with Ad-SUMO1 prevented the exendin 4-induced Na+ current inhibition (n = 8, τ = 6.0 ± 1.8 ms) (Fig. 4A and B) and leftward shift in voltage-dependent inactivation (Fig. 4C and D).
Figure 4. SUMO1 prevents glucagon-like peptide-1 (GLP-1) receptor-dependent inhibition of α-cell Na+ currents.
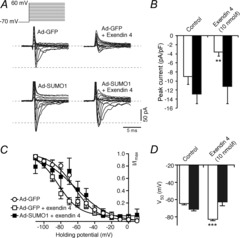
We measured voltage-dependent Na+ currents in mouse α-cells, positively identified by glucagon immunostaining, in the whole-cell configuration. A, representative responses to a series of membrane depolarizations from a holding potential of −70 mV in α-cells infected with green fluorescent protein (Ad-GFP) and exposed to 0 nm or 10 nm exendin 4 during the experiment, and in α-cells infected with SUMO1 (Ad-SUMO1) then treated with 0 nm or 10 nm exendin 4. B, quantification of peak Na+ currents in cells infected with Ad-GFP (open bars) or Ad-SUMO1 (black bars). C, exendin 4 (10 nm) inhibits α-cell Na+ currents by causing a leftward shift of voltage-dependent inactivation (open squares). This is prevented by upregulation of SUMO1 (black squares). D, half-maximal current inactivation (V50) is shown for cells infected with Ad-GFP (open bars) or Ad-SUMO1 (black bars). *P < 0.01, ***P < 0.001 for comparisons with the control.
Activation of the GLP-1 receptor inhibits α-cell exocytosis (De Marinis et al. 2010). Indeed, exendin 4 (10 nm) suppressed exocytosis elicited by membrane depolarization in mouse α-cells infected with Ad-GFP (Fig. 5). This inhibitory effect was observable when intracellular cAMP levels were clamped at 0.1 mm (n = 7–11; P < 0.05) or 1 mm (n = 8–11; P < 0.01). Consistent with the results above, we find that SUMO1 alone (n = 14) was sufficient to enhance the α-cell exocytosis beyond that observed in control cells (n = 23; P < 0.05). Following upregulation of SUMO1, exendin 4 (10 nm) was no longer able to suppress α-cell exocytosis (n = 6, n = 7) (Fig. 5). Together, our data suggest that the ability of SUMO1 to prevent the effects of exendin 4 on Na+ currents and exocytotic responsiveness does not reflect an impairment of cAMP responses (which were clamped in these experiments), but, rather, an as yet unappreciated signalling mechanism.
Figure 5. SUMO1 enhances α-cell exocytosis and prevents glucagon-like peptide-1 (GLP-1) receptor-mediated inhibition.
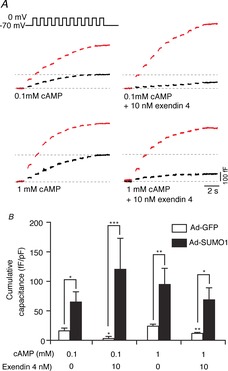
We measured exocytosis as increases in membrane capacitance in mouse α-cells, positively identified by glucagon immunostaining, in response to membrane depolarization. A, representative capacitance responses of mouse α-cells infected with green fluorescent protein (Ad-GFP; black traces) or SUMO1 (Ad-SUMO1; red traces). B, cumulative capacitance responses from mouse α-cells expressing Ad-GFP (open bars) and Ad-SUMO1 (black bars). Treatment with exendin 4 in the bath solution suppressed the exocytotic response, regardless of the presence of 0.1 mm or 1 mm cAMP in the patch pipette, but could not suppress the elevated responses observed in α-cells infected with Ad-SUMO1. *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons with the respective control, or as indicated.
The ability of SUMO1 to enhance α-cell exocytosis is cAMP-dependent
To further explore the interaction between SUMO1 and cAMP in α-cell exocytotic responses, we monitored α-cell capacitance without including cAMP in our patch pipette (Fig. 6). In the absence of cAMP, the exocytotic responses of α-cells infected with Ad-GFP or Ad-SUMO1 were similar (n = 19–29). This contrasts with the robust increase in exocytosis seen in α-cells infected with Ad-SUMO1 at 0.1 mm or 1 mm cAMP (Fig. 5). Indeed, upregulation of SUMO1 significantly increased the α-cell exocytotic response to cAMP-raising agents forskolin (10 μm; n = 25–29; P < 0.01) and adrenaline (10 μm; n = 21, n = 22; P < 0.01) (Fig. 6). Cells were pre-treated with these agents in the bath solution immediately prior to and during the patch clamp experiment.
Figure 6. SUMO1 increases α-cell exocytosis in a cAMP-dependent manner.
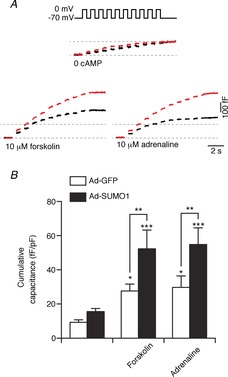
We measured exocytosis as increases in membrane capacitance in mouse α-cells, identified by glucagon immunostaining, in response to membrane depolarization in the whole-cell configuration. A, representative capacitance responses of α-cells infected with green fluorescent protein (Ad-GFP; black traces) or SUMO1 (Ad-SUMO1; red traces) without cAMP included in the patch pipette and following treatment with adrenaline or forskolin as indicated. B, cumulative capacitance response of α-cells infected with Ad-GFP (open bars) or Ad-SUMO1 (black bars). Ad-SUMO1 was unable to increase α-cell exocytosis in the absence of cAMP, but this response was restored following treatment with forskolin or adrenaline. *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons with the respective control, or as indicated.
SUMO1 enhances glucagon secretion stimulated by adrenaline
To assess the effect of SUMO1 on glucagon secretion per se, we performed perifusion analysis of mouse islets infected with either Ad-GFP or Ad-SUMO1. Interestingly, upregulation of SUMO1 generally had little effect on glucagon secretion at either 1 mm or 6 mm glucose (Fig. 7) and had no effect on islet glucagon content (not shown). In one set of experiments (Fig. 7B), the slight increase in glucagon release with Ad-SUMO1 was significant (n = 5; P < 0.05) at only one time-point. In addition, the glucagon response to 20 mm KCl was modestly increased (n = 4; P < 0.05) (AUC in Fig. 7A). This is consistent with the lack of effect of SUMO1 on α-cell exocytosis in the absence of elevated cAMP. Somewhat surprisingly, exendin 4 (10 nm) remained effective in inhibiting glucagon secretion at 1 mm glucose in islets infected with Ad-SUMO1 (n = 5) (Fig. 7B). Finally, consistent with the cAMP dependence of the effect of SUMO1 on α-cell exocytosis, glucagon secretion in response to 10 μm adrenaline was significantly elevated in islets infected with Ad-SUMO1 compared with Ad-GFP (n = 5; P < 0.01) and remained elevated following the removal of adrenaline (P < 0.001), although this latter effect may in part be related to the slow perifusion rate (50 μl min−1) used in these experiments.
Figure 7. SUMO1 increases adrenaline-stimulated glucagon secretion from intact islets.
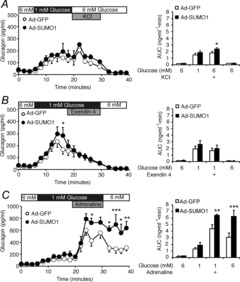
We measured glucagon secretion from mouse islets infected with green fluorescent protein (Ad-GFP; open circles/bars) or SUMO1 (Ad-SUMO1; black circles/bars) during perifusion. A, glucagon secretion in response to 1 mm glucose followed by 20 mm KCl. Areas under the curve (AUCs) for the various conditions are shown on the right. B, glucagon secretion in response to 1 mm glucose followed by 10 nm exendin 4. AUCs for the various conditions are shown on the right. C, glucagon secretion in response to 1 mm glucose followed by 10 μm adrenaline. AUCs for the various conditions are shown on the right. *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons with the Ad-GFP control.
Discussion
The importance of glucagon secretion in the physiologic control of glucose and the pathophysiology of diabetes is receiving renewed interest (Cryer, 2012). However, we know surprisingly little about the cellular machinery that determines α-cell function, and how this is regulated by metabolic and hormonal inputs. Significant parallels can be drawn between the functions of β-cells and α-cells; in fact they express much of the same downstream secretory machinery that includes similar complements of ion channels and exocytotic proteins (Andersson et al. 2011; Rorsman et al. 2012). It is thus no surprise that the final events of glucagon secretion, including AP firing and Ca2+-dependent exocytosis, are quite similar to those of insulin secretion. The control of this machinery by both internal and external cues, however, differs strikingly between β and α-cells. For example, insulin and glucagon secretion are regulated in opposite manners by intracellular PAS kinase (da Silva Xavier et al. 2004, 2011; Fontés et al. 2009), adrenergic signals (Dunning & Taborsky, 1991; Straub & Sharp, 2012), and GLP-1 receptor signalling (Gromada & Rorsman, 2004).
SUMO1 increases AP amplitudes and frequency in α-cells. This is likely to result from a combination of increases in Kv and L-type Ca2+ currents. Effects of SUMOylation on ion channels (Rajan et al. 2005; Benson et al. 2007; Dai et al. 2009; Plant et al. 2010, 2011), and Ca2+ handling (Feligioni et al. 2009; Kho et al. 2011) have been demonstrated, and a recent abstract suggests the SUMO-dependent inhibition of P/Q-type Ca2+ channels (Davila et al. 2010); however, a SUMO1-dependent upregulation of L-type Ca2+ current has not been demonstrated previously. This upregulated Ca2+ current cannot be explained by the changes in voltage-dependent inactivation or by acute effects of SUMO1 on the channels (as dialysis had no effect). Thus, given the known roles for SUMOylation in gene transcription (Yeh, 2009), SUMOylation in α-cells may increase Ca2+ currents primarily by upregulating L-type Ca2+ channel density.
We wondered if the SUMO1-dependent increase in L-type Ca2+ current results in enhanced depolarization-induced exocytosis. Indeed, we find that SUMO1 (or knockdown of SENP1) enhances depolarization-induced exocytosis in α-cells. This is mediated entirely by the upregulated Ca2+ current, as evidenced by the inability of SUMO1 to affect exocytotic responses to direct intracellular Ca2+ dialysis. This is in stark contrast with the effects of SUMOylation in β-cells, where exocytosis is inhibited directly by SUMOylation with no effect on VDCC activity or intracellular Ca2+ responses (Dai et al. 2011). Furthermore, a key role for the L-type Ca2+ channel in upregulated α-cell exocytosis is supported by the sensitivity of this response to block by isradipine. Our observation that SUMO1 promotes a shift from ω-conotoxin- to isradipine-sensitive exocytosis in α-cells is reminiscent of recent work demonstrating a similar response of glucagon secretion to stimulation with adrenaline (De Marinis et al. 2010), where cAMP was proposed to underlie the shift from non-L-type to L-type channel-dependent glucagon secretion.
Inhibition of the non-L-type Ca2+ channels that are coupled to glucagon exocytosis under basal conditions are proposed to underlie the suppression of glucagon secretion by GLP-1 (De Marinis et al. 2010). These Ca2+ currents, which are probably mediated by N- or P/Q-type channels, are generally quite small in α-cells (Rorsman et al. 2012). As noted above, we do find that a large proportion of α-cell exocytosis under control conditions is blocked by ω-conotoxin; however, in our hands exendin 4 remains able to inhibit α-cell exocytotic responses in the presence of 1 mm intracellular cAMP. This argues against ‘low cAMP’ as the mediator, and is consistent with the suggestion that additional cAMP-independent signals contribute to the inhibition of glucagon secretion by GLP-1 (Tian et al. 2011). Nonetheless, we cannot exclude the possibility that a small change in Ca2+ current, or perhaps an uncoupling of glucagon granules from the non-L-type VDCCs, may contribute to the suppression of glucagon secretion by exendin 4.
We also now provide evidence for an exendin 4-dependent inhibition of α-cell Nav channels, resulting from a hyperpolarizing shift in the voltage dependence of inactivation. This is likely to contribute to reduced AP height and would serve to reduce the overall activation of high-voltage-activated N- or P/Q-type Ca2+ channels on which glucagon secretion depends under basal conditions. Although in apparent contrast with the suggestion that transient α-cell hyperpolarization by GLP-1 leads to increases in AP amplitude in two of six α-cells examined from intact mouse islets (De Marinis et al. 2010), our experiments examined APs elicited by current injection (to allow us to study the AP characteristics, rather than glucose dependence per se) following a longer exposure to exendin 4 (10–30 min) in isolated α-cells. As such, the methodological differences may account for this, and our results are consistent with a decreased AP firing frequency reported in the steady-state following GLP-1 treatment and the lack of effect of GLP-1 on AP duration in the earlier study.
Based upon the ability of SUMOylation to downregulate GLP-1 receptor signalling in β-cells (Rajan et al. 2012), we investigated whether SUMO1 would prevent α-cell responses to the GLP-1 receptor agonist exendin 4. Indeed, upregulation of SUMO1 prevented the inhibition of α-cell Na+ currents and exocytosis by GLP-1 receptor activation. Although this may appear to be consistent with an inhibition of GLP-1 receptor signalling, our finding that SUMO1 fails to prevent exendin 4-mediated suppression of glucagon secretion suggests that GLP-1 receptor signalling is preserved in intact islets. This discrepancy can be explained by a key experimental difference between the single-cell patch clamp and whole-islet secretion studies: our initial patch clamp studies were performed with a high level of intracellular cAMP, which is likely to be much lower (if increased at all) following GLP-1 receptor activation in α-cells of the intact islet (De Marinis et al. 2010; Tian et al. 2011). Thus SUMO1 is unlikely to block GLP-1 receptor signalling directly in α-cells, but may have a more direct cAMP-dependent effect to increase glucagon secretion.
We find that the facilitation of α-cell exocytosis by SUMO1 is indeed cAMP-dependent. This is likely to explain the inability of Ad-SUMO1 to enhance glucagon secretion stimulated by low glucose or exendin 4 because GLP-1 receptor activation produces only a small rise in α-cell cAMP, if any (De Marinis et al. 2010; Tian et al. 2011). By contrast, signalling of β- and α1-adrenergic receptors is generally considered to produce a robust increase in α-cell cAMP (Schuit & Pipeleers, 1986; Vieira et al. 2004; Tian et al. 2011). We found that stimulation of single α-cells with adrenaline facilitated depolarization-induced α-cell exocytosis to a degree similar to that of forskolin or direct infusion of cAMP, and enabled a further SUMO1-dependent increase in exocytosis. Thus, although upregulation of SUMO1 results in increased α-cell L-type Ca2+ channel currents and enhanced excitability, a rise in cAMP and shift to L-type-dependent glucagon exocytosis is required to realize an enhancement of glucagon secretion such as that observed upon stimulation with adrenaline (Fig. 8).
Figure 8. Schematic hypothesis for the interaction between SUMOylation and cAMP on glucagon secretion.
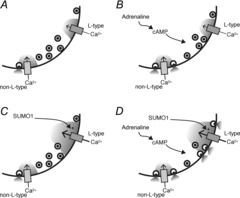
A, in the absence of elevated cAMP or SUMO1, glucagon exocytosis is triggered by Ca2+ entry through non-L-type Ca2+ channels. B, in response to elevated cAMP, such as that stimulated by adrenaline, glucagon granules become more coupled to L-type channels. However, the majority of glucagon exocytosis remains mediated by the non-L-type Ca2+ channels. C, increased SUMOylation enhances L-type Ca2+ channel currents in α-cells, but in the absence of the increased coupling of glucagon granules to these channels conferred by elevated cAMP, this does not result in increased glucagon exocytosis. D, only when cAMP is elevated, and the coupling of glucagon granules to L-type Ca2+ channels is increased, does SUMOylation increase glucagon exocytosis as a result of increased L-type Ca2+ current.
Thus, we now demonstrate that SUMO1 regulates glucagon secretion in a manner opposite to its control of insulin secretion. SUMO1 enhances glucagon secretion by increasing L-type Ca2+ currents in α-cells, increasing exocytosis under conditions of elevated cAMP in which glucagon granules become more dependent on Ca2+ entry through L-type channels. The result is that glucagon secretion in response to adrenergic stimulation is enhanced by SUMO1. Interestingly, although our data suggest that GLP-1 receptor inhibition of α-cell Nav channels and exocytosis is lost following SUMO1 upregulation, this is insufficient to prevent exendin 4-mediated suppression of glucagon secretion where α-cell cAMP levels may be low. Further understanding of the interaction of the SUMOylation and cAMP pathways may reveal novel approaches to the control of insulin and glucagon secretion in diabetes. The demonstration that SUMOylation regulates key components of the α-cell secretory mechanism in a manner opposite to that of the β-cell indicates a novel mechanism underlying the differential regulation of glucagon and insulin secretion.
Acknowledgments
The Human Organ Procurement and Exchange (HOPE) Program and the Trillium Gift of Life Network (TGLN) are gratefully acknowledged for their efforts in procuring human donor pancreas, as are Drs Tatsuya Kin and James Shapiro and the Clinical Islet Laboratory at the University of Alberta for providing access to human research islets.
Glossary
- AP
action potential
- GFP
green fluorescent protein
- GLP-1
glucagon-like peptide-1
- GST
glutathione S-transferase
- Kv
voltage-dependent K+
- Nav
voltage-dependent Na+
- SENP
sentrin/SUMO-specific protease
- SUMO
small ubiquitin-like modifier
- VDCC
voltage-dependent Ca2+ channel
Additional information
Competing interests
None declared.
Author contributions
X.-Q.D., A.F.S. and S.K. contributed to data acquisition, analysis and interpretation. X.-Q.D., M.B., J.E.M.F. and P.E.M. designed the study. P.E.M. drafted the manuscript. All authors contributed to the revision of the manuscript and approved the final version.
Funding
Human islet isolation was funded in part by the Alberta Diabetes Foundation. This work was supported by research grants from the Canadian Institutes of Health Research (MOP244739) and the Merck Investigator Study Program of Merck Frosst Canada Ltd. The opinions expressed in this paper are those of the authors and do not necessarily represent those of Merck Frosst Canada Ltd or its affiliates and related companies. P.E.M. is supported by a scholarship from Alberta-Innovates Health Solutions, a Juvenile Diabetes Research Foundation Career Development Award, and holds the Canada Research Chair in Islet Biology.
References
- Andersson SA, Pedersen MG, Vikman J, Eliasson L. Glucose-dependent docking and SNARE protein-mediated exocytosis in mouse pancreatic α-cell. Pflugers Arch. 2011;462:443–454. doi: 10.1007/s00424-011-0979-5. [DOI] [PubMed] [Google Scholar]
- Aukrust I, Bjørkhaug L, Negahdar M, Molnes J, Johansson BB, Müller Y, Haas W, Gygi SP, Søvik O, Flatmark T, Kulkarni RN, Njølstad PR. SUMOylation of pancreatic glucokinase regulates its cellular stability and activity. J Biol Chem. 2013;288:5951–5962. doi: 10.1074/jbc.M112.393769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Barg S, Galvanovskis J, Göpel SO, Rorsman P, Eliasson L. Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting α-cells. Diabetes. 2000;49:1500–1510. doi: 10.2337/diabetes.49.9.1500. [DOI] [PubMed] [Google Scholar]
- Benson MD, Li Q-J, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iñiguez-Lluhí JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci U S A. 2007;104:1805–1810. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryer PE. Minireview: glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology. 2012;153:1039–1048. doi: 10.1210/en.2011-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Xavier G, Farhan H, Kim H, Caxaria S, Johnson P, Hughes S, Bugliani M, Marselli L, Marchetti P, Birzele F, Sun G, Scharfmann R, Rutter J, Siniakowicz K, Weir G, Parker H, Reimann F, Gribble FM, Rutter GA. Per-Arnt-Sim (PAS) domain-containing protein kinase is downregulated in human islets in type 2 diabetes and regulates glucagon secretion. Diabetologia. 2011;54:819–827. doi: 10.1007/s00125-010-2010-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Xavier G, Rutter J, Rutter GA. Involvement of Per-Arnt-Sim (PAS) kinase in the stimulation of preproinsulin and pancreatic duodenum homeobox 1 gene expression by glucose. Proc Natl Acad Sci U S A. 2004;101:8319–8324. doi: 10.1073/pnas.0307737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XQ, Kolic J, Marchi P, Sipione S, MacDonald PE. SUMOylation regulates Kv2.1 and modulates pancreatic β-cell excitability. J Cell Sci. 2009;122:775–779. doi: 10.1242/jcs.036632. [DOI] [PubMed] [Google Scholar]
- Dai XQ, Plummer G, Casimir M, Kang Y, Hajmrle C, Gaisano HY, Manning Fox JE, MacDonald PE. SUMOylation regulates insulin exocytosis downstream of secretory granule docking in rodents and humans. Diabetes. 2011;60:838–847. doi: 10.2337/db10-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila MA, Chan H, Piedras-Renteria ES. Sumoylation of voltage-gated αA1a calcium channels. Biophysical J. 2010;98:692a–693a. [Google Scholar]
- De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, Habib AM, Moritoh Y, Zhang E, Reimann F, Rosengren AH, Shibasaki T, Gribble F, Renström E, Seino S, Eliasson L, Rorsman P. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010;11:543–553. doi: 10.1016/j.cmet.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning BE, Taborsky GJ. Neural control of islet function by norepinephrine and sympathetic neuropeptides. Adv Exp Med Biol. 1991;291:107–127. doi: 10.1007/978-1-4684-5931-9_10. [DOI] [PubMed] [Google Scholar]
- Ernst SJ, Aguilar-Bryan L, Noebels JL. Sodium channel β1 regulatory subunit deficiency reduces pancreatic islet glucose-stimulated insulin and glucagon secretion. Endocrinology. 2009;150:1132–1139. doi: 10.1210/en.2008-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feligioni M, Nishimune A, Henley JM. Protein SUMOylation modulates calcium influx and glutamate release from presynaptic terminals. Eur J Neurosci. 2009;29:1348–1356. doi: 10.1111/j.1460-9568.2009.06692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontés G, Semache M, Hagman DK, Tremblay C, Shah R, Rhodes CJ, Rutter J, Poitout V. Involvement of Per-Arnt-Sim kinase and extracellular-regulated kinases-1/2 in palmitate inhibition of insulin gene expression in pancreatic β-cells. Diabetes. 2009;58:2048–2058. doi: 10.2337/db08-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpel SO, Kanno T, Barg S, Weng XG, Gromada J, Rorsman P. Regulation of glucagon release in mouse cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol (Lond) 2000;528:509–520. doi: 10.1111/j.1469-7793.2000.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Rorsman P. New insights into the regulation of glucagon secretion by glucagon-like peptide-1. Horm Metab Res. 2004;36:822–829. doi: 10.1055/s-2004-826169. [DOI] [PubMed] [Google Scholar]
- Hare KJ, Vilsbøll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes. 2010;59:1765–1770. doi: 10.2337/db09-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RR, Smith SR, Schwartz SL, Mudaliar SR, Deacon CF, Holst JJ, Duan RY, Chen RS, List JF. Effects of saxagliptin on β-cell stimulation and insulin secretion in patients with type 2 diabetes. Diabetes Obes Metab. 2011;13:850–858. doi: 10.1111/j.1463-1326.2011.01417.x. [DOI] [PubMed] [Google Scholar]
- Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262–269. doi: 10.1038/nrendo.2009.48. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, De Marinis YZ, Ramracheya R, Salehi A, Ma X, Johnson PRV, Cox R, Eliasson L, Rorsman P. A K ATP channel-dependent pathway within α cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol. 2007;5:e143. doi: 10.1371/journal.pbio.0050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning Fox JE, Hajmrle C, MacDonald PE. Novel roles of SUMO in pancreatic β-cells: thinking outside the nucleus. Can J Physiol Pharmacol. 2012;90:765–770. doi: 10.1139/y11-134. [DOI] [PubMed] [Google Scholar]
- Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SAN. One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci U S A. 2010;107:10743–10748. doi: 10.1073/pnas.1004712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SAN. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol. 2011;137:441–454. doi: 10.1085/jgp.201110604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SAN. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell. 2005;121:37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Rajan S, Torres J, Thompson MS, Philipson LH. SUMO downregulates GLP-1-stimulated cAMP generation and insulin secretion. Am J Physiol Endocrinol Metab. 2012;302:E714–E723. doi: 10.1152/ajpendo.00486.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, Johnson PR, Rorsman P, Braun M. Membrane potential-dependent inactivation of voltage-gated ion channels in α-cells inhibits glucagon secretion from human islets. Diabetes. 2010;59:2198–2208. doi: 10.2337/db09-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch T, Graefe-Mody U, Deacon CF, Ring A, Holst JJ, Woerle H-J, Dugi KA, Heise T. Linagliptin increases incretin levels, lowers glucagon, and improves glycemic control in type 2 diabetes mellitus. Diabetes Ther. 2012;3:10. doi: 10.1007/s13300-012-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium. 2012;51:300–308. doi: 10.1016/j.ceca.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit FC, Pipeleers DG. Differences in adrenergic recognition by pancreatic A and B cells. Science. 1986;232:875–877. doi: 10.1126/science.2871625. [DOI] [PubMed] [Google Scholar]
- Spigelman AF, Dai X, MacDonald PE. Voltage-dependent K+ channels are positive regulators of α cell action potential generation and glucagon secretion in mice and humans. Diabetologia. 2010;53:1917–1926. doi: 10.1007/s00125-010-1759-z. [DOI] [PubMed] [Google Scholar]
- Straub SG, Sharp GWG. Evolving insights regarding mechanisms for the inhibition of insulin release by norepinephrine and heterotrimeric G proteins. Am J Physiol Cell Physiol. 2012;302:C1687–C1698. doi: 10.1152/ajpcell.00282.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Sandler S, Gylfe E, Tengholm A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes. 2011;60:1535–1543. doi: 10.2337/db10-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira E, Liu Y-J, Gylfe E. Involvement of α1 and β-adrenoceptors in adrenaline stimulation of the glucagon-secreting mouse α-cell. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:179–183. doi: 10.1007/s00210-003-0858-5. [DOI] [PubMed] [Google Scholar]
- Vignali S, Leiss V, Karl R, Hofmann F, Welling A. Characterization of voltage-dependent sodium and calcium channels in mouse pancreatic A- and B-cells. J Physiol (Lond) 2006;572:691–706. doi: 10.1113/jphysiol.2005.102368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt D, Malik R, Vesecky AC, Marchese A. Small ubiquitin-like modifier modification of arrestin-3 regulates receptor trafficking. J Biol Chem. 2011;286:3884–3893. doi: 10.1074/jbc.M110.152116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ETH. SUMOylation and de-SUMOylation: wrestling with life's processes. J Biol Chem. 2009;284:8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]