

Abstract
We investigated the impact of cardiac reactive oxygen species (ROS) during the development of pressure overload-induced heart failure. We used our previously described rat model where transverse aortic constriction (TAC) induces compensated hypertrophy after 2 weeks, heart failure with preserved ejection fraction at 6 and 10 weeks, and heart failure with systolic dysfunction after 20 weeks. We measured mitochondrial ROS production rates, ROS damage and assessed the therapeutic potential of in vivo antioxidant therapies. In compensated hypertrophy (2 weeks of TAC) ROS production rates were normal at both mitochondrial ROS production sites (complexes I and III). Complex I ROS production rates increased with the appearance of diastolic dysfunction (6 weeks of TAC) and remained high thereafter. Surprisingly, maximal ROS production at complex III peaked at 6 weeks of pressure overload. Mitochondrial respiratory capacity (state 3 respiration) was elevated 2 and 6 weeks after TAC, decreased after this point and was significantly impaired at 20 weeks, when contractile function was also impaired and ROS damage was found with increased hydroxynonenal. Treatment with the ROS scavenger α-phenyl-N-tert-butyl nitrone or the uncoupling agent dinitrophenol significantly reduced ROS production rates at 6 weeks. Despite the decline in ROS production capacity, no differences in contractile function between treated and untreated animals were observed. Increased ROS production occurs early in the development of heart failure with a peak at the onset of diastolic dysfunction. However, ROS production may not be related to the onset of contractile dysfunction.
Key points
Pressure overload induces cardiac hypertrophy developing into heart failure.
During pressure overload-induced heart failure development in the rat, mitochondrial capacity to produce reactive oxygen species (ROS) increased significantly with the onset of diastolic functional changes.
Treatment to reduce ROS production was able to diminish mitochondrial ROS production but was not able to prevent or delay heart failure development.
The results question a primary role of ROS in the mechanism causing contractile dysfunction under pressure overload.
Introduction
Pressure overload is considered a major contributor to, and a cause of, heart failure (Ozkan et al. 2011). However, mechanisms for the development of heart failure are still a matter of debate. Mitochondrial dysfunction has been suggested as a potential mechanism (Caldeira da Silva et al. 2008; Marín-García & Goldenthal, 2008; Rosca & Hoppel, 2009). Mitochondrial function has been the focus of many previous studies on heart failure, using both different species and different methods for the induction of heart failure (Murray et al. 2008; Rosca et al. 2008). Heterogeneous results suggest a model-dependent alteration of mitochondrial function in heart failure, which makes it difficult to compare the outcome of different studies (Estabrook, 1967; Baudet et al. 1994; Nojiri et al. 2006; Rosca & Hoppel, 2009).
Besides their role in ATP production, mitochondria are also a major site for reactive oxygen species (ROS) production in the heart (Chance & Williams, 1955; Hansford et al. 1997; Maltecca & Casari, 2010). Production of ROS has been suggested as another potential mechanism for the development of heart failure (Dai et al. 2011). In mitochondria, ROS are produced at a basal level during normal respiration and their generation has been described on complexes I and III of the respiratory chain (Chen et al. 2003). ROS are known to be important signalling molecules involved in different cellular pathways (Finkel, 2011).
Despite their important role in molecular signalling, ROS may also be deleterious, especially at higher concentrations. Increased rates of ROS production may overwhelm antioxidant defences and ROS may react directly with lipids, proteins and DNA, thereby compromising their function (Gomez et al. 2009). ROS production has been found to be elevated with impaired mitochondrial function (Chen et al. 2008). Mitochondrial ROS production may particularly affect the activity of proteins of the citric acid cycle and the respiratory chain (Berlett & Stadtman, 1997; Paradies et al. 2004; Bulteau et al. 2005; Heather et al. 2010) possibly leading to further deterioration of mitochondrial function and elevated ROS production (Ide et al. 2001). ROS may thus aggravate the impairment in mitochondrial ATP generation contributing to the progression of heart failure.
Since most studies in this field have focused on the stage of overt heart failure only, ROS production and mitochondrial function have not been assessed over the time course of development of cardiac hypertrophy and heart failure. We investigated the impact of cardiac ROS production on pressure overload-induced heart failure. We used our previously described rat model where pressure overload induces compensated hypertrophy after 2 weeks, heart failure with preserved ejection fraction at 6 and 10 weeks, and heart failure with systolic dysfunction after 20 weeks (Doenst et al. 2010; Schrepper et al. 2012; Shingu et al. 2013). We measured the production of ROS on complexes I and III individually and assessed the therapeutic potential of antioxidant therapies to evaluate if ROS production may be causally involved in the development of heart failure. We provide for the first time a detailed analysis of mitochondrial ROS production capacity during the development of pressure overload.
Methods
Animals
Male Sprague–Dawley rats were obtained from Janvier (Genest, France). The animals were 3 weeks of age (40–50 g), fed ad libitum and housed with a light cycle of 12 h at 21°C. The use of animals was consistent with the Guide for the Care and Use of Laboratory Animals as published by the US National Institutes of Health (NIH publication No. 85–23, revised 1996). All experimental protocols were approved by the local Animal Welfare Committee of the Universities of Leipzig and Jena, Germany (AZ: 24-9168.11 – TVV 36/06, 02/10).
Materials
Chemicals were obtained from Sigma Aldrich (Deisenhofen), Merck (Darmstadt), Serva (Heidelberg) and BIO-RAD (München) in Germany.
Surgical interventions
Pressure overload was induced by aortic constriction (TAC) of the transverse aorta as described before (Doenst et al. 2010). Briefly, rats of 40–50 g (3 weeks of age) were anaesthetized with ketamine/xylazine (100/5 mg kg−1, i.p.) and intubated for a partial sternotomy. A titanium clip was placed around the aortic arch between the brachiocephalic trunk and the left carotid artery. The clip had a remaining internal diameter of 0.35 mm. Age-matched sham-operated rats underwent the same procedure without application of a clip. Animals were allowed to recover and were followed for the next 20 weeks when they reached end-stage heart failure. Analgesic treatment was performed for 3 days with metamizole (275 mg kg−1 day−1) orally. Animals were assessed at 2, 6, 10 and 20 weeks after TAC.
Clinical assessment and echocardiography
Rats were inspected weekly. Echocardiographic examination was performed after 2, 6, 10 and 20 weeks postoperatively. The animals were anaesthetized with fentanyl/midazolam hydrochloride/medetomidine hydrochloride (0.005/2/0.15 mg kg−1, i.m.). Chests were shaved and the rats were examined in supine position with a 12 MHz phased array transducer (Agilent/Philips, Germany). Two-dimensional short-axis views of the left ventricle at papillary muscle level were obtained. 2-D guided M-mode tracings were recorded with a sweep speed of 100 mm s−1. The following parameters were measured: heart rate (HR), left ventricular (LV) end-diastolic dimension (LVEDD), and LV posterior wall thickness in diastole (LVPWD). Based on the measurements, ejection fraction (EF), left ventricular endocardial fractional shortening (FS), and calculated LV mass were determined as described before (Doenst et al. 2010). LV mass index (LVMI) was determined as described in Salemi et al. (2004). At the end of echocardiographic assessment anaesthesia was antagonized with naloxone/flumazenil/atipamezol hydrochloride (0.12/0.2/0.75 mg kg−1, i.p.) and the animals allowed to recover.
Organ harvesting
At 2, 6, 10 and 20 weeks, animals were weighed and killed. Deep anaesthesia was induced using thiopental (150 mg (kg body weight)−1) and hearts were explanted, weighed and prepared for isolation of mitochondria. Both lungs and liver were excised and weighed. Lung to body weight index (LBI) and liver to body weight index (LiverBI) were calculated as lung wet weight (g) and liver wet weight (g), respectively, to body weight (kg). Length of the left tibia was measured and heart weight to tibia length calculated.
Isolation of mitochondria
Cardiac interfibrillar mitochondria were isolated from freshly explanted hearts as described before (Doenst et al. 2010) with a modified Chappell–Perry buffer (containing 100 mm KCl, 50 mm Mops, 1 mm EGTA, 5 mm MgSO4.7H2O, and 1 mm ATP, pH 7.4, 4°C). Mitochondria were harvested after treating the homogenate with 5 mg trypsin (g wet weight of heart)–1 for 10 min at 4°C. Mitochondrial protein content was determined by the Bradford method using bovine serum albumin as a standard. Citrate synthase activity was measured in fresh heart homogenate and isolated mitochondria according to the protocol of Srere (1969).
Assessment of mitochondrial respiratory rates
Oxygen consumption of isolated mitochondria was measured using a Clark-type oxygen electrode (Strathkelvin, North Lanarkshire, Scotland) at 25°C (Tomec & Hoppel, 1975; Mogensen et al. 2007; Schwarzer et al. 2013) as linearity of respiratory measurements has been described between 22 and 41°C (Samartsev et al. 2003). Mitochondria were incubated in a solution consisting of 100 mm KCl, 50 mm Mops, 1 mm EGTA, 5 mm KH2PO4 and 1 mg ml−1 fatty acid-free bovine serum albumin at pH 7.4. The rate of oxidative phosphorylation was measured using 10 mm glutamate, 20 μm palmitoylcarnitine/2.5 mm malate, 5 mm pyruvate/2.5 mm malate, or 10 mm succinate/3.75 μm rotenone as substrates and ADP as stimulus. The ADP-stimulated oxygen consumption (state 3) and the ADP-limited oxygen consumption (state 4) in the respiratory chamber and the ADP/O ratio (ADP added per oxygen consumed) were determined as previously described (Chance & Williams, 1955; Estabrook, 1967). Respiratory control ratio was calculated as state 3 respiration to state 4 respiration. To determine uncoupled respiration, state 3 respiration was measured in the presence of 0.1 mm 2,4-dinitrophenol (DNP).
Determination of isolated complex activities
Mitochondria were treated with 1 mg cholate (mg mitochondrial protein)–1 and further prepared according to Rosca et al. (2008). After one cycle of freeze/thaw (−80°C to +25°C), electron transport chain complex activities were measured as specific donor–acceptor oxidoreductase activities. Complex I was measured as rotenone-sensitive reduction of 2,6-dichloroindophenol (DCIP) with NADH as substrate as described by Janssen et al. (2007). Reduction of DCIP with succinate as substrate assesses complex II (Krahenbuhl et al. 1991). Complex III activity was determined as antimycin-A-sensitive reduction of cytochrome c (Krahenbuhl et al. 1991), using decylubiquinol as substrate, which was prepared as previously described (Kirby et al. 2007). Cytochrome c oxidase (complex IV) was measured as the oxidation of reduced cytochrome c according to the method of Wharton & Tzagoloff (1967). The combined activities of complexes I + III and complexes II + III were determined as described before (Rosca et al. 2008).
Hydroxynonenal (HNE) assay
For analysis of hydroxynonenal adducts in cardiac tissue a commercially available rat 4-hydroxynonenal (4-HNE) enzyme-linked immunosorbent assay (ELISA) kit was used (Biotrend, Cologne, Germany). Frozen tissue was powdered under liquid nitrogen. Small samples (∼50 mg) were dissolved in PBS and homogenized with a glass homogenizer on ice. The suspension was subjected to ultrasonication and centrifuged for 15 min at 4°C with 1500 g. Supernatant was used to assay on HNE ELISA following the manufacturer's instructions.
Protein carbonylation assay
Protein carbonylation by oxidative stress was detected in cardiac tissue by a spectrophotometric assay (Levine et al. 1990; Reznick & Packer, 1994; Wehr & Levine, 2013). Frozen tissue was powdered under liquid nitrogen. Homogenization was performed and carbonyl groups were derivatized using diphenylhydrazone. After washing with ethylacetate to remove any excess of reactants, absorption was measured at 370 nm to quantify carbonyl derivates and at 276 nm to quantify protein.
Assessment of reactive oxygen species production
Mitochondrial superoxide generation was assessed on fresh, intact mitochondria and was measured using the homovanillic acid assay at 37°C (Barja, 2002). Briefly, the assay is based on superoxide´s dismutation into hydrogen peroxide which subsequently oxidizes homovanillic acid in the presence of horseradish peroxidase. The following increase in fluorescence is monitored kinetically with a fluorometer (BioTek, Bad Friedrichshall, Germany). Mitochondria (0.1 mg ml−1), homovanillic acid (0.1 mm) and horseradish peroxidase (3 U ml−1) are added to the reaction buffer (145 mm KCl, 0.1 mm EGTA, 0.1% albumin, 30 mm Hepes, 5 mm KH2PO4, 3 mm MgCl2, pH 7.4). Oligomycin (1 μg ml−1), an inhibitor of the ATP synthase, was present in all experiments to prevent ATP synthesis and maintain the high proton gradient across the inner mitochondrial membrane. Pyruvate/malate (2.5 mm/2.5 mm) and succinate (5 mm) were used as substrates. Rotenone (10 μm) was used to block electron flow at complex I. To evaluate whether ROS are released into the matrix or the intermembrane space (in the presence of complex inhibitors), external superoxide dismutase (50 U ml−1) was added. Standard curves were obtained by adding known amounts of H2O2 to the reaction medium.
At the level of the respiratory chain, ROS are predominantly formed at complex I and complex III (Poyton et al. 2009). We therefore used the following strategy to assess mitochondrial ROS production. First, basal ROS production was monitored with pyruvate/malate as complex I substrate (Fig. 1A). Under these conditions, ROS are produced at complex I and at complex III. In a second step, succinate was used as complex II substrate (Fig. 1B). In this setting, the simultaneous addition of rotenone inhibits reverse electron flow to complex I, thereby preventing ROS production at this site (Selivanov et al. 2011). Thus, after the addition of succinate and rotenone, ROS are generated at complex III only. In a third step, complex I substrate pyruvate/malate was administered together with rotenone for complex I inhibition to assess maximal ROS production capacity at complex I (Fig. 1C). Under these circumstances electron transfer from complex I to the electron-transferring protein ubiquinone is inhibited, resulting in reduced flow of electrons and increased reduction of complex I, subsequently increasing the formation of ROS at complex I (Turrens, 2003).
Figure 1. Scheme for analysis of ROS production.
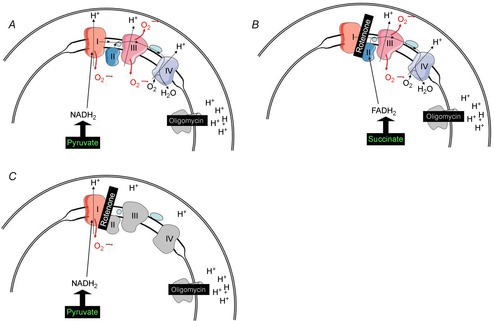
Basal ROS production with pyruvate/malate as complex I substrate (A). Under these conditions, ROS are produced at complex I and at complex III. Succinate as complex II substrate (B), rotenone inhibiting reverse electron flow to complex I, preventing ROS production at this site. Maximal ROS production capacity at complex I with substrate pyruvate/malate and rotenone for complex I inhibition (C).
Antioxidant treatment
Animals with transverse aortic constriction were treated with the radical scavenger α-phenyl-N-tert-butyl nitrone (PBN) or the mild uncoupling agent 2,4-dinitrophenol (DNP). Administration of PBN has been linked to reduced mitochondrial ROS release previously (Wen et al. 2006). Uncoupling mitochondrial respiration from oxidative phosphorylation lowers the proton-motive force, leading to reduced mitochondrial ROS production (Skulachev, 1996). In a previous study, DNP has been shown to extend the lifespan (of mice) when administered at the concentration used in the present study (Caldeira da Silva et al. 2008). This suggests that the use of DNP at such a low concentration is not associated with significant ATP loss or with adverse effects on physiological function. PBN was applied intraperitoneally twice weekly at a dose of 50 mg (kg body weight)−1 in a mixture of 80% saline and 20% distilled water from day 5 after TAC (Bolli et al. 1988; Wen et al. 2006). DNP was given in the drinking water adjusted for a dose of 100 μg kg−1 day−1 (Caldeira da Silva et al. 2008).
Statistical analysis
Data are presented as mean ± SEM. Data were analysed using Student's t test or one-way analysis of variance where appropriate. Post hoc comparisons among the groups were performed using the Holm–Sidak test. Differences among groups were considered statistically significant if P < 0.05.
Results
Pressure overload induces development of cardiac hypertrophy and heart failure
We used our previously described model of aortic constriction for the induction of pressure overload (Doenst et al. 2010). In this model compensated hypertrophy is present after 2 weeks, heart failure with preserved ejection fraction after 6 and 10 weeks, and heart failure with contractile dysfunction after 20 weeks (Doenst et al. 2010; Shingu et al. 2013). Table 1 shows basal data of animals subjected to pressure overload. The increase in heart weight in animals with pressure overload indicates cardiac hypertrophy at all investigated time points. This finding is confirmed by increased heart to body weight and heart weight to tibia length ratios. Increased lung to body weight ratios indicate cardiac congestion which correlates well with reduced diastolic function from 6 weeks of pressure overload (Mandinov et al. 2000; Doenst et al. 2010; Shingu et al. 2013). The presence of cardiac hypertrophy was verified echocardiographically by increased left ventricular posterior wall diameter (LVPWD) shown in Table 2. Echocardiography further demonstrated dilatation of the left ventricle after 10 weeks of pressure overload with increased left ventricular end-diastolic diameter (LVEDD). Systolic function (left ventricular ejection fraction and fractional shortening) was preserved up to 6 weeks of pressure overload, but was decreased at 20 weeks of pressure overload. We aimed to relate contractile function to function of the ATP-producing apparatus. Thus, we isolated mitochondria and assessed their respiratory capacity.
Table 1.
Heart and body weights as well as tibia lengths of rats after 2, 6, 10 and 20 weeks of pressure overload compared to control
| 2 weeks | 6 weeks | 10 weeks | 20 weeks | |||||
|---|---|---|---|---|---|---|---|---|
| Sham | AoB | Sham | AoB | Sham | AoB | Sham | AoB | |
| BW (g) | 132.5 ± 6.6 | 116.5 ± 2.4 | 237.8 ± 6.7 | 235.9 ± 17.3 | 357.5 ± 10.8 | 349.8 ± 15.1 | 454.1 ± 22.7 | 419.5 ± 11.0 |
| HW (mg) | 494 ± 15 | 779 ± 81** | 827 ± 18 | 1701 ± 63*** | 1130 ± 25 | 2053 ± 85*** | 1282 ± 43 | 2273 ± 125*** |
| HW/BW (mg g−1) | 3.75 ± 0.14 | 6.67 ± 0.62** | 3.49 ± 0.09 | 7.31 ± 0.40*** | 3.18 ± 0.08 | 5.90 ± 0.19*** | 2.84 ± 0.11 | 5.43 ± 0.28*** |
| TL (mm) | 25.0 ± 0.5 | 24.0 ± 0.5 | 30.1 ± 0.3 | 31.0 ± 0.7 | 34.9 ± 0.5 | 36.7 ± 0.4** | 40.2 ± 0.7 | 41.1 ± 0.8 |
| HW/TL (mg mm−1) | 19.7 ± 0.5 | 32.4 ± 3.2** | 27.5 ± 0.5 | 54.9 ± 1.3*** | 32.4 ± 0.7 | 56.4 ± 2.4*** | 31.8 ± 0.8 | 55.7 ± 3.5*** |
| Lung weight (mg) | 829 ± 23 | 926 ± 83 | 1121 ± 30 | 2106 ± 116*** | 1399 ± 43 | 3312 ± 338*** | 1586 ± 60 | 3751 ± 459** |
| LBI (mg g−1) | 6.29 ± 0.22 | 7.95 ± 0.70 | 4.72 ± 0.06 | 9.05 ± 0.61*** | 3.93 ± 0.10 | 9.73 ± 1.30*** | 3.54 ± 0.23 | 8.85 ± 1.01** |
| Liver weight (g) | 5.63 ± 0.35 | 5.11 ± 0.13 | 10.50 ± 0.55 | 9.43 ± 0.76 | 13.00 ± 0.83 | 16.02 ± 1.23 | 13.91 ± 1.16 | 15.97 ± 1.04 |
| LiverBI (mg g−1) | 42.4 ± 0.7 | 43.9 ± 1.2 | 44.4 ± 1.3 | 40.2 ± 2.5 | 36.4 ± 2.2 | 45.8 ± 2.8* | 30.5 ± 1.4 | 38.1 ± 2.5* |
Data are mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, compared to age-matched controls after 2, 6, 10 and 20 weeks of pressure overload by aortic constriction. Sham, sham-operated animals; AoB, animals subjected to aortic banding; BW, body weight; HW, heart weight; TL, tibia length; LBI, lung to body weight index; LiverBI, liver to body weight index; n = 5–8.
Table 2.
Echocardiographic parameters of hearts subjected to pressure overload compared to control
| 2 weeks | 6 weeks | 10 weeks | 20 weeks | |||||
|---|---|---|---|---|---|---|---|---|
| Sham | AoB | Sham | AoB | Sham | AoB | Sham | AoB | |
| HR (beats min–1) | 288 ± 8 | 276 ± 17 | 267 ± 11 | 274 ± 14 | 243 ± 15 | 264 ± 13 | 229 ± 15 | 242 ± 6 |
| LVPWD (mm) | 1.21 ± 0.06 | 1.69 ± 0.17* | 1.67 ± 0.17 | 2.21 ± 0.11* | 1.49 ± 0.09 | 2.37 ± 0.12*** | 1.98 ± 0.24 | 2.71 ± 0.19* |
| LVEDD (mm) | 5.81 ± 0.32 | 4.80 ± 0.35 | 6.46 ± 0.12 | 7.00 ± 0.34 | 7.31 ± 0.09 | 8.16 ± 0.24** | 7.98 ± 0.27 | 9.44 ± 0.46* |
| LVEF (%) | 76.9 ± 2.8 | 88.6 ± 4.6 | 75.7 ± 2.1 | 64.7 ± 4.6 | 74.8 ± 2.1 | 65.6 ± 3.1* | 68.2 ± 2.0 | 47.8 ± 1.6*** |
| FS (%) | 46.6 ± 2.5 | 64.6 ± 5.7 | 45.9 ± 2.0 | 37.6 ± 3.7 | 45.6 ± 2.1 | 37.9 ± 2.5* | 39.8 ± 1.6 | 25.4 ± 1.0*** |
| LVMI | 2.64 ± 0.12 | 4.24 ± 0.30** | 3.13 ± 0.22 | 4.64 ± 0.32** | 2.46 ± 0.11 | 4.80 ± 0.38*** | 2.56 ± 0.15 | 573 ± 0.41*** |
Data are mean ± SEM; *P < 0.05, ***P < 0.001, compared to sham-operated animals after 2, 6, 10 and 20 weeks of pressure overload by aortic constriction. Sham, sham-operated animals; AoB, animals subjected to aortic banding; HR, heart rate; LVPWD, left ventricular posterior wall thickness in diastole; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; FS, fractional shortening; LVMI, left ventricular mass index; n = 5–11.
Mitochondrial respiration
Figure 2 shows maximal mitochondrial respiration (state 3 respiration) in cardiac mitochondria of rats with aortic constriction. At 2 weeks of pressure overload, an increase in state 3 respiration was observed with all substrates used. Elevated state 3 respiration also persisted at 6 weeks of pressure overload. After 10 weeks, maximal respiratory capacity was no longer different from respiratory capacity of control animals who experienced a known increase in respiratory capacity at this time (Schrepper et al. 2012). Age-dependent changes in respiratory capacity have been described before (Iossa et al. 2004). According to this, the increase in respiratory capacity in our control rats between 6 and 10 weeks after sham operation can be explained by their simultaneous transition from a juvenile to an adult rat. As animals in the corresponding groups are age-matched, rats in the TAC group are undergoing this transition as well.
Figure 2. Mitochondrial respiration.
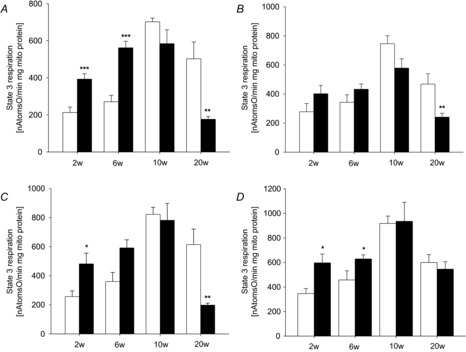
State 3 respiration of cardiac mitochondria after 2, 6, 10 and 20 weeks (w) of pressure overload induced by transverse aortic constriction (filled bars) compared to sham-operated control (open bars) with glutamate (A), pyruvate/malate (B), palmitoylcarnitine/malate (C) or succinate (D) as substrates. Data are mean ± SEM; n = 5–10; *P < 0.05, **P < 0.01, ***P < 0.001 compared to sham-operated animals.
Citrate synthase activity of the heart was normal at 2, 6 and 10 weeks of pressure overload (control vs. aortic banding (AoB) (U (g wet weight)−1): 2 weeks, 124 ± 22 vs. 92 ± 12; 6 weeks, 128 ± 21 vs. 104 ± 12; 10 weeks, 155 ± 19 vs. 116 ± 21). After 20 weeks of pressure overload, when systolic contractile function was impaired, citrate synthase activity was reduced (135 ± 12 vs. 84 ± 11 U (g wet weight)−1 and maximal mitochondrial respiratory capacity with complex I substrates (glutamate, pyruvate and palmitoylcarnitine) was significantly impaired. However, respiration with complex II substrate succinate was not affected at this time point. Uncoupled mitochondrial respiration showed the same pattern of respiratory changes (see online Supporting information, Table S1). This indicates that ATP production on complex V was not the rate-limiting step in our investigation. ADP-limited respiration (state 4) remained unchanged at most time points assessed and ADP/O ratios were not reduced (not shown). Thus, our results indicate a possible defect in the respiratory chain on complex I.
Isolated respiratory chain complex activities
Figure 3 shows individual complex activities of the respiratory chain. Despite the observed initial increase in mitochondrial respiratory capacity, all complexes showed normal activities at 2 and 6 weeks of pressure overload. Activities of complexes II, III and IV remained normal even with the onset of contractile dysfunction. However, complex I activity was found to be slightly reduced at 2 and 10 weeks, and significantly impaired at 20 weeks of pressure overload displaying a 25% reduction of total activity. Combined complex I and III activities were reduced about 45%. Complexes I and III are possible sites of mitochondrial ROS production, rendering them particularly vulnerable to ROS-induced damage. As complex I and combined complex I + III activities were impaired, we assessed mitochondrial ROS production on complexes I and III.
Figure 3. Isolated respiratory chain complex activities.
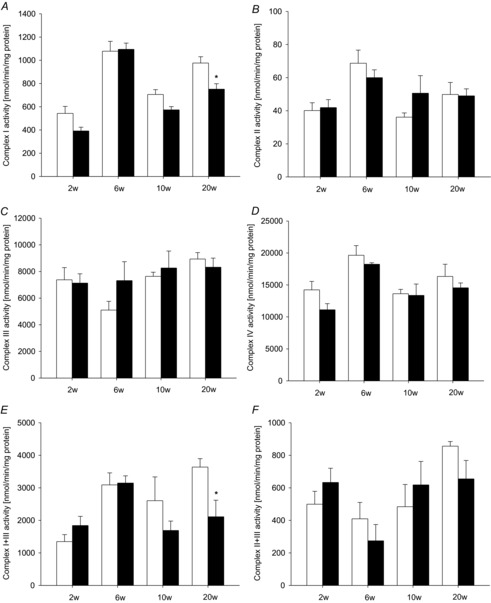
Activities for individual respiratory chain complexes I (A), II (B), III (C) and IV (D) and the combination of complexes I + III (E) and II + III (F) measured in isolated cardiac mitochondria after 2, 6, 10 and 20 weeks of pressure overload (filled bars) and sham-operated control groups (open bars). Data are mean ± SEM; n = 5–9; *P < 0.05 compared to sham-operated animals.
ROS production
In Fig. 4, ROS production rates on complexes I and III are shown. Maximal ROS production was not affected by pressure overload at 2 weeks. At 6 weeks of pressure overload, an increase in ROS production rates with pyruvate as substrate was observed. Using rotenone as inhibitor to prevent electron transfer to complex III, we were able to locate this increase to complex I. ROS production rates on complex I remained elevated at 10 and 20 weeks of pressure overload. Elevated ROS production rates on complex I may be causally related to impairment in the function of complex I.
Figure 4. ROS production.
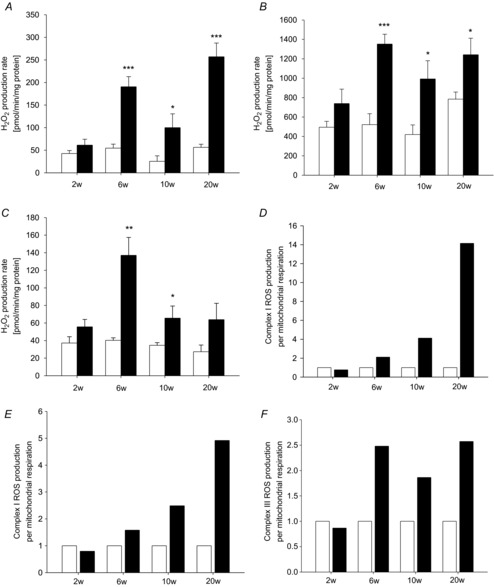
ROS production capacity after 2, 6, 10 and 20 weeks of pressure overload (filled bars), compared to sham-operated control (open bars) with substrates pyruvate/malate (A), pyruvate/malate and rotenone (B), or succinate and rotenone (C). ROS production capacity in relation to respiratory capacity: complex I ROS production with pyruvate/malate as substrate compared to pyruvate/malate-based respiration (D), complex I ROS production with pyruvate/malate and rotenone compared to pyruvate/malate-based respiration (E), and complex III ROS production with succinate and rotenone related to succinate-based respiration (F). Data are mean ± SEM; n = 6–13; *P < 0.05; **P < 0.01, ***P < 0.001 compared to sham-operated animals.
Using succinate as a substrate and rotenone to inhibit reverse flow, an increase in ROS production capacity on complex III could be observed at 6 weeks of pressure overload. In contrast to ROS production rates on complex I, maximal ROS generation on complex III was elevated at 6 and 10 weeks of pressure overload only. Then, the difference was no longer significant at 20 weeks. The sequence of events indicated that elevated ROS production, specifically a peak at 6 weeks of pressure overload, may be involved in the onset of contractile and mitochondrial dysfunction.
ROS damage
Figure 5A shows the amount of 4-hydroxynonenal adducts (HNE) as a measure for oxidative damage in cardiac tissue. The amount of HNE at 2, 6 or 10 weeks of pressure overload was not significantly different to control. At 20 weeks of pressure overload a significant increase in 4-HNE was found, indicating accumulation of ROS damage at this time point. However, variation in TAC groups was relatively high, most likely due to differences in the individual response of each animal to the procedure of aortic banding. This may conceal a potentially significant increase in 4-HNE beginning as early as 2 weeks after aortic banding. We further assessed protein carbonylation as another marker of oxidative damage. As shown in Fig. 5B, protein carbonylation was not different to control animals at any time point. This seems to be in contrast to other studies describing increased protein carbonylation and may be due to different assay techniques used. Taken together, these data indicate that ROS damage occurs in the late phase of decompensated hypertrophy and heart failure and seems to primarily lead to lipid peroxidation.
Figure 5. ROS damage.
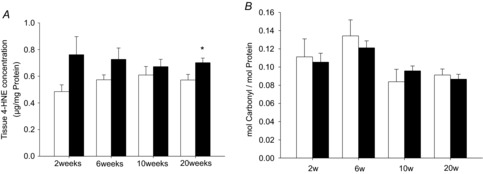
Accumulation of 4-hydroxynonenal (A) adducts (4-HNE) and carbonylated proteins (B) as a measure for oxidative damage in pressure overload after 2, 6, 10 and 20 weeks of pressure overload (filled bars), compared to sham-operated control (open bars). Data are mean ± SEM; n = 4–6; *P < 0.05.
Antioxidative therapy
Thus, we next assessed the effect of reducing the concentration of reactive oxygen species or limiting their production. The radical scavenger PBN and the mild uncoupler DNP were administered to animals subjected to pressure overload. Animals treated with antioxidative therapy were examined at 2, 6 and 10 weeks of pressure overload. Therapy had no influence on animal weight, heart weight or heart to body weight ratio (see online Supporting information, Table S2). Additionally, lung to body weight index was similarly elevated in PBN- and DNP-treated animals as in untreated pressure overload, suggesting an identical degree of contractile dysfunction. Echocardiographic data are shown in Table 3. At 10 weeks of pressure overload increased posterior wall thickness could be detected with PBN treatment. A significant increase in LVMI was found in both treatment groups at 2 and 6 weeks. Figure 6 shows mitochondrial H2O2 production as a measure for ROS production rates in cardiac mitochondria of animals treated with PBN or DNP. Both PBN and DNP efficiently prevented the peak in mitochondrial ROS production capacity on complexes I and III at 6 weeks. At 10 weeks, both treatments were unable to prevent the increase in ROS production rates completely and at that time, ROS production rates were comparable between treated and untreated animals with pressure overload. We further assessed the relation of antioxidant treatment and complex I activity. In Fig. 6D complex I activity is shown in control, pressure overload- and antioxidant-treated animals. Antioxidant treatment was associated with an increase in isolated complex I activity at 2 weeks of pressure overload. At 6 weeks of pressure overload such increase was only visible with PBN. The slight decrease in complex I activity found after 10 weeks of pressure overload did not appear with antioxidative treatment.
Table 3.
Echocardiographic parameters of hearts subjected to pressure overload and antioxidative therapy
| 2 weeks | 6 weeks | 10 weeks | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AoB | PBN | DNP | AoB | PBN | DNP | AoB | PBN | DNP | |
| HR (beats min–1) | 276 ± 17 | 321 ± 16* | 425 ± 8* | 274 ± 14 | 285 ± 14 | 276 ± 11 | 264 ± 13 | 259 ± 29 | 296 ± 11 |
| LVPWD (mm) | 1.69 ± 0.17 | 1.50 ± 0.12 | 2.06 ± 0.14 | 2.21 ± 0.11 | 2.56 ± 0.20 | 2.39 ± 0.10 | 2.37 ± 0.12 | 3.25 ± 0.21* | 2.60 ± 0.14 |
| LVEDD (mm) | 4.80 ± 0.35 | 5.70 ± 0.31 | 4.09 ± 0.17 | 7.00 ± 0.34 | 7.86 ± 0.65 | 8.06 ± 0.33* | 8.16 ± 0.24 | 7.52 ± 0.91 | 8.16 ± 0.62 |
| LVEF (%) | 88.6 ± 4.6 | 78.4 ± 4.2 | 98.1 ± 0.8 | 64.7 ± 4.6 | 61.3 ± 3.0 | 56.6 ± 2.3 | 65.6 ± 3.1 | 69.3 ± 4.6 | 65.5 ± 9.8 |
| FS (%) | 64.6 ± 5.7 | 50.8 ± 5.4 | 83.0 ± 3.4* | 37.6 ± 3.7 | 34.6 ± 2.2 | 31.2 ± 1.6 | 37.9 ± 2.4 | 43.4 ± 3.6 | 40.9 ± 9.2 |
| LVMI | 4.24 ± 0.30 | 6.09 ± 0.58* | 5.71 ± 0.18* | 4.64 ± 0.32 | 8.28 ± 0.72* | 7.78 ± 0.23* | 4.80 ± 0.38 | 6.25 ± 0.72 | 6.29 ± 0.75 |
Data are mean ± SEM; *significant different compared to untreated animals after 2, 6 and 10 weeks of pressure overload by aortic constriction. AoB, animals subjected to aortic banding without treatment; PBN, animals subjected to aortic banding with PBN (α-phenyl-N-tert-butyl nitrone) treatment; DNP, animals subjected to aortic banding with DNP (2,4-dinitrophenol) treatment. Abbreviations in first column as in Table 2. n = 5–9.
Figure 6. Antioxidative therapy.
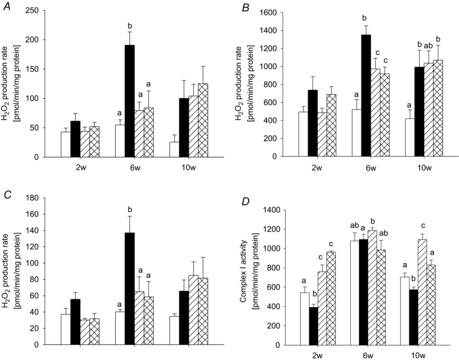
ROS production capacity after 2, 6 and 10 weeks of pressure overload in cardiac mitochondria from PBN-treated (hatched bars) and DNP-treated animals (cross-hatched bars), compared to pressure overload without antioxidant supplement (filled bars) and sham animals (open bars) with substrates pyruvate/malate (A), pyruvate/malate and rotenone (B), or succinate and rotenone (C). D, activities for individual respiratory chain complex I in these animals measured in isolated cardiac mitochondria. Data are mean ± SEM; n = 5–9; different superscripts indicate significant differences.
Discussion
We found in this study that an increase in maximal ROS production occurs early in the development of heart failure with a peak concomitant with the onset of diastolic dysfunction. However, in our setup this increase may not be directly related to the onset of contractile dysfunction.
ROS are discussed as a potential mechanism for the development of heart failure (Dai et al. 2011). Activation of ROS production has been demonstrated in the failing heart (Chance et al. 1979). Studies with genetically modified animals with increased oxidative stress indicated that ROS may cause heart failure and mitochondrial dysfunction (Nojiri et al. 2006) while increased expression of antioxidant enzymes has been shown to ameliorate cardiac abnormalities (Giordano, 2005). In general, ROS are produced at various sites inside the cell, including NADPH oxidase, nitric oxide synthase and xanthine oxidase. In addition, mitochondria are a major site of cardiac ROS production and the largest producers of ROS in the cell (Turrens, 2003; Balaban et al. 2005) and an increase in mitochondrial ROS production has been linked to various cardiac pathologies. Mitochondrial ROS production has been described to mediate angiotensin II-induced cardiac hypertrophy and Gαq overexpression-induced heart failure (Dai et al. 2011). However, the time course of ROS production in heart failure development has not been investigated. Thus, we present for the first time an analysis of mitochondrial ROS production during the development of contractile dysfunction and heart failure and we used a non-transgenic animal model. Mitochondrial ROS production increased with the onset of diastolic dysfunction but before the onset of systolic dysfunction. Increased ROS production led to increased amounts of HNE with contractile dysfunction. The sequence of events suggested that mitochondrial ROS production may be involved in the development of heart failure. However, neither ROS scavenging with a spin trap agent, nor mild uncoupling affected the onset of contractile dysfunction and our results thus do not support a potential causal relationship. In this context it is important to consider that not all antioxidants are equal and other antioxidants may have different effects.
Epidemiological data suggest that antioxidants reduce cardiovascular disease but clinical trials have failed thus far to reproduce such an effect (Jha et al. 1995; Giordano, 2005). In experimental models, treatments with antioxidants, albeit with inconsistent results, have found beneficial effects (Giordano, 2005). However, in pressure overload only a few investigations have been published. Treatment of guinea pigs with vitamin E was found to have no influence on hypertrophy (Dhalla et al. 1996). In pressure overload in mice, antioxidant treatment with edaravone reduced early hypertrophy but long-term treatment has not been investigated (Tsujimoto et al. 2005). Date et al. applied N-2-mercaptopropionyl glycine and attenuated hypertrophy in pressure overload slightly (Date et al. 2002). However, neither study tested an antioxidant effect on the development of heart failure. The superoxide dismutase mimetic M40401 attenuated cardiac oxidative stress in pressure overload in mice with TAC. Although the treatment reduced hypertrophy, it was not able to prevent cardiac hypertrophy or contractile dysfunction (Lu et al. 2010). Nevertheless, the authors suggested that antioxidative treatment protected against chronic heart failure. Transgenic overexpression of mitochondrial catalase in mice partially attenuated heart failure parameters but was not able to prevent heart failure. The effect may have been mediated by ROS scavenging, but effects on gene expression could not be excluded (a limitation in all transgenic animal models) (Dai et al. 2012). One other study in mice was able to prevent pressure overload-induced reduction of contractile function using antioxidants (SS20 or SS31) (Dai et al. 2013). In other words, a causal relationship has been suggested frequently but it has so far been rarely shown. We now present data, showing that our applied antioxidative treatment reduced ROS production capacity, but was not able to prevent the development of contractile dysfunction. Two independent treatments with a spin trap agent scavenging oxygen radicals and a mild uncoupler reducing mitochondrial ROS generation led to a decline in maximal ROS production at 2 and 6 weeks of pressure overload, demonstrating efficacy of antioxidant treatment. A tendency for reduced cardiac hypertrophy could be observed at 6 weeks of pressure overload. However, despite these observations the selected antioxidant treatments were neither able to delay nor prevent the onset of contractile dysfunction. Thus, our results support the notion that ROS do not play a causal role in the development of heart failure. In line with these results, measurements revealed no changes of protein carbonylation and changes of lipid peroxidation with heart failure only. While our assays tested for global oxidative damage, they do not exclude oxidative damage to specific proteins or lipids. Our observation that mild uncoupling with DNP temporarily reduced ROS production without preventing the onset of systolic dysfunction may shed new light on whether uncoupling in the development of heart failure is beneficial or detrimental. The role of uncoupling proteins in cardiac metabolism and heart failure has been discussed extensively (Laskowski & Russell, 2008; Nabben & Hoeks, 2008).
Changes in substrate oxidation and energy metabolism in heart failure have been demonstrated (Stanley et al. 2005) and mitochondrial dysfunction has been discussed as a mechanism for heart failure development (Stanley et al. 2005). Lemieux et al. assessed mitochondrial function in human patients with heart disease and chronic heart failure and found a progressive decrease of phosphorylation and electron transport chain capacities, irrespective of the site of substrate entry into the electron transport chain (Lemieux et al. 2011). Although multiple mitochondrial defects occurred early in the progression of heart failure, it remains unclear if mitochondrial dysfunction is causally related to contractile dysfunction. Rosca et al. (2008) found reduced mitochondrial respiratory capacity with normal complex function and suggested impaired supercomplex formation as a potential mechanism for impaired mitochondrial function. In contrast, in our study, reduced mitochondrial respiratory capacity with complex I-dependent substrates correlated with reduced complex I and III activity. Thus, impaired supercomplex assembly seems an unlikely mechanism for mitochondrial dysfunction in our model as the impairment is fully explainable with reduced complex activities. We also did not find increased uncoupling or reduced ADP/O ratios as described by Murray et al. for heart failure after myocardial infarction (Murray et al. 2008). These studies present different mitochondrial defects in heart failure. The reason for the reduction of complex I activity in pressure overload is not clear and may be caused by the modification of subunits of complex I by defective assembly or by changes in gene expression of subunits. We suggest that the development of mitochondrial dysfunction seems to be dependent on the model used.
Mitochondrial respiration in pressure overload heart failure was reduced by about 50% with complex I substrates. In contrast, reduction of isolated complex I activity was only about 20%, while all other isolated single complex activities remained normal. We subsequently measured combined complex activity on complexes I and III and observed a reduction in activity which was comparable in extent to the reduction in mitochondrial respiration. Although these numerical comparisons may not be fully adequate, the findings support the conclusions that a small change in individual complex activity may have a much larger impact on the function of the entire system if investigated in the proper context.
Pressure overload induces cardiac hypertrophy, which requires a concomitant increase in oxygen supply to maintain cellular homeostasis and contractile function (Hilfiker-Kleiner et al. 2006). Impaired angiogenesis may therefore contribute to the development of contractile dysfunction during pressure overload-induced hypertrophy. Interestingly, decreased oxygen levels were reported to be associated with both increased and decreased ROS production rates (Acker et al. 2006). As we have not measured angiogenesis in our model of pressure overload, it cannot be ruled out that our findings are at least partially due to hypoxia. In a recent study, Heather et al. evaluated the function of cardiac mitochondria in rats subjected to chronic hypoxia (Heather et al. 2012). In their study, oxygen shortage was characterized by reduced rates of mitochondrial respiration and unchanged or reduced mitochondrial ROS production after 3 weeks of hypoxia. In contrast, we show that mitochondrial respiration is increased or unchanged during the development of systolic dysfunction, accompanied by elevated levels of ROS production. Thus, it seems unlikely that relevant hypoxia occurs during the first 10 weeks of hypertrophy in our model of pressure overload-induced heart failure. Hypoxia has also been shown to reduce complex I activity by downregulating the expression of ISCU1/2 (Chan et al. 2009), which is required for the assembly of iron–sulfur clusters that are integral parts of the mitochondrial complexes I, II and III (Rouault & Tong, 2008). However, if hypoxic repression of ISCU1/2 would occur in our model, one might expect to see changes in the activities of complex II and complex III as well, which is not the case. Therefore, the specific reduction of complex I activity in our model is unlikely to be due to reduced ISCU1/2 expression under hypoxic conditions.
We found in this study that an increase in mitochondrial ROS production capacity occurs early in the development of heart failure with a peak at the onset of diastolic dysfunction. Subsequently, mitochondrial dysfunction appeared simultaneously with systolic dysfunction. Antioxidative treatment reduced mitochondrial ROS production rates. However, antioxidative treatment did not affect the development of contractile dysfunction. Thus, ROS production may not be related to the onset of contractile dysfunction.
The investigation is limited by the use of inhibitors which may not be fully specific. The methods used to detect reactive oxygen species may not be sensitive enough to fully exclude a causal relationship. However, these limitations are common shortcomings in this field.
Acknowledgments
None declared.
Glossary
- ADP/O
ratio of consumed ADP to oxygen consumed
- AoB
aortic banding
- DNP
2,4-dinitrophenol
- HNE
hydroxynonenal
- LV
left ventricle
- LVMI
LV mass index
- PBN
α-phenyl-N-tert-butyl nitrone
- ROS
reactive oxygen species
- TAC
transverse aortic constriction
Additional information
Competing interests
None declared.
Author contributions
The work was performed at the University of Leipzig Heart Center and at the Department of Cardiothoracic Surgery of Jena University Hospital, Friedrich Schiller University of Jena. M.S. and A.L. designed the study; M.S., M.O., A.L., A.S. and P.A. collected and analysed the data; M.S., M.O., A.L., A.S. and T.D. interpreted the data; M.O. and M.S. drafted the manuscript; M.O., A.S. and T.D. revised the manuscript. All authors qualify for authorship; all persons qualifying for authorship are listed and all authors approved the final version of the manuscript.
Funding
This study was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to T.D. (DO 602/4-1, 6-1, and 8-1). T.D. was Heisenberg professor of the DFG at the University of Leipzig.
Supporting Information
The following supporting information is available in the online version of this article
Disclaimer: Supplementary materials have been peer-reviewed but not copyedited.
Table S1. Cardiac mitochondrial respiratory properties of rats subjected to pressure overload compared to control.
Table S2. Heart and body weights as well as tibia lengths of animals after 2, 6 and 10 weeks of pressure overload with antioxidative therapy.
References
- Acker T, Fandrey J, Acker H. The good, the bad and the ugly in oxygen-sensing: ROS, cytochromes and prolyl-hydroxylases. Cardiovasc Res. 2006;71:195–207. doi: 10.1016/j.cardiores.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Barja G. The quantitative measurement of H2O2 generation in isolated mitochondria. J Bioenerg Biomembr. 2002;34:227–233. doi: 10.1023/a:1016039604958. [DOI] [PubMed] [Google Scholar]
- Baudet S, Kuznetsov A, Merciai N, Gorza L, Ventura-Clapier R. Biochemical, mechanical and energetic characterization of right ventricular hypertrophy in the ferret heart. J Mol Cell Cardiol. 1994;26:1573–1586. doi: 10.1006/jmcc.1994.1177. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bolli R, Patel BS, Jeroudi MO, Lai EK, McCay PB. Demonstration of free radical generation in ‘stunned’ myocardium of intact dogs with the use of the spin trap α-phenyl Ntert-butyl nitrone. J Clin Invest. 1988;82:476–485. doi: 10.1172/JCI113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulteau AL, Lundberg KC, Ikeda-Saito M, Isaya G, Szweda LI. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc Natl Acad Sci U S A. 2005;102:5987–5991. doi: 10.1073/pnas.0501519102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7:552–560. doi: 10.1111/j.1474-9726.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation: III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- Dai D-F, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCoss MJ, Rabinovitch PS. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013;6:1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai D-F, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Gαq overexpression-induced heart failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date MO, Morita T, Yamashita N, Nishida K, Yamaguchi O, Higuchi Y, Hirotani S, Matsumura Y, Hori M, Tada M, Otsu K. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J Am Coll Cardiol. 2002;39:907–912. doi: 10.1016/s0735-1097(01)01826-5. [DOI] [PubMed] [Google Scholar]
- Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol. 1996;28:506–514. doi: 10.1016/0735-1097(96)00140-4. [DOI] [PubMed] [Google Scholar]
- Doenst T, Pytel G, Schrepper A, Amorim P, Färber G, Shingu Y, Mohr FW, Schwarzer M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res. 2010;86:461–470. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- Estabrook RW. Mitochondrial respiratory control and the polarographic measurement of ADP:O ratios. Methods Enzymol. 1967;10:41–47. [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez J, Caro P, Sanchez I, Naudi A, Jove M, Portero-Otin M, Lopez-Torres M, Pamplona R, Barja G. Effect of methionine dietary supplementation on mitochondrial oxygen radical generation and oxidative DNA damage in rat liver and heart. J Bioenerg Biomembr. 2009;41:309–321. doi: 10.1007/s10863-009-9229-3. [DOI] [PubMed] [Google Scholar]
- Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- Heather LC, Carr CA, Stuckey DJ, Pope S, Morten KJ, Carter EE, Edwards LM, Clarke K. Critical role of complex III in the early metabolic changes following myocardial infarction. Cardiovasc Res. 2010;85:127–136. doi: 10.1093/cvr/cvp276. [DOI] [PubMed] [Google Scholar]
- Heather LC, Cole MA, Tan JJ, Ambrose LJ, Pope S, Abd-Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ, Clarke K. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol. 2012;107:268. doi: 10.1007/s00395-012-0268-2. [DOI] [PubMed] [Google Scholar]
- Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure: focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol. 2006;48:A56–66. [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Iossa S, Mollica MP, Lionetti L, Crescenzo R, Tasso R, Liverini G. A possible link between skeletal muscle mitochondrial efficiency and age-induced insulin resistance. Diabetes. 2004;53:2861–2866. doi: 10.2337/diabetes.53.11.2861. [DOI] [PubMed] [Google Scholar]
- Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- Jha P, Flather M, Lonn E, Farkouh M, Yusuf S. The antioxidant vitamins and cardiovascular disease. A critical review of epidemiologic and clinical trial data. Ann Intern Med. 1995;123:860–872. doi: 10.7326/0003-4819-123-11-199512010-00009. [DOI] [PubMed] [Google Scholar]
- Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- Krahenbuhl S, Chang M, Brass EP, Hoppel CL. Decreased activities of ubiquinol:ferricytochrome c oxidoreductase (complex III) and ferrocytochrome c:oxygen oxidoreductase (complex IV) in liver mitochondria from rats with hydroxycobalamin[c-lactam]-induced methylmalonic aciduria. J Biol Chem. 1991;266:20998–21003. [PubMed] [Google Scholar]
- Laskowski KR, Russell RR., 3rd Uncoupling proteins in heart failure. Curr Heart Fail Rep. 2008;5:75–79. doi: 10.1007/s11897-008-0013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux H, Semsroth S, Antretter H, Hofer D, Gnaiger E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int J Biochem Cell Biol. 2011;43:1729–1738. doi: 10.1016/j.biocel.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, Fassett J, Tao Y, Zhang P, dos Remedios C, Pritzker M, Hall JL, Garry DJ, Chen Y. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation. 2010;121:1474–1483. doi: 10.1161/CIRCULATIONAHA.109.906818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Casari G. In vivo detection of oxidized proteins: a practical approach to tissue-derived mitochondria. Methods Mol Biol. 2010;648:257–267. doi: 10.1007/978-1-60761-756-3_17. [DOI] [PubMed] [Google Scholar]
- Mandinov L, Eberli FR, Seiler C, Hess OM. Diastolic heart failure. Cardiovasc Res. 2000;45:813–825. doi: 10.1016/s0008-6363(99)00399-5. [DOI] [PubMed] [Google Scholar]
- Marín-García J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- Mogensen M, Sahlin K, Fernström M, Glintborg D, Vind BF, Beck-Nielsen H, Højlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Cole MA, Lygate CA, Carr CA, Stuckey DJ, Little SE, Neubauer S, Clarke K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J Mol Cell Cardiol. 2008;44:694–700. doi: 10.1016/j.yjmcc.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Nabben M, Hoeks J. Mitochondrial uncoupling protein 3 and its role in cardiac- and skeletal muscle metabolism. Physiol Behav. 2008;94:259–269. doi: 10.1016/j.physbeh.2007.11.039. [DOI] [PubMed] [Google Scholar]
- Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem. 2006;281:33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- Ozkan A, Kapadia S, Tuzcu M, Marwick TH. Assessment of left ventricular function in aortic stenosis. Nat Rev Cardiol. 2011;8:494–501. doi: 10.1038/nrcardio.2011.80. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signalling. Trends Endocrinol Metab. 2009;20:332–340. doi: 10.1016/j.tem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick AZ, Packer L. Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. 1994;233:357–363. doi: 10.1016/s0076-6879(94)33041-7. [DOI] [PubMed] [Google Scholar]
- Rosca MG, Hoppel CL. New aspects of impaired mitochondrial function in heart failure. J Bioenerg Biomembr. 2009;41:107–112. doi: 10.1007/s10863-009-9215-9. [DOI] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi VM, Pires MD, Cestari IN, Cestari IA, Picard MH, Leirner AA, Mady C. Echocardiographic assessment of global ventricular function using the myocardial performance index in rats with hypertrophy. Artif Organs. 2004;28:332–337. doi: 10.1111/j.1525-1594.2004.47350.x. [DOI] [PubMed] [Google Scholar]
- Samartsev VN, Chezganova SA, Polishchuk LS, Paydyganov AP, Vidyakina OV, Zeldi IP. Temperature dependence of rat liver mitochondrial respiration with uncoupling of oxidative phosphorylation by fatty acids. Influence of inorganic phosphate. Biochemistry (Mosc) 2003;68:618–626. doi: 10.1023/a:1024657507814. [DOI] [PubMed] [Google Scholar]
- Schrepper A, Schwarzer M, Schope M, Amorim PA, Doenst T. Biphasic response of skeletal muscle mitochondria to chronic cardiac pressure overload – role of respiratory chain complex activity. J Mol Cell Cardiol. 2012;52:125–135. doi: 10.1016/j.yjmcc.2011.10.022. [DOI] [PubMed] [Google Scholar]
- Schwarzer M, Schrepper A, Amorim PA, Osterholt M, Doenst T. Pressure overload differentially affects respiratory capacity in interfibrillar and subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2013;304:H529–H537. doi: 10.1152/ajpheart.00699.2012. [DOI] [PubMed] [Google Scholar]
- Selivanov VA, Votyakova TV, Pivtoraiko VN, Zeak J, Sukhomlin T, Trucco M, Roca J, Cascante M. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput Biol. 2011;7:e1001115. doi: 10.1371/journal.pcbi.1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingu Y, Amorim PA, Nguyen TD, Osterholt M, Schwarzer M, Doenst T. Echocardiography alone allows the determination of heart failure stages in rats with pressure overload. Thorac Cardiovasc Surg. 2013;61:718–725. doi: 10.1055/s-0032-1326775. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–11. [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Tomec RJ, Hoppel CL. Carnitine palmitoyltransferase in bovine fetal heart mitochondria. Arch Biochem Biophys. 1975;170:716–723. doi: 10.1016/0003-9861(75)90169-1. [DOI] [PubMed] [Google Scholar]
- Tsujimoto I, Hikoso S, Yamaguchi O, Kashiwase K, Nakai A, Takeda T, Watanabe T, Taniike M, Matsumura Y, Nishida K, Hori M, Kogo M, Otsu K. The antioxidant edaravone attenuates pressure overload-induced left ventricular hypertrophy. Hypertension. 2005;45:921–926. doi: 10.1161/01.HYP.0000163461.71943.e9. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr NB, Levine RL. Quantification of protein carbonylation. Methods Mol Biol. 2013;965:265–281. doi: 10.1007/978-1-62703-239-1_18. [DOI] [PubMed] [Google Scholar]
- Wen JJ, Bhatia V, Popov VL, Garg NJ. Phenyl-α-tert-butyl nitrone reverses mitochondrial decay in acute Chagas’ disease. Am J Pathol. 2006;169:1953–1964. doi: 10.2353/ajpath.2006.060475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Cardiac mitochondrial respiratory properties of rats subjected to pressure overload compared to control.
Table S2. Heart and body weights as well as tibia lengths of animals after 2, 6 and 10 weeks of pressure overload with antioxidative therapy.