

Abstract
Sympathoexcitation elicited by central command, a parallel activation of the motor and autonomic neural circuits in the brain, has been shown to become exaggerated in chronic heart failure (CHF). The present study tested the hypotheses that oxidative stress in the medulla in CHF plays a role in exaggerating central command-elicited sympathoexcitation, and that exercise training in CHF suppresses central command-elicited sympathoexcitation through its antioxidant effects in the medulla. In decerebrate rats, central command was activated by electrically stimulating the mesencephalic locomotor region (MLR) after neuromuscular blockade. The MLR stimulation at a current intensity greater than locomotion threshold in rats with CHF after myocardial infarction (MI) evoked larger (P < 0.05) increases in renal sympathetic nerve activity and arterial pressure than in sham-operated healthy rats (Sham) and rats with CHF that had completed longterm (8–12 weeks) exercise training (MI + TR). In the Sham and MI + TR rats, bilateral microinjection of a superoxide dismutase (SOD) mimetic Tempol into the rostral ventrolateral medulla (RVLM) had no effects on MLR stimulation-elicited responses. By contrast, in MI rats, Tempol treatment significantly reduced MLR stimulation-elicited responses. In a subset of MI rats, treatment with Tiron, another SOD mimetic, within the RVLM also reduced responses. Superoxide generation in the RVLM, as evaluated by dihydroethidium staining, was enhanced in MI rats compared with that in Sham and MI + TR rats. Collectively, these results support the study hypotheses. We suggest that oxidative stress in the medulla in CHF mediates central command dysfunction, and that exercise training in CHF is capable of normalizing central command dysfunction through its antioxidant effects in the medulla.
Key points
In heart failure, sympathoexcitation elicited by central command, a parallel activation of the motor and autonomic neural circuits in the brain, is exaggerated.
Mechanisms underlying central command dysfunction in heart failure were unexplored, and effects of exercise training on central command dysfunction in heart failure were not determined.
Data presented here suggest that oxidative stress in the medulla in heart failure mediates central command dysfunction, and that exercise training in heart failure is capable of normalizing central command dysfunction through its antioxidant effects in the medulla.
The present study contributes to our understanding of brain mechanisms underlying abnormal autonomic adjustments to exercise in heart failure.
Introduction
Supervised exercise training interventions in patients with chronic heart failure (CHF) have now been accepted as therapeutic treatment. Such treatment not only improves quality of life and functional class, but also decreases resting sympathetic overactivity, alleviates peripheral inflammation, and reverses morphometric and histochemical features of skeletal myopathy (Middlekauff, 2010; Downing & Balady, 2011; Piepoli et al. 2011). However, sympathoexcitation seen during a bout of exercise is augmented in patients with CHF in comparison with healthy individuals (Sterns et al. 1991; Negrão et al. 2001). The augmented sympathoexcitation during exercise is a possible cause of exercise intolerance, a hallmark of CHF (Notarius et al. 1999). Thus, understanding regulatory mechanisms of sympathetic nerve activity (SNA) during exercise in CHF is clinically important.
Central command is a feedforward neural mechanism that evokes parallel modifications of motor and autonomic functions during exercise (Goodwin et al. 1972). This neural activation elicits sympathoexcitation by exciting the medullary cardiovascular pathway containing the rostral ventrolateral medulla (RVLM), in which sympathetic premotor neurones are located (Nolan & Waldrop, 1997; Padley et al. 2007). Central command function in patients with CHF has been suggested to become exaggerated, thereby contributing to augmented sympathoexcitation during exercise (Sterns et al. 1991; Negrão et al. 2001). This suggestion was supported by the findings of a subsequent rat study which showed that sympathoexcitatory responses to central command activation, caused by electrical stimulation of the mesencephalic locomotor region (MLR) after neuromuscular blockade (Eldridge et al. 1985; Bedford et al. 1992), were larger in rats with CHF than in healthy controls (Koba et al. 2006a). However, the mechanisms underlying central command dysfunction in CHF have so far remained unknown.
Oxidative stress developing in CHF may contribute to central command dysfunction. Superoxide overproduction in the central nervous system has been suggested to lead to neurocardiovascular dysregulation, such as resting sympathetic overactivity (Gao et al. 2004, 2005, 2007; Kishi et al. 2004, 2011; Lindley et al. 2004; Zimmerman et al. 2004; Nishi et al. 2013). It was reported that in conscious CHF rabbits in which the RVLM was exposed to oxidative stress, intracerebroventricular administration of a superoxide dismutase (SOD) mimetic Tempol decreased resting SNA (Gao et al. 2004). Moreover, in vitro studies have suggested that superoxide increases the sensitivity of neuronal cells, which respond to excitatory input by regulating membrane ion channels and controlling action potential generation. For example, voltage-gated potassium current in neuronal cells was shown to be inhibited by superoxide (Sun et al. 2005; Yin et al. 2010). These findings led us to hypothesize that oxidative stress in the medulla of CHF may play a role in sensitizing the RVLM neurones which respond to central command activation, thereby exaggerating central command-elicited sympathoexcitation (Hypothesis 1).
As stated, longterm exercise training has beneficial effects on various clinical parameters in CHF. However, there has been no information about the effect of exercise training on central command dysfunction. Longterm exercise training in rabbits with CHF reportedly had an antioxidant effect in the RVLM, thereby decreasing resting sympathetic overactivity (Gao et al. 2007). Given these findings, and if the present Hypothesis 1 is correct, it is reasonable to hypothesize that exercise training in CHF suppresses central command-elicited sympathoexcitation through its antioxidant effects in the medulla (Hypothesis 2).
The purposes of this study were to test Hypotheses 1 and 2 in rats. In the experiments, we employed three rat groups: (i) rats with CHF after myocardial infarction (MI); (ii) rats with CHF that had completed longterm exercise training, and (iii) sham-operated control rats. In these rats, renal SNA (RSNA) and cardiovascular responses to electrical stimulation of the MLR were examined. The effects of an antioxidant treatment within the rat RVLM on MLR stimulation-elicited responses were also investigated. Moreover, in situ superoxide production in the rat medulla was studied.
Methods
All procedures outlined in the present study complied with the Guiding Principles for the Care and Use of Animals in the Fields of Physiological Sciences of the Physiological Society of Japan, and were approved by the Animal Care Committee of Tottori University. The present experiments were performed in male Sprague–Dawley rats (n = 70). Rats were housed in standard rodent cages in a temperature-controlled room (24°C) and regulated on a 12 : 12 h light–dark schedule. Food and water were made available ad libitum.
Ligation surgery, exercise training and echocardiography
Coronary artery ligation surgery to induce MI was performed as described previously (Koba et al. 2006a, 2009). Rats (aged 5–6 weeks, 170–220 g, n = 47) were anaesthetized with a mixture of isoflurane (<4%) and oxygen, intubated and artificially ventilated with a respirator (model SN-480-7; Shinano Co., Tokyo, Japan). An incision between the fourth and fifth ribs was made, and the left ventricular wall was exposed through left thoracotomy. The left coronary artery was then ligated (MI rats). In another set of rats (n = 18), sham operations were performed without ligation of the coronary artery (Sham rats). The thorax was closed, the tracheal tube was removed, and the rat was allowed to recover from anaesthesia.
At 6–9 weeks after the ligation surgery, a subset of MI rats was randomly assigned to a training group (MI + TR rats). MI + TR rats (n = 17) were treadmill-trained for 8–12 weeks according to a progressive exercise protocol adapted from a previous study (Musch et al. 1986). In the present study, the treadmill exercise was conducted five times per week between 09.00 hours and 11.00 hours. On the first day of the protocol, the rats were acclimatized to a custom-built treadmill (MK-680C; Muromachi Kikai Co. Ltd, Tokyo, Japan) by running at 20 m min−1 on a 5% incline for 5 min. The duration of running was then increased by 0–10 min per day over a 3 week period until rats ran for 60 min per day. Rats that exhibited refusal to run during the training programme were excluded from further training. The protocol employed in the present study is considered sufficient to have exercise training effects; exercise training protocols previously employed by others in which the training period was equivalent to or shorter than that used in the present study had significant effects in terms of increasing maximal oxygen consumption, skeletal muscle succinate dehydrogenase activities, and/or skeletal muscle citrate synthase activities in rats with CHF (Musch et al. 1986; Kleiber et al. 2008). Sedentary MI or Sham rats had limited activity in the cages in which they were housed during the exercise training for MI + TR rats.
Transthoracic echocardiography (model 5189002; GE Healthcare Ltd, Little Chalfont, UK) was performed in rats anaesthetized with 1.5% isoflurane in oxygen to assess left ventricular function, prior to the experiments described below. Rats that underwent ligation were excluded from the study if they did not meet the present criteria for CHF [i.e. left ventricular fractional shortening (FS) of <35%].
Experiment preparation
The surgery and experiments to observe MLR stimulation-elicited responses were conducted as reported previously (Koba et al. 2006a,b2006b). In rats anaesthetized with a mixture of isoflurane (<4%) and oxygen, the trachea was cannulated and the lungs were mechanically ventilated with a respirator with 6 ml kg−1 tidal volume at a frequency of 70 per min. The left jugular vein and common carotid artery were cannulated to administer drugs and to measure arterial pressure (AP), respectively. The arterial catheter was attached to a pressure transducer (P23XL; Becton, Dickinson & Co., Newark, DE, USA). Two needle electrodes were placed on the chest for the purposes of electrocardiography (ECG), the signals of which were amplified with an AC amplifier (P511; Grass Instruments, Natus Neurology, Inc., Warwick, RI, USA). Heart rate (HR) was calculated beat to beat with detection of the time between successive R waves in the ECG. Arterial pH was monitored with a pH meter (B-212; Horiba Corp., Kyoto, Japan) and maintained within normal limits (pH 7.4) by an i.v. infusion of a sodium bicarbonate solution (8.4%). Rectal body temperature was monitored with a digital thermometer, and adequately maintained at 37.5–38.5°C with an external heating lamp. To measure RSNA, a bipolar electrode made of a Teflon-insulated stainless steel wire (790600; A-M Systems, Inc., Sequim, MA, USA) was connected to the renal nerve directed to the left kidney. The RSNA signal was amplified with an amplifier (MEG-5200; Nihon Kohden Corp., Tokyo, Japan) with a bandpass low-frequency filter of 150 Hz and a high-frequency filter of 3 kHz, and made audible. The rats were held in a stereotaxic head unit (900LS; David Kopf Instruments, Inc., Tujunga, CA, USA). The dorsal surface of the rat brainstem was exposed by a middle incision made at the back of the head, a dissection of muscles overlaying the base of the skull, and an incision made through the atlanto-occipital membrane. For the decerebration procedures, a parietal craniotomy was performed and cortical tissue was removed by aspiration. Then, the brain was sectioned coronally with a blade at the precollicular level, and all neural tissue rostral to the section and the cortical tissues covering the cerebellum were aspirated. Anaesthesia was withdrawn. A recovery period of at least 60 min after decerebration was allowed before the identification of brain sites of the MLR and RVLM.
The site of the rat MLR was functionally identified as described previously (Bedford et al. 1992; Koba et al. 2006a,b2006b). A needle-type bipolar microelectrode (CBBPE75; FHC, Inc., Bowdoin, ME, USA) connected to an electronic stimulator (SEN-7103; Nihon Kohden Corp.) through an isolator (SS-202J; Nihon Kohden Corp.) was inserted into the midbrain at an angle vertical to the surface of the superior and inferior colliculus with the aid of an operating microscope. Initial stereotaxic coordinates for the MLR were 0.7 mm rostral and 1.9–2.1 mm lateral to the border of the inferior and superior colliculi, and 3.5–4.5 mm ventral to the dorsal surface of the colliculi. Determination of the site of the MLR was confirmed by the physiological criteria observed when the site was electrically stimulated (60 Hz frequency, 1 ms duration) as follows: (i) threshold of locomotion with reciprocal limb movement at <35 μA; (ii) stimulus-bound locomotion, and (iii) graded activity of locomotion and gait changes with increased stimulation current. It should be noted that electrical stimulation of the rat MLR at 35 μA, which initially evoked rhythmic alternating locomotor activity, gradually resulted in a wide variation in the expression of motor activity including tonic contraction, short bursts of locomotor activity which started, stopped and started again, and odd forms of locomotion such as ‘goose stepping’ as the stimulating current was applied. Having identified the site of the MLR, we subjected the rats to neuromuscular blockade with an i.v. infusion of pancuronium bromide (0.5 mg kg−1 body weight).
The bilateral sites of the rat RVLM were then functionally identified as described previously (Kishi et al. 2004; Mueller 2007). A glass micropipette was inserted into the brainstem at an angle vertical to the dorsal surface of the brainstem with the aid of the microscope. Initial stereotaxic coordinates for the RVLM were 1.0 mm rostral and 1.9–2.1 mm lateral to the calamus scriptorius, and 3.5–3.8 mm ventral to the dorsal surface of the medulla. In all studies, the RVLM was identified functionally by observing a pressor response (>10 mmHg) when glutamate (10 mm, 50.6 nl) was pressure ejected using a calibrated microinjection system (Nanoject II; Drummond Scientific, Co., Broomall, PA, USA). A period of at least 20 min was allowed to elapse after the identification of the MLR and RVLM prior to the initiation of the experimental protocols.
Experimental protocols to observe MLR stimulation-elicited responses
RSNA and cardiovascular responses to electrical stimulation of the MLR before and after bilateral microinjection into the RVLM of Tempol, a compound that mimics the enzymatic activity of SOD, were examined in 11 Sham, 12 MI and 10 MI + TR rats. The MLR of decerebrated rats under neuromuscular blockade was electrically stimulated at 35 μA for 30 s. The MLR was also electrically stimulated at 20 μA for 30 s. The order of current intensity was random and intervals of at least 5 min were allowed between manoeuvres. In the present study, the MLR was not stimulated at 50 μA, a level we had previously employed (Koba et al. 2006b), in order not to evoke huge pressor responses that may accidentally cause brain bleeding. Subsequently, Tempol diluted in saline [10 mm, 92 nl (23.0 nl × 4)] was microinjected into the RVLM bilaterally using the Nanoject II. At 30–40 min after the Tempol microinjection, the MLR was again electrically stimulated at 20 μA or 35 μA for 30 s. The amount of Tempol microinjected into the rat RVLM was based on findings in a previous rat study (Kishi et al. 2004).
In a subset of MI rats (n = 7), the effect on MLR stimulation-elicited responses of microinjection into the RVLM of Tiron, which is not chemically related to Tempol but has a similar superoxide scavenging activity, was examined. The MLR was electrically stimulated at 20 μA or 35 μA for 30 s before and 30–40 min after bilateral microinjection of Tiron [10 mm, 92 nl (23.0 nl × 4)] into the RVLM. The amount of Tiron was equivalent to that of Tempol because the same amounts of Tempol and Tiron bilaterally injected into the RVLM in conscious rabbits reportedly caused equivalent reductions in the pressor response to air-jet stress (Mayorov et al. 2004).
In cases in which stimulation of the MLR induced actual locomotion during the experiments, supplemental doses of pancuronium bromide (0.25 mg kg−1 body weight) were given i.v. At the end of data collection, the MLR was stimulated at 150 μA for 10 s in order to obtain maximal values of RSNA (Koba et al. 2006a). After all of the observations had been conducted, the renal nerve was cut between the electrode and the neural axis in order to measure the background noise of RSNA. The microinjection sites for the RVLM were marked using India ink for histological verification. At the conclusion of the experiment, the rats were humanely killed with an i.v. infusion of sodium pentobarbital (75 mg kg−1) followed by an i.v. infusion of saturated potassium chloride solution (1 ml).
In another set of experiments using normal control rats (n = 5, 361 ± 30 g body weight), we tested if electrical stimulation of the MLR at 35 μA would increase α-motoneurone discharge. In rats anaesthetized with a mixture of isoflurane (<4%) and oxygen, a laminectomy exposing the lower lumbar portion of the spinal cord (L1–L6) was performed. The meningial layers surrounding the cord were cut and reflected laterally. Two nerve bundles obtained from L4 and L5, or L5 and L6 ventral roots on the left side were carefully isolated and sectioned. The exposed neural tissue was immersed in mineral oil. The central end of the roots was placed on an insulated bipolar recording electrode. The neural activity signal was amplified with the amplifier with a bandpass low-frequency filter of 150 Hz and a high-frequency filter of 3 kHz, and made audible. In decerebrate rats after the withdrawal of anaesthesia, the site of the MLR was identified with the procedure described above. In decerebrated rats under neuromuscular blockade, electrical stimulation of the MLR at 35 μA, greater than the locomotion threshold, continuously increased the ventral root discharge as long as the stimulating current was applied; Fig. 1 indicates the typical response. These observations are consistent with the findings of previous studies (Eldridge et al. 1985; Degtyarenko & Kaufman, 2000; Koba et al. 2006b).
Figure 1. Neural discharge from the cut ends of the left lumbar ventral roots and arterial pressure changes in a healthy rat.
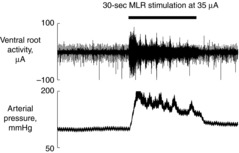
Continuous increases in neural discharge recorded from the cut ends of the left lumbar ventral roots (L5 and L6) and arterial pressure changes during 30 s of electrical stimulation of the mesencephalic locomotor region (MLR) at 35 μA in a decerebrate healthy rat under neuromuscular blockade in which the locomotion threshold was 12 μA. In the other four rats tested, continuous increases in motoneurone discharge during MLR stimulation were also observed.
In situ superoxide production in the rat medulla
In other subsets of Sham (n = 7), MI (n = 7) and MI + TR (n = 7) rats, in which neither Tempol nor Tiron were microinjected into the RVLM, intracellular superoxide generation in the medulla was evaluated with dihydroethidium (DHE) staining (Zimmerman et al. 2004; Nishi et al. 2013). The brains including the medulla of rats anaesthetized with isoflurane (<4% in oxygen) were quickly removed, embedded in optimal cutting temperature compounds, and cryostat-sectioned (30 μm, coronal). To exclude the possible effects of a bout of exercise acutely increasing superoxide generation in the brain, the brains of MI + TR rats were harvested 2 days after the cessation of the exercise training programme. However, acute exercise (treadmill running to exhaustion) in rats reportedly had little effect on brain oxidative stress biomarkers (Liu et al. 2000a). Brain sections containing the bilateral RVLM obtained from a set of one Sham, one MI and one MI + TR rat were prepared on the same day, and two sections from each rat brain were used in the following process. Sections were incubated with DHE (1 μm) in the dark for 30 min at 37°C. Images of red-fluorescent ethidium, which result from the oxidation of DHE, were then obtained using an epifluorescence microscope system with a camera (DMRB; Leica Microsystems AG, Wetzlar, Germany). The RGB confocal images were loaded into ImageJ Version 1.47 (National Institutes of Health, Bethesda, MD, USA), and converted to 8-bit grey scale. Fluorescence intensity was then measured. For each sample, an average fluorescence intensity was obtained from bilateral measurements within the RVLM of the two sections. Another in situ experiment was also performed to determine if Tempol/Tiron has antioxidant effects on brain tissues. Sections obtained in another subset of MI rats (n = 4) were pretreated in saline, Tempol (10 mm diluted in saline) or Tiron (10 mm diluted in saline) for 30 min, and incubated with DHE (1 μm) in the dark for 1 h at 37°C. Then, fluorescence intensity was measured using the procedure described above.
Statistics
In the experiments to observe MLR stimulation-elicited responses, all measured variables were displayed continuously on a computer monitor and stored on a hard disk at a sampling rate of 1 kHz through an analog–digital interface (PowerLab/8 s; AD Instruments, Dunedin, New Zealand). Baseline data were obtained from averaged values for 30 s immediately prior to electrical stimulation of the MLR. RSNA was expressed as a percentage of maximum RSNA induced by electrical stimulation of the MLR at 150 μA.
Data are expressed as means ± s.e.m. To assess significant differences, data were analysed with a paired t test or one-way or two-way repeated-measures ANOVA followed by the appropriate post hoc test (Dunnet's or Tukey's method). The level of significance was set at P < 0.05.
Results
Characterization of the model of CHF
All the Sham rats had FS of >40%, whereas all the MI and MI + TR rats had FS of <35%. Averaged FS values in the MI and MI + TR groups were significantly less than that in the Sham group (Table 1). No significant differences were found in FS between the MI and MI + TR groups (Table 1). Moreover, left ventricular end-diastolic and end-systolic dimensions were significantly increased in ligated rats irrespective of exercise training (Table 1). These results indicate that ligation developed a left ventricular dilatation, and that, as previously reported (Musch et al. 1986), longterm exercise training had no therapeutic effects on deterioration of heart function in CHF.
Table 1.
Morphometric and echocardiographic characteristics of rats used for this study
| Sham | MI | MI + TR | |
|---|---|---|---|
| Rats, n | 18 | 30 | 17 |
| Body weight, g | 490 ± 10 | 503 ± 14 | 477 ± 16 |
| Heart weight/body weight, mg g−1 | 2.87 ± 0.08 | 3.40 ± 0.06* | 3.41 ± 0.14* |
| Left ventricular end-diastolic diameter, mm | 7.8 ± 0.2 | 10.5 ± 0.2* | 10.1 ± 0.4* |
| Left ventricular end-systolic diameter, mm | 4.2 ± 0.2 | 8.5 ± 0.2* | 8.0 ± 0.4* |
| Fractional shortening, % | 46.9 ± 1.1 | 20.2 ± 1.2* | 20.7 ± 1.5* |
Values are means ± SEM. Abbreviations: Sham, sham-operated rats; MI, rats with myocardial infarction; MI + TR, MI and exercise-trained rats.
P < 0.05 versus Sham detected by Tukey's post hoc test following one-way ANOVA.
MLR stimulation-elicited RSNA and cardiovascular responses
We examined the effect of exercise training on central command dysfunction in CHF by comparing RSNA and cardiovascular responses to stimulation of the MLR among Sham (n = 11), MI (n = 12) and MI + TR (n = 10) rats, in which neither Tempol nor Tiron had yet been microinjected into the RVLM. The MLR in decerebrate rats was electrically stimulated after neuromuscular blockade with an i.v. infusion of pancuronium bromide (0.5 mg kg−1 body weight). The locomotion threshold, the minimum current intensity at which MLR stimulation evoked locomotion, was <35 μA in all rats and averaged values did not differ significantly among the groups (Table 2). Neither were there any significant differences among the groups in baseline RSNA, MAP and HR (Table 2), as previously reported (Koba et al. 2006a). Electrical stimulation of the MLR at either 20 μA or 35 μA evoked significant increases in RSNA and MAP in these rats (Figs 2 and 3). This stimulation had no significant effects on HR except in MI rats in which the MLR was stimulated at 35 μA and MI + TR rats in which the MLR was stimulated at 20 μA (Fig. 3). Little effect on HR of MLR stimulation has been reported previously (Koba et al. 2006a,b). In MI rats, stimulation of the MLR at 35 μA evoked significantly larger RSNA and MAP responses, as evaluated by mean changes in RSNA and MAP from baseline during stimulation, than those in Sham and MI + TR rats (Fig. 4). There were no significant differences in responses to MLR stimulation at 35 μA between Sham and MI + TR rats. RSNA and MAP responses to stimulation of the MLR at 20 μA did not differ significantly among the rat groups (Fig. 4).
Table 2.
Baseline data prior to electrical stimulation of the mesencephalic locomotor region (MLR) before and 30–40 min after Tempol microinjection into the rostral ventrolateral medulla (RVLM) in sham-operated rats (Sham; n = 11), rats with myocardial infarction (MI; n = 12), and MI and exercise-trained rats (MI + TR; n = 10). The locomotion threshold, the minimum current at which electrical stimulation of the MLR will evoke locomotion, is also presented. Baseline data were obtained from 30 s averaged values immediately prior to MLR stimulation
| Sham | MI | MI + TR | ||||
|---|---|---|---|---|---|---|
| Before Tempol | After Tempol | Before Tempol | After Tempol | Before Tempol | After Tempol | |
| Baseline data prior to the MLR stimulation at 20 μA | ||||||
| RSNA, % of max | 30 ± 4 | 31 ± 4 | 26 ± 2 | 33 ± 5 | 28 ± 4 | 34 ± 3 |
| Signal-to-noise ratio for RSNA | 4.0 ± 0.8 | 4.1 ± 0.9 | 3.8 ± 0.8 | 4.4 ± 1.1 | 3.3 ± 0.4 | 4.1 ± 0.6 |
| MAP, mmHg | 104 ± 9 | 105 ± 7 | 107 ± 6 | 102 ± 6 | 116 ± 6 | 121 ± 5 |
| HR, beats per min | 382 ± 11 | 372 ± 13 | 380 ± 14 | 348 ± 13* | 392 ± 9 | 381 ± 10 |
| Baseline data prior to the MLR stimulation at 35 μA | ||||||
| RSNA, % of max | 30 ± 4 | 31 ± 3 | 24 ± 2 | 32 ± 5 | 27 ± 3 | 35 ± 3 |
| Signal-to-noise ratio for RSNA | 4.0 ± 0.8 | 4.2 ± 0.8 | 3.5 ± 0.6 | 4.4 ± 1.2 | 3.4 ± 0.4 | 4.1 ± 0.6 |
| MAP, mmHg | 107 ± 8 | 106 ± 7 | 108 ± 5 | 107 ± 7 | 120 ± 5 | 120 ± 5 |
| HR, beats per min | 381 ± 12 | 371 ± 14 | 381 ± 13 | 345 ± 12* | 391 ± 8 | 382 ± 10 |
| Locomotion threshold, μA | 23.7 ± 1.8 | 26.1 ± 1.5 | 22.2 ± 3.2 | |||
Values are means ± s.e.m. Abbreviations: RSNA, renal sympathetic nerve activity; MAP, mean arterial pressure; HR, heart rate.
P < 0.05 versus before Tempol in each group, detected by Tukey's post hoc test following two-way repeated-measures ANOVA.
Figure 2. Representative recordings of rectified renal sympathetic nerve activity (RSNA), mean arterial pressure (MAP) and heart rate (HR).
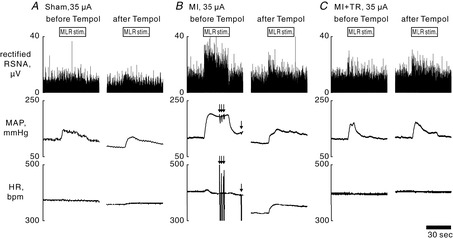
Rectified RSNA, MAP and HR during 30 s of electrical stimulation of the mesencephalic locomotor region (MLR) at 35 μA, before and 30–40 min after bilateral microinjection of Tempol (10 mm, 92 nl) into the rostral ventrolateral medulla of sham-operated [Sham; fractional shortening (FS) = 44%, locomotion threshold (LT) = 32 μA] (A), myocardial infarction (MI; FS = 16%, LT = 31 μA) (B), and MI and exercise-trained (MI + TR; FS = 16%, LT = 22 μA) (C) rats. Arrows in B indicate fluctuations of MAP/HR induced by arrhythmia during stimulation of the MLR.
Figure 3. Time course changes in renal sympathetic nerve activity (RSNA), mean arterial pressure (MAP) and heart rate (HR).
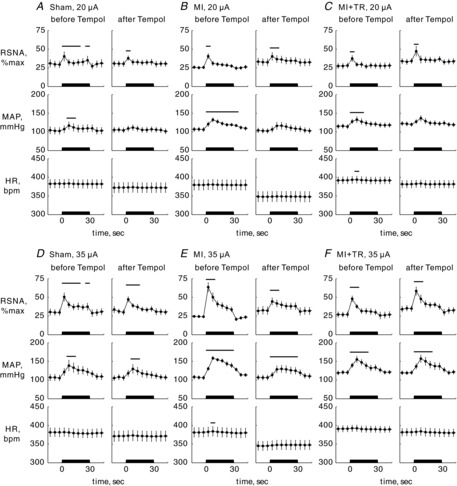
Time course changes averaged for 5 s in RSNA per maximal activity (RSNAperMax), MAP and HR during 30 s of electrical stimulation of the mesencephalic locomotor region at 20 μA (A–C) or 35 μA (D–F) before and 30–40 min after bilateral microinjection of Tempol (10 mm, 92 nl) into the rostral ventrolateral medulla in 11 sham-operated (Sham), 12 myocardial infarction (MI) and 10 MI and exercise-trained (MI + TR) rats. Values are means ± s.e.m. Abnormal values of HR caused by arrhythmia were discarded in the analyses. Thick horizontal bars located on the x-axis indicate the 30 s stimulation periods. Horizontal bars indicate significant (P < 0.05) differences from baseline values, detected by a Dunnett's post hoc test following one-way repeated-measures ANOVA.
Figure 4. Mean changes from baseline in renal sympathetic nerve activity (RSNA) and mean arterial pressure (MAP).
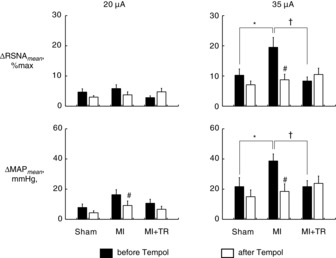
Mean changes in RSNA and MAP from baseline (ΔRSNAmean and ΔMAPmean) during the 30 s periods of stimulation at 20 μA (left) or 35 μA (right). Filled and open bars represent data before and 30–40 min after, respectively, bilateral microinjection of Tempol into the rostral ventrolateral medulla. Test animals included Sham-operated rats (Sham; n = 11), rats with myocardial infarction (MI; n = 12), and MI and exercise-trained rats (MI + TR; n = 10). Values are means ± s.e.m. *P < 0.05, Sham versus MI; †P < 0.05, MI versus MI + TR; #P < 0.05 versus before Tempol injection. Significant differences were detected by Tukey's post hoc test following two-way repeated-measures ANOVA.
We also investigated the role played by brain oxidative stress in central command dysfunction in CHF by comparing MLR stimulation-elicited RSNA and cardiovascular responses before and 30–40 min after bilateral microinjection of Tempol into the RVLM of Sham, MI and MI + TR rats. As previously observed (Kishi et al. 2004), Tempol microinjection in these rats acutely decreased RSNA, MAP and HR, and the decreases in RSNA and MAP returned to pre-microinjection levels within 10–15 min. At 30–40 min after Tempol microinjection, this chemical had no effect on RSNA or MAP in any rat group, or on HR in Sham and MI + TR rats, but reduced HR from baseline in MI rats (Table 2). In all rat groups, Tempol administration abolished increases in MAP from baseline during stimulation of the MLR at 20 μA (Fig. 3). In MI rats, Tempol administration significantly reduced MAP responses to stimulation of the MLR at 20 μA and both RSNA and MAP responses to stimulation of the MLR at 35 μA (Fig. 4). In Sham and MI + TR rats, by contrast, Tempol administration had no significant effect on RSNA and MAP responses to MLR stimulation at either 20 μA or 35 μA.
In a subset of MI rats (n = 7), we compared MLR stimulation-elicited responses before and 30–40 min after bilateral microinjection of Tiron into the RVLM. The locomotion threshold in this rat group was 24.4 ± 0.7 μA. Tiron administration had no effects on baseline RSNA, MAP or HR at 30–40 min after microinjection (Table 3). Tiron administration significantly reduced MAP responses to stimulation of the MLR at 20 μA and both RSNA and MAP responses to stimulation of the MLR at 35 μA (Fig. 5), as did Tempol administration.
Table 3.
Baseline data prior to electrical stimulation of the mesencephalic locomotor region (MLR) before and 30–40 min after Tiron microinjection into the rostral ventrolateral medulla (RVLM) of rats with myocardial infarction (MI; n = 7). A paired t test was employed to test the effects of Tiron on baseline values and found no significant differences
| MI rats | ||
|---|---|---|
| Before Tiron | After Tiron | |
| Baseline data prior to MLR stimulation at 20 μA | ||
| RSNA, % of max | 30 ± 5 | 26 ± 4 |
| Signal-to-noise ratio for RSNA | 3.6 ± 0.8 | 3.4 ± 0.7 |
| MAP, mmHg | 119 ± 5 | 117 ± 8 |
| HR, beats per min | 429 ± 15 | 399 ± 22 |
| Baseline data prior to MLR stimulation at 35 μA | ||
| RSNA, % of max | 28 ± 5 | 27 ± 5 |
| Signal-to-noise ratio for RSNA | 3.5 ± 0.7 | 3.3 ± 0.7 |
| MAP, mmHg | 114 ± 8 | 114 ± 6 |
| HR, beats per min | 426 ± 16 | 395 ± 21 |
Values are means ± s.e.m. Abbreviations: RSNA, renal sympathetic nerve activity; MAP, mean arterial pressure; HR, heart rate.
Figure 5. Effects of Tiron on renal sympathetic nerve activity (RSNA) and cardiovascular responses to electrical stimulation.
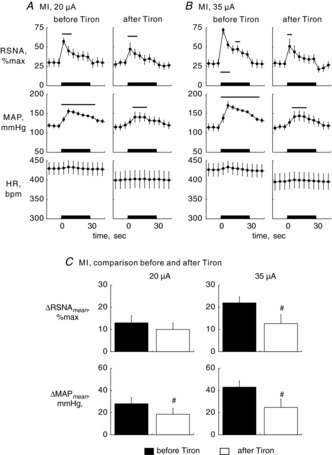
Time course changes averaged for 5 s in RSNAperMax, mean arterial pressure (MAP) and heart rate (HR) during 30 s of electrical stimulation of the mesencephalic locomotor region in rats with myocardial infarction (MI; n = 7) at 20 μA (A) or 35 μA (B) before and 30–40 min after bilateral microinjection of Tiron (10 mm, 92 nl) into the rostral ventrolateral medulla. Values are means ± s.e.m. Of note, abnormal values of HR caused by arrhythmia were discarded in this analysis. Thick horizontal bars located on the x-axis indicate the 30 s stimulation periods. Horizontal bars indicate significant (P < 0.05) differences from baseline values, detected by Dunnett's post hoc test following one-way repeated-measures ANOVA. C, ΔRSNAmean and ΔMAPmean at 20 μA (left) and 35 μA (right). Filled and open bars demonstrate the data before and after, respectively, bilateral injection of Tiron. #P < 0.05 versus before Tiron injection, paired t test.
Verification of the RVLM sites
We identified the RVLM functionally by observing pressor responses (increases of >10 mmHg) to microinjections of glutamate in all rats used in the experiments in which Tempol or Tiron would be administered. In addition, we confirmed microinjection sites histologically by examining locations at which India ink was injected. This verified that the pipette tips were located within the regions of the RVLM that have been demonstrated in previous studies (Kishi et al. 2004; Mueller et al. 2007; Nishi et al. 2013) (Fig. 6).
Figure 6. Distribution of injection sites.
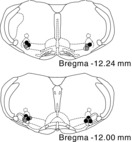
Injection sites in randomly chosen representative sham-operated rats (black circles), rats with myocardial infarction (MI; white circles), and MI and exercise-trained rats (MI + TR; grey circles) used in the Tempol experiments (n = 4 for each), mapped on standard sections from Paxinos & Watson (2007). Injection sites in other rats were located within the area of the rostral ventrolateral medulla shown.
In situ superoxide production in the medulla
In cryosections of the medulla including the RVLM, ethidium fluorescence was significantly enhanced in MI rats compared with that in Sham or MI + TR rats (Fig. 7). There were no significant differences in ethidium fluorescence between Sham and MI + TR rats (Fig. 7). Another set of DHE staining experiments indicated the antioxidant effects of Tempol/Tiron on superoxide production in rat brain tissue by showing that pretreatment with either Tempol or Tiron on cryosections of the medulla of MI rats (n = 4) significantly reduced the intensity of ethidium fluorescence in the RVLM compared with that of normal saline (Tempol: −42 ± 10%, Tiron: −55 ± 4%, versus saline, respectively).
Figure 7. In situ superoxide detection in dihydroethidium (DHE)-treated brainstem slices.
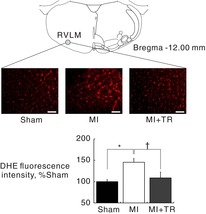
A, schematic section adapted from Paxinos & Watson (2007) showing sites of DHE intensity measurements in the rostral ventrolateral medulla (RVLM) and representative confocal images of the RVLM in sham-operated rats (Sham), rats with myocardial infarction (MI), and MI and exercise-trained rats (MI + TR). Scale bars: 100 μm. B, data are expressed as means ± s.e.m. for each group relative to Sham rats. *P < 0.05, Sham versus MI; †P < 0.05, MI versus MI + TR. Significant differences were detected by Tukey's post hoc test following one-way ANOVA.
Discussion
To test Hypotheses 1 and 2, as stated in the Introduction, RSNA and cardiovascular responses to stimulation of the MLR in healthy rats, rats with CHF, and rats with CHF that had completed longterm exercise training were examined before and after an antioxidant treatment within the RVLM. Before the antioxidant treatment, electrical stimulation of the MLR at 35 μA evoked larger RSNA and pressor responses in MI rats than in Sham or MI + TR rats. Responses in MI + TR rats were similar to those in Sham rats. Moreover, in Sham and MI + TR rats, the SOD mimetic Tempol microinjected bilaterally into the RVLM did not modulate MLR stimulation-elicited responses. In MI rats, by contrast, microinjected Tempol reduced RSNA and pressor responses to stimulation of the MLR. The effect of Tiron, another SOD mimetic, mirrored that of Tempol; when microinjected bilaterally into the RVLM of MI rats, Tiron also reduced MLR stimulation-elicited responses. Furthermore, ethidium fluorescence in the RVLM of MI rats was enhanced in comparison with that in Sham and MI + TR rats, and fluorescence intensity in MI + TR rats was similar to that in Sham rats. These results, indicating superoxide overproduction in the RVLM of CHF and the antioxidant effects of exercise training in the RVLM of CHF, support previous findings (Gao et al. 2004, 2005, 2007; Guggilam et al. 2011). Taken together, the present observations support Hypotheses 1 and 2 by demonstrating that superoxide overproduction in the RVLM of MI rats played a role in amplifying the sympathoexcitatory response to electrical stimulation of the MLR, and that antioxidant effects in the RVLM of MI + TR rats underlay the normalization of MLR stimulation-elicited responses. We suggest that oxidative stress in the medulla of CHF mediates central command dysfunction. We also suggest that exercise training in CHF is capable of normalizing central command-elicited sympathoexcitation through its antioxidant effects in the medulla.
Autonomic nervous system dysfunction in patients with CHF includes not only resting sympathetic overactivity (Leimbach et al. 1986), but also augmented sympathoexcitation during volitional exercise (Sterns et al. 1991; Negrão et al. 2001). It is suggested that central command dysfunction arising from oxidative stress in the medulla of CHF may contribute to the augmentation of sympathoexcitation during exercise. Moreover, supervised longterm exercise training in patients with CHF has been shown not only to decrease resting sympathetic overactivity (Roveda et al. 2003), but also to suppress the sympathoexcitatory response to handgrip exercise towards normal levels (Soares-Miranda et al. 2011). The present study suggests that central command function normalized by antioxidant effects in the medulla after exercise training of CHF may have a role in restoring abnormal sympathetic regulation during exercise in this disease.
Sympathoexcitation during exercise is modulated by two principal neural mechanisms: central command, and a reflex originating in exercising skeletal muscle. The muscle-based reflex, termed the ‘exercise pressor reflex’, is activated by the increase in discharge of mechanically and metabolically sensitive skeletal muscle afferents caused by contraction (Kaufman & Hayes, 2002). Central command dysfunction in CHF has been suggested by human studies in which it was found to be augmented in patients with CHF in whom muscle metaboreceptor responsiveness was impaired in comparison with muscle sympathoexcitation seen during volitional handgrip exercise in healthy subjects (Sterns et al. 1991; Negrão et al. 2001). However, the human studies could not exclude the possibility that augmented sympathoexcitation during exercise in patients with CHF was not mediated by central command, but instead reflected the activation of the muscle mechanoreceptor reflex, which is accentuated in this disease (Middlekauff et al. 2001). The concept of central command dysfunction in CHF was subsequently supported by findings in our rat study, in which we found that rats with CHF displayed larger increases in renal and lumbar SNAs in response to electrical stimulation of the MLR after neuromuscular blockade than healthy rats (Koba et al. 2006a). Such experimental preparation allowed us to exclude the effects of muscle afferent engagement and enabled us to focus on roles for central command (Eldridge et al. 1985; Bedford et al. 1992). The current study utilized this preparation to test the present Hypotheses 1 and 2.
The present study proposes a novel role played by oxidative stress in the RVLM of CHF in central command dysfunction, as well as a role played by the antioxidant effects of exercise training in the RVLM of CHF in normalizing central command dysfunction. Previous studies have shown that oxidative stress in the RVLM has a role in resting sympathetic overactivity in pathological conditions. In conscious rabbits with CHF in which the RVLM was exposed to oxidative stress, resting sympathetic overactivity was reportedly decreased by acute intracerebroventricular infusion of Tempol (Gao et al. 2004) and suppressed towards normal levels by chronic intracerebroventricular infusion of an antioxidant simvastatin (Gao et al. 2005). Roles played by oxidative stress in the RVLM in stroke-prone spontaneously, obesity-induced and renovascular hypertensive rat models in resting sympathetic overactivity have also been suggested (Kishi et al. 2004, 2004; Nishi et al. 2013). Further, molecular mechanisms by which oxidative stress is induced in the RVLM of CHF have been previously investigated. A series of studies by Gao et al. (2004, 2005, 2007) showed upregulation of NADPH oxidase subunits and downregulation of CuZnSOD and MnSOD in the RVLM of rabbits with CHF. The molecular basis that causes prooxidant effects in the RVLM of CHF may mediate not only resting sympathetic overactivity, but also central command dysfunction. Moreover, longterm exercise training in CHF rabbits has been demonstrated to lead to downregulation of NADPH oxidase subunits and upregulation of CuZnSOD and MnSOD (Gao et al. 2007). The molecular basis that causes antioxidant effects after training in the RVLM of CHF may be responsible for not only the decrease in resting SNA but also the normalization of central command dysfunction. However, the mechanisms underlying CHF-induced and training-induced protein expression changes in the brain remain unknown. Further studies are required to address this issue.
Angiotensin II (Ang II), which is increased in CHF, increases NADPH oxidase-derived superoxide production through stimulation of Ang II type 1 receptors (AT1R) in various tissues, including the brain (Bedard & Krause, 2007; Chan & Chan, 2012). Further, previous studies showed that AT1R expression was increased in the RVLM of CHF (Gao et al. 2004, 2005, 2007, 2008), and that AT1R stimulation upregulated NADPH oxidase subunits and vice versa (Bedard & Krause, 2007; Liu et al. 2008). Thus, increased renin angiotensin system (RAS) activity in the RVLM of CHF may contribute to central command dysfunction by causing superoxide overproduction. Moreover, exercise training in rabbits with CHF has been shown to suppress the increase in RAS activity in the RVLM (Mousa et al. 2008; Kar et al. 2010). This suppressive effect may be part of the normalization of central command dysfunction.
Mechanisms by which superoxide plays a role in exaggerating MLR stimulation-elicited sympathoexcitation must be elucidated. Experiments employing whole-cell configuration of the patch clamp technique applied to neuronal cells showed that NADPH oxidase-derived (Sun et al. 2005) and mitochondria-produced (Yin et al. 2010) superoxide inhibited voltage-gated potassium current. Moreover, NADPH oxidase-derived superoxide reportedly increased the intracellular concentration of Ca2+ by inducing an influx of extracellular Ca2+ through voltage-gated Ca2+ channels in cultured neuronal cells (Wang et al. 2004; Zimmerman et al. 2005). Thus, superoxide is considered to play a role in sensitizing neuronal cells responding to excitatory input by regulating membrane ion channels. In the present study, superoxide overproduction in the medulla of MI rats may have modulated the functions of those ion channels located on RVLM sympathetic premotor neurones. In turn, the RVLM neurones stimulated by central command activation may have been sensitized, and thus sympathoexcitation in response to stimulation of the MLR may have been amplified in MI rats. Moreover, antioxidant effects in the RVLM of MI + TR rats may have played a role in restoring dysfunctions of these ion channels, thereby normalizing central command dysfunction.
Although resting SNA is overactive in CHF (Leimbach et al. 1986; Gao et al. 2004, 2005) and resting sympathetic overactivity in CHF is reduced by exercise training (Liu et al. 2000b; Roveda et al. 2003), the present results showed no difference in baseline RSNA among the Sham, MI and MI + TR rats, as reported previously (Koba et al. 2006a). In the experimental preparation, the hypothalamus, in which the paraventricular nucleus contains sympathetic premotor neurones, was removed for the decerebration. This procedure may buffer resting sympathetic overactivity in MI rats. Other research employing the decerebrate rat preparation showed no difference in resting RSNA between spontaneously hypertensive rats, which would have resting sympathetic overactivity, and normotensive rats (Mizuno et al. 2011).
In the present study, microinjection of Tempol into the RVLM decreased HR from baseline in MI rats at 30–40 min after its administration. Tiron mirrored this effect, although the result did not reach statistical significance. We speculate that the prolonged decrease from baseline HR after Tempol or Tiron administration may be attributable to their effects of increasing baroreflex sensitivity because an antioxidant treatment in the RVLM of rabbits with CHF reportedly improved baroreflex function (Gao et al. 2005).
The present findings identifying a cause of central command dysfunction in CHF suggest that an antioxidant treatment in central cardiovascular pathways may hold therapeutic potential for autonomic dysfunction during exercise in patients with this disease. In this regard, Deo et al. (2012) have reported that, in patients with CHF, 1 month of oral treatment with capsules of simvastatin, which crosses the blood–brain barrier, decreased resting sympathetic overactivity directed to skeletal muscle. Whether this treatment in patients with CHF will also normalize sympathetic regulation during a bout of exercise remains to be determined.
Several limitations should be noted in the interpretation of the findings of this study. Firstly, although we evaluated superoxide generation within the RVLM in situ by DHE staining, we did not measure it in vivo. Neither do we know how much oxidative stress in vivo was indeed reduced by antioxidant treatments with Tempol or Tiron microinjection into the RVLM of the same animal. An experimental approach to determine the amount of in vivo superoxide generation in rats is demanded for quantitative evaluation of prooxidant/antioxidant effects within the medulla to modulate central command function. It is noted that transgenic mice expressing the oxidative stress indicator in which oxidative stress can be monitored in vivo are currently available (Oikawa et al. 2012).
Secondly, evidence has not been provided to support the speculation described above, that superoxide which mediates central command function might be produced, at least in part, by NADPH oxidases in Ang II signalling in the medulla. In order to examine this hypothetical mechanism, experiments which determine the effects on central command function of administration within the medulla of an AT1R blocker (e.g. losartan) and/or NADPH oxidase inhibitors such as apocynin are considered likely to provide some clues and should be conducted in the future.
Thirdly, although central command functionally interacts with baroreflex (Matsukawa, 2012), the present study did not examine sympathetic baroreflex sensitivity during stimulation of the MLR. Baroreflex sensitivity is attenuated in CHF and enhanced after exercise training in this disease (Liu et al. 2000b; Mousa et al. 2008). It is suggested that attenuated baroreflex sensitivity may contribute to the exaggeration of the MLR stimulation-elicited responses in MI rats and that enhanced baroreflex sensitivity after exercise training may be part of the normalization of the responses in MI + TR rats.
In conclusion, the present results demonstrate that superoxide overproduction in the RVLM of MI rats played a role in amplifying the sympathoexcitatory response to electrical stimulation of the MLR, and that exercise training alleviated superoxide overproduction in the medulla of MI rats, thereby suppressing MLR stimulation-elicited responses. We suggest that oxidative stress in the medulla of CHF mediates central command dysfunction. We also suggest that exercise training in CHF is capable of normalizing central command dysfunction through its antioxidant effects in the medulla.
Acknowledgments
We thank Yumiko Inoue (Tottori University Graduate School of Medical Science) for assistance in rat echocardiography, and Dr Sayaka Fujita, Hiroshi Onishi, Ryosuke Watanabe, Naoko Kano, Kumiko Kono, Naoki Sumi, Hirokazu Tsuchie, Shunsuke Yahata and Go Yoshino (Tottori University Faculty of Medicine) for assistance in rat coronary artery ligation surgery and/or rat exercise training. We also thank Dr Nobuhiro Honda and Dr Yoshitaka Hirooka (Kyushu University Graduate School of Medical Sciences) for advice on the rostral ventrolateral medulla microinjection technique.
Glossary
- Ang II
angiotensin II
- AT1R
angiotensin II type 1 receptor
- CHF
chronic heart failure
- FS
fractional shortening
- HR
heart rate
- MAP
mean arterial pressure
- MI
myocardial infarction
- MLR
mesencephalic locomotor region
- NADPH
nicotinamide adenine dinucleotide phosphate
- RAS
renin angiotensin system
- RSNA
renal sympathetic nerve activity
- RVLM
rostral ventrolateral medulla
- SNA
sympathetic nerve activity
- SOD
superoxide dismutase
Additional information
Competing interests
None declared.
Author contributions
S.K. designed the research, performed the experiments, analysed the data, prepared the figures, interpreted the results, and drafted and edited the manuscript. I.H. and T.W. interpreted the results and edited the manuscript. All authors approved the manuscript. All experiments were performed in the Division of Integrative Physiology, Tottori University Faculty of Medicine.
Funding
This study was supported by research grants from the Uehara Memorial Foundation (to S.K.), the Nakatomi Foundation (to S.K.), the Takeda Science Foundation (to S.K.) and the Mitsui Life Social Foundation (to S.K.).
References
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bedford TG, Loi PK, Crandall CC. A model of dynamic exercise: the decerebrate rat locomotor preparation. J Appl Physiol. 1992;72:121–127. doi: 10.1152/jappl.1992.72.1.121. [DOI] [PubMed] [Google Scholar]
- Chan SH, Chan JY. Brain stem oxidative stress and its associated signaling in the regulation of sympathetic vasomotor tone. J Appl Physiol. 2012;113:1921–1928. doi: 10.1152/japplphysiol.00610.2012. [DOI] [PubMed] [Google Scholar]
- Degtyarenko AM, Kaufman MP. Fictive locomotion and scratching inhibit dorsal horn neurons receiving thin fiber afferent input. Am J Physiol Regul Integr Comp Physiol. 2000;279:R394–R403. doi: 10.1152/ajpregu.2000.279.2.R394. [DOI] [PubMed] [Google Scholar]
- Deo SH, Fisher JP, Vianna LC, Kim A, Chockalingam A, Zimmerman MC, Zucker IH, Fadel PJ. Statin therapy lowers muscle sympathetic nerve activity and oxidative stress in patients with heart failure. Am J Physiol Heart Circ Physiol. 2012;303:H377–H385. doi: 10.1152/ajpheart.00289.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing J, Balady GJ. The role of exercise training in heart failure. J Am Col Cardiol. 2011;58:561–569. doi: 10.1016/j.jacc.2011.04.020. [DOI] [PubMed] [Google Scholar]
- Eldridge FL, Millhorn DE, Kiley JP, Waldrop TG. Stimulation by central command of locomotion, respiration and circulation during exercise. Respir Physiol. 1985;59:313–337. doi: 10.1016/0034-5687(85)90136-7. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res. 2004;95:937–944. doi: 10.1161/01.RES.0000146676.04359.64. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Simvastatin therapy normalizes sympathetic neural control in experimental heart failure: roles of angiotensin II type 1 receptors and NAD(P)H oxidase. Circulation. 2005;112:1763–1770. doi: 10.1161/CIRCULATIONAHA.105.552174. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang W, Liu D, Zucker IH. Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation. 2007;115:3095–3102. doi: 10.1161/CIRCULATIONAHA.106.677989. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang WZ, Wang W, Zucker IH. Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension. 2008;52:708–714. doi: 10.1161/HYPERTENSIONAHA.108.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin GM, McCloskey DI, Mitchell JH. Cardiovascular and respiratory responses to changes in central command during isometric exercise at constant muscle tension. J Physiol. 1972;226:173–190. doi: 10.1113/jphysiol.1972.sp009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggilam A, Cardinale JP, Mariappan N, Sriramula S, Haque M, Francis J. Central TNF inhibition results in attenuated neurohumoral excitation in heart failure: a role for superoxide and nitric oxide. Basic Res Cardiol. 2011;106:273–286. doi: 10.1007/s00395-010-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol. 2010;108:923–932. doi: 10.1152/japplphysiol.00840.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MP, Hayes SG. The exercise pressor reflex. Clin Auton Res. 2002;12:429–439. doi: 10.1007/s10286-002-0059-1. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Ogawa K, Konno S, Sunagawa K. Calorie restriction inhibits sympathetic nerve activity via anti-oxidant effect in the rostral ventrolateral medulla of obesity-induced hypertensive rats. Clin Exp Hypertens. 2011;33:240–245. doi: 10.3109/10641963.2011.583969. [DOI] [PubMed] [Google Scholar]
- Kleiber AC, Zheng H, Schultz HD, Peuler JD, Patel KP. Exercise training normalizes enhanced glutamate-mediated sympathetic activation from the PVN in heart failure. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1863–R1872. doi: 10.1152/ajpregu.00757.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koba S, Gao Z, Sinoway LI. Oxidative stress and the muscle reflex in heart failure. J Physiol. 2009;587:5227–5237. doi: 10.1113/jphysiol.2009.177071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koba S, Gao Z, Xing J, Sinoway LI, Li J. Sympathetic responses to exercise in myocardial infarction rats: a role of central command. Am J Physiol Heart Circ Physiol. 2006a;291:H2735–H2742. doi: 10.1152/ajpheart.00522.2006. [DOI] [PubMed] [Google Scholar]
- Koba S, Yoshida T, Hayashi N. Differential sympathetic outflow and vasoconstriction responses at kidney and skeletal muscles during fictive locomotion. Am J Physiol Heart Circ Physiol. 2006b;290:H861–H868. doi: 10.1152/ajpheart.00640.2005. [DOI] [PubMed] [Google Scholar]
- Leimbach WN, Jr, Wallin BG, Victor RG, Aylward PE, Sundlöf G, Mark AL. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation. 1986;73:913–919. doi: 10.1161/01.cir.73.5.913. [DOI] [PubMed] [Google Scholar]
- Lindley TE, Doobay MF, Sharma RV, Davisson RL. Superoxide is involved in the central nervous system activation and sympathoexcitation of myocardial infarction-induced heart failure. Circ Res. 2004;94:402–409. doi: 10.1161/01.RES.0000112964.40701.93. [DOI] [PubMed] [Google Scholar]
- Liu D, Gao L, Roy SK, Cornish KG, Zucker IH. Role of oxidant stress on AT1 receptor expression in neurons of rabbits with heart failure and in cultured neurons. Circ Res. 2008;103:186–193. doi: 10.1161/CIRCRESAHA.108.179408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yeo HC, Overvik-Douki E, Hagen T, Doniger SJ, Chyu DW, Brooks GA, Ames BN. Chronically and acutely exercised rats: biomarkers of oxidative stress and endogenous antioxidants. J Appl Physiol. 2000a;89:21–28. doi: 10.1152/jappl.2000.89.1.21. [DOI] [PubMed] [Google Scholar]
- Liu JL, Irvine S, Reid IA, Patel KP, Zucker IH. Chronic exercise reduces sympathetic nerve activity in rabbits with pacing-induced heart failure: a role for angiotensin II. Circulation. 2000b;102:1854–1862. doi: 10.1161/01.cir.102.15.1854. [DOI] [PubMed] [Google Scholar]
- Matsukawa K. Central command: control of cardiac sympathetic and vagal efferent nerve activity and the arterial baroreflex during spontaneous motor behaviour in animals. Exp Physiol. 2012;97:20–28. doi: 10.1113/expphysiol.2011.057661. [DOI] [PubMed] [Google Scholar]
- Mayorov DN, Head GA, De Matteo R. Tempol attenuates excitatory actions of angiotensin II in the rostral ventrolateral medulla during emotional stress. Hypertension. 2004;44:101–106. doi: 10.1161/01.HYP.0000131290.12255.04. [DOI] [PubMed] [Google Scholar]
- Middlekauff HR. Making the case for skeletal myopathy as the major limitation of exercise capacity in heart failure. Curr Heart Fail. 2010;3:537–546. doi: 10.1161/CIRCHEARTFAILURE.109.903773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlekauff HR, Nitzsche EU, Hoh CK, Hamilton MA, Fonarow GC, Hage A, Moriguchi JD. Exaggerated muscle mechanoreflex control of reflex renal vasoconstriction in heart failure. J Appl Physiol. 2001;90:1714–1719. doi: 10.1152/jappl.2001.90.5.1714. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Murphy MN, Mitchell JH, Smith SA. Antagonism of the TRPv1 receptor partially corrects muscle metaboreflex overactivity in spontaneously hypertensive rats. J Physiol. 2011;589:6191–6204. doi: 10.1113/jphysiol.2011.214429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousa TM, Liu D, Cornish KG, Zucker IH. Exercise training enhances baroreflex sensitivity by an angiotensin II-dependent mechanism in chronic heart failure. J Appl Physiol. 2008;104:616–624. doi: 10.1152/japplphysiol.00601.2007. [DOI] [PubMed] [Google Scholar]
- Mueller PJ. Exercise training attenuates increases in lumbar sympathetic nerve activity produced by stimulation of the rostral ventrolateral medulla. J Appl Physiol. 2007;102:803–813. doi: 10.1152/japplphysiol.00498.2006. [DOI] [PubMed] [Google Scholar]
- Musch TI, Moore RL, Leathers DJ, Bruno A, Zelis R. Endurance training in rats with chronic heart failure induced by myocardial infarction. Circulation. 1986;74:431–441. doi: 10.1161/01.cir.74.2.431. [DOI] [PubMed] [Google Scholar]
- Negrão CE, Rondon MU, Tinucci T, Alves MJ, Roveda F, Braga AM, Reis SF, Nastari L, Barretto AC, Krieger EM, Middlekauff HR. Abnormal neurovascular control during exercise is linked to heart failure severity. Am J Physiol Heart Circ Physiol. 2001;280:H1286–H1292. doi: 10.1152/ajpheart.2001.280.3.H1286. [DOI] [PubMed] [Google Scholar]
- Nishi EE, Bergamaschi CT, Oliveira-Sales EB, Simon KA, Campos RR. Losartan reduces oxidative stress within the rostral ventrolateral medulla of rats with renovascular hypertension. Am J Hypertens. 2013;26:858–865. doi: 10.1093/ajh/hpt037. [DOI] [PubMed] [Google Scholar]
- Nolan PC, Waldrop TG. Integrative role of medullary neurons of the cat during exercise. Exp Physiol. 1997;82:547–558. doi: 10.1113/expphysiol.1997.sp004046. [DOI] [PubMed] [Google Scholar]
- Notarius CF, Ando S, Rongen GA, Floras JS. Resting muscle sympathetic nerve activity and peak oxygen uptake in heart failure and normal subjects. Eur Heart J. 1999;20:880–887. doi: 10.1053/euhj.1998.1447. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Akai R, Tokuda M, Iwawaki T. A transgenic mouse model for monitoring oxidative stress. Sci Rep. 2012;2:229. doi: 10.1038/srep00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padley JR, Kumar NN, Li Q, Nguyen TB, Pilowsky PM, Goodchild AK. Central command regulation of circulatory function mediated by descending pontine cholinergic inputs to sympathoexcitatory rostral ventrolateral medulla neurons. Circ Res. 2007;100:284–291. doi: 10.1161/01.RES.0000257370.63694.73. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Burlington, MA: Elsevier; 2007. [Google Scholar]
- Piepoli MF, Conraads V, Corrà U, Dickstein K, Francis DP, Jaarsma T, McMurray J, Pieske B, Piotrowicz E, Schmid JP, Anker SD, Solal AC, Filippatos GS, Hoes AW, Gielen S, Giannuzzi P, Ponikowski PP. Exercise training in heart failure: from theory to practice. A consensus document of the Heart Failure Association and the European Association for Cardiovascular Prevention and Rehabilitation. Eur J Heart Fail. 2011;13:347–357. doi: 10.1093/eurjhf/hfr017. [DOI] [PubMed] [Google Scholar]
- Roveda F, Middlekauff HR, Rondon MU, Reis SF, Souza M, Nastari L, Barretto AC, Krieger EM, Negrão CE. The effects of exercise training on sympathetic neural activation in advanced heart failure: a randomized controlled trial. J Am Coll Cardiol. 2003;42:854–860. doi: 10.1016/s0735-1097(03)00831-3. [DOI] [PubMed] [Google Scholar]
- Soares-Miranda L, Franco FG, Roveda F, Martinez DG, Rondon MU, Mota J, Brum PC, Antunes-Correa LM, Nobre TS, Barretto AC, Middlekauff HR, Negrão CE. Effects of exercise training on neurovascular responses during handgrip exercise in heart failure patients. Int J Cardiol. 2011;146:122–125. doi: 10.1016/j.ijcard.2010.09.091. [DOI] [PubMed] [Google Scholar]
- Sterns DA, Ettinger SM, Gray KS, Whisler SK, Mosher TJ, Smith MB, Sinoway LI. Skeletal muscle metaboreceptor exercise responses are attenuated in heart failure. Circulation. 1991;84:2034–2039. doi: 10.1161/01.cir.84.5.2034. [DOI] [PubMed] [Google Scholar]
- Sun C, Sellers KW, Sumners C, Raizada MK. NAD(P)H oxidase inhibition attenuates neuronal chronotropic actions of angiotensin II. Circ Res. 2005;96:659–666. doi: 10.1161/01.RES.0000161257.02571.4b. [DOI] [PubMed] [Google Scholar]
- Wang G, Anrather J, Huang J, Speth RC, Pickel VM, Iadecola C. NADPH oxidase contributes to angiotensin II signaling in the nucleus tractus solitarius. J Neurosci. 2004;24:5516–5524. doi: 10.1523/JNEUROSCI.1176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JX, Yang RF, Li S, Renshaw AO, Li YL, Schultz HD, Zimmerman MC. Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current. Am J Physiol Cell Physiol. 2010;298:C857–C865. doi: 10.1152/ajpcell.00313.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Sharma RV, Davisson RL. Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension. 2005;45:717–723. doi: 10.1161/01.HYP.0000153463.22621.5e. [DOI] [PubMed] [Google Scholar]