

Review of how cells can sense their own infection, and their attempts to eliminate pathogens.
Keywords: PAMP, DAMP, pathogen sensing
Abstract
Historically, once a cell became infected, it was considered to be beyond all help. By this stage, the invading pathogen had breached the innate defenses and was beyond the reach of the humoral arm of the adaptive immune response. The pathogen could still be removed by cell-mediated immunity (e.g., by NK cells or cytotoxic T lymphocytes), but these mechanisms necessitated the destruction of the infected cell. However, in recent years, it has become increasingly clear that many cells possess sensor and effector mechanisms for dealing with intracellular pathogens. Most of these mechanisms are not restricted to professional immune cells nor do they all necessitate the destruction of the host. In this review, we examine the strategies that cells use to detect and destroy pathogens once the cell membrane has been penetrated.
Introduction
For many years, immune recognition was attributed solely to professional cells, circulating guardians that detect pathogens through extracellular receptors, such as the BCRs and TCRs. This assumption formed the basis of the clonal-selection theory [1], in which lymphocytes had randomly produced antigen receptors that were selected on the basis of their ability to lead to enough activation to allow clonal expansion should the cognate antigen appear but not overactivation, which would lead to the deletion of the cell in negative selection. Whereas this theory was able to explain issues of immune memory and tolerance, it was unable to describe fully the requirement for two signals in immune activation [2]. It was at this stage in our understanding two decades ago that Charles A. Janeway, Jr., described a new theory [3], in which there were two arms of the immune recognition: an adaptive arm that comprised the clonally selected lymphocytes and an innate arm that used germline-encoded receptors to sense conserved molecular patterns. These defensive strategies cooperate to ensure that a rapid and correct response is formed to any intruders. Much work has followed since then [4, 5], with the identification of many of these receptors, which suggested that it is not only the leukocytes that can detect pathogens, but also, cells that are not part of professional immunity have a limited capability to sense infection. To follow Janeway's description of the development of immunology, the latest evolution in the pattern recognition theory is that extracellular identification is not the only method of sensing, but there is also intracellular detection of pathogens, forming the basis of the first half of this review. However, sensing of infection is useless unless the organism attempts to rectify the problem, and we focus on these effector mechanisms in the remainder of this review article. If professional immune cells were not the only cell types that could detect pathogens, then it might be expected that these professional immune cells were also not the only method to remove the pathogen. In addition to the recruitment of cytotoxic T cells, NK cells, neutrophils, macrophages, and other leukocytes, which are well-reviewed elsewhere [6–8], recent evidence suggests that to some level, infected cells themselves are able to eliminate intracellular pathogens in a new paradigm, called cell-autonomous immunity [9]. Since the discovery of IFN as an inhibitory factor produced by cells that lead to the establishment of an antiviral state over one-half of a century ago [10], it has been shown that individual cells have the ability to restrict pathogens by themselves. However, only recently has this “black box” of inhibitory activity been explained with some possible mechanisms by which cells react to intracellular microbes.
This review does not attempt to be an exhaustive guide on all of the intracellular factors that a pathogen may encounter but provides an overview of concepts and classes of sensors and effector mechanisms, with an emphasis on antiviral immunity.
INTRACELLULAR PATHOGEN DETECTION
The pattern recognition theory required the identification of germline-encoded receptors (PRRs), which can detect pathogens that have entered the cells, along with the substances that are naturally identified by the cell for immune activation without the education of a selection process. The substances to be detected must be highly conserved and can be derived from self-molecules that are normally hidden from the PRRs by compartmentalization, becoming exposed during infection or cellular damage (DAMPs), or from the pathogens themselves (PAMPs) and could take the form of nucleic acids, pathogen surface constituents (e.g., LPS), or other small molecules [11]. These PRRs would have to differentiate among pathogen, danger, and host, not just by the presence of the molecule itself but by its location and/or modifications. It was one decade after publication of Janeway's initial theory that the evidence for some of these receptors surfaced. An overview of these receptors is shown in Fig. 1.
Figure 1. Localization and classification of intracellular sensors.
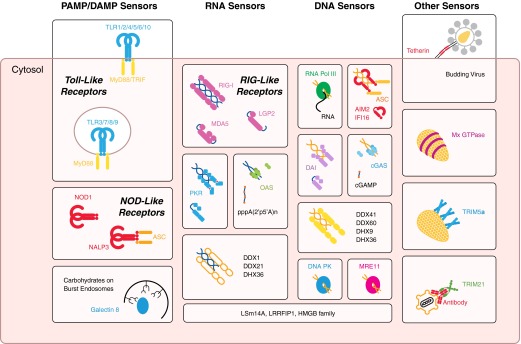
Intracellular pathogen sensors can be classified according to their mechanism of recognition, be it via PAMPs and DAMPs, RNA, DNA, or other features of a pathogen-replication cycle. Most of these sensors are soluble, cytoplasmic proteins. TLR, NLR, and galectin-8 sensing of burst endosomes detect danger associated with infection. RNA is detected by RLRs, PKR, OAS, DDX1 and -21, and DHX36. DNA is sensed by transcription by RNA Pol III to RNA products, ALRs, DAI, cGAS, DNA PK, MRE11, DDX41 and -60, and DHX9 and -36. Other proteins—LSm14A, LRRFIP1, and the HMGB family—have been found to be important in the detection of both types of nucleic acids. Other detection methods for pathogens, based on their replication cycle, are present. Mx GTPases and TRIM5α sense the virus capsid coat, with TRIM21 detecting antibody carried into the cytosol on incoming pathogens. Tetherin is able to sense budding events from the plasma membrane. NALP3, NLR family, PYD-containing 3.
TLRs
Famously, the TLRs were the first of the PRRs to be discovered [12, 13], with over 10 described, and are well-reviewed elsewhere [7, 14]. TLRs are largely restricted to professional immune cells [15], such as plasmacytoid DCs, although some TLRs, e.g., TLR4, are expressed on nonprofessional cells. TLR1, -2, -4, and -6 recognize lipids (such as TLR4-sensing LPS), TLR5 recognizes flagellin, whereas TLR3 senses dsRNA; TLR7 and -8 detect ssRNA, and TLR9 recognizes dsDNA [16]. In addition, humans have TLR10 [17], although the ligand for this is yet to be determined, with mice also possessing TLR11 [18], TLR12 [19] against Toxoplasma gondii profilin, and TLR13 recognizes viral [20] and bacterial ribosomal RNA [21]. As a result, TLR1, -2, -3, -5, and -6 predominantly sense bacteria, whereas TLR3, -7, -8, -9, and -13 can sense bacterial, viral, or protozoan nucleic material. However, TLRs are found on the plasma membrane or in endosomes [14], suggesting that they may be more important in the detection of pathogens that have not yet entered into the cell cytosol, either extracellular or on an incoming path of infection [22].
TLRs consist of a TM protein that contains extracytosolic, LRR domains to bind their respective ligands; TLR3, -7, -8, and -9 bind to nucleic acids, with the remainder binding other DAMPs or PAMPs, such as the surface components of bacteria. Activation of these TLRs leads to dimerization, revealing their TIR domain for the use of adaptors MyD88 or TRIF, setting off intracellular transduction cascades, leading to the phosphorylation of transcription factors.
NLRs
The other large class of PRRs are the NLRs, and the details are beyond the scope of this review but are well-described elsewhere [23–25]. Like the TLRs, the NLRs use LRRs to form binding domains but are present intracellularly. Based on homology, >20 of these have been discovered in humans and seem to be able to detect a variety of different insults, from bacterial glycans and lipid moieties to self-molecules, such as ATP or monosodium urate, to chemical irritants, e.g., silica, asbestos. Most PAMP ligands have yet to be linked to their respective NLRs, although NOD1 and -2 have been found to be stimulated by bacterial peptidoglycans, and NLRP3 responds to RNA and pore-forming toxins [26]. Most possess a CARD domain on the N-terminus for protein–protein interactions, with others having PYDs for recruitment of ASC, which contains a CARD domain for the downstream function. Depending on this N-terminal domain, these NLRs have different functions, which can range from activation of transcription factors, activation of the inflammasome, or modulation of MHC class II presentation.
Nucleic acid receptors
From these large groups of PRRs, there are also the intracellular, cytosolic nucleic acid sensors. The RNA sensors are the best-described out of this group, although much interest has occurred recently with the discovery of several novel DNA sensors. In theory, for nondividing eukaryotic cells, with well-defined nuclei to contain DNA, detection of cytosolic DNA should be relatively simple, whereas the RNA sensors require the detection of certain motifs that expose the sequence as being of nonhost origin.
RIG-I (also known as DDX58) is a dsRNA helicase that is the most-studied in the family of RLRs [27], which also contain MDA5 and LGP2/DHX58. These possess DEAD or DEXH (where X can be any amino acid) containing helicase domains for dsRNA recognition and a RD, with RIG-I and MDA5 also having CARD domains to lead to the activation of MAVS. RIG-I and MDA5 rely on different properties of the dsRNA strands for detection; RIG-I detects the 5′ triphosphate in a manner that is consistent with that found in pan-handle dsRNA [28], as well as being activated with a much shorter dsRNA sequence than MDA5 [29]. LGP2 is believed to detect to similar sequences as RIG-I, even having a higher affinity for binding to dsRNA sequences [30], but as a result of the lack of the CARD domain, it is believed to function as a positive regulator of RNA-induced antiviral immunity [31]. Upon binding of these helicases to RNA, conformation changes are induced that lead to the displacement of the RD, such that the helicase can multimerize and bind to MAVS on the mitochondrial surface [30, 32].
PKR is the most well-studied member of the eIF2α-specific kinase subfamily [33, 34]. PKR is able to detect long stretches of dsRNA, with a preference for hairpin loops, followed by a ssRNA sequence [35]. It can also be activated by heparin or the stress-induced protein activator of the IFN-induced protein kinase [36]. These ligands are detected via a dsRNA-binding domain, allowing dimerization and autophosphorylation. PKR is then able to feed into several downstream pathways, such as phosphorylation of the transcription factors, p53, IRF, AP-1, and NF-κB [33], as well as the direct modification of factors involved in translation, such as eIF2α [34].
Other RNA sensors, such as the OAS family of receptors, are also present in the cell as 10 isoforms [37, 38], although are only weakly induced by IFN in human cells [39]. These bind to dsRNA, recognizing the sugar backbone, and lead to the synthesis of atypical 2′-5′ chains of adenosine from ATP, mainly forming 5′-triphosphorylated triadenylate or tetradenylate [38, 40, 41]. This acts as a second messenger, leading to the activation of a specific partner, RNase L, which is discussed later in this review. No other binding partner could be found in mice with RNase L knocked out [42], so it is likely that RNase L is the only pathway that is activated by the OAS family. As a second messenger is required in the method of detection, it is important that the signal is removed when no longer necessary. As genomic DNA strands are 3′-5′, usual phosphodiesterases would be predicted not to be able to function on these unique 2′-5′ linkage chains. This was found to be the case, with an IFN-inducible enzyme that rapidly degraded the 2′-5′ oligoadenylate strands [43] and much later identified as a 2′ phosphodiesterase [44], found to be responsible for signal dampening.
DNA sensors in the cell have been reviewed well elsewhere [45–47], with a few noticeable examples provided here. Unterholzner [47] also provides an interesting discussion on the probable reasons as to why so many DNA sensors have been proposed.
RNA sensors can also be used in the detection of DNA viruses—through the properties of RNA Pol III. Cytosolic DNA, if A:T-rich, leads to the recruitment of RNA Pol III, which functions by transcribing this sequence into RNA [48]. This phenomenon has been demonstrated for adenovirus [49] and for intracellular bacteria, such as Legionella [48]. The resultant RNA can then be detected by the RNA sensors described previously, in particular, RIG-I [48, 50].
DAI was the first cytosolic receptor to be discovered that sensed DNA directly [51]. Subsequent work has identified the role of DAI in initiating transcription factor activation, such as that of the IRF and NF-κB families [51–53]. The full role of DAI in the immune response is unknown, however, as mice lacking DAI have normal responses to infection [54], and depletion in human cells has no effect on IFN production [55]. DAI may have a role at a certain time of infection or possibly in a cell type-specific manner.
Paralogues IFI16 and AIM2 are both HIN200-containing proteins with additional PYDs in a family called the ALRs [56–58]. IFI16 appears to have a greater affinity to ssDNA than dsDNA [59], although it is believed that both are able to signal in response to dsDNA in the cytosol by detection of the sugar-phosphate backbone [60]. As they contain PYDs, they have features similar to those of the NLRs, signaling through the protein ASC in the inflammasome, leading to the oligomerization of STING through CARD–CARD interactions between STING and ASC. Recently, IFI16 has been implicated in positive and negative roles during HIV infection, both in the detection of the virus [61], as well as leading to the death of the quiescent, nonproductively infected CD4 T cells [62].
Much interest has been placed in the recently identified cGAS as a receptor for cytosolic DNA [63]. Like the other DNA sensors, it appears to bind dsDNA by the backbone, although cGAS interacts with DNA through two binding sites [64], leading to activation of the enzyme. The bound form is similar to the structure of the OAS family members when bound to dsRNA [65], and activation also leads to the production of the second messenger, cGAMP. Interestingly, cGAMP is arranged in a cyclical fashion, whereas GMP is attached in an atypical 2′-5′ manner, and the adenosine is in the usual 3′-5′ linkage [66]. This second messenger is then detected by STING, before activation of immune pathways. Bacteria produce a form of cGAMP that is 3′-5′-linked in both directions, which has a slightly different affinity for STING but is able to elicit similar immune pathways [67]. The cGAS–STING pathway has been shown to be important in the detection of adenovirus [68] and HIV [69], with the recruitment of specific host factors (cleavage and polyadenylation specificity factor subunit 6 and cyclophilins, Nup358 and CypA) leading to immune evasion from cGAS [70]. Currently, unlike the OAS family and the 2′-phosphodiesterase [44], no enzyme involved in the breakdown of cGAMP has been reported, although it is almost certain that it must exist. It is possible that the 2′-phosphodiesterase may also possess activity over cGAMP, as a result of the presence of the atypical 2′-5′ linkage.
In addition, recently, the DNA damage response has been linked with sensing DNA in the cytosol, with sensors MRE11 [71] and DNA PK [72] described to possess roles in this process. All of these are believed to feed into immune pathways through the activation of STING, which appears to be central to the recognition of DNA [73].
Other RNA and DNA sensors in the cytosol may be present. It was discovered that other DEAD and DEXH helicases, such as DHX9 and DHX36, are able to sense DNA in plasmacytoid DCs [74], with DDX41 found to signal through STING [75] and DDX60 leading to antiviral signaling [76].
Some of these sensors have been found to be able to detect DNA and RNA. For example, LSm14A, a protein in processing bodies that matures RNA, has been found to initiate responses to viral RNA and DNA [77], although requiring the assistance of RIG-I, MDA5, or STING. LRRFIP1 is a cytosolic protein that has been found to bind to DNA and RNA [78] and mediates the production of cytokines in a manner independent of usual immune transcription factors, such as NF-κB or IRFs, instead, functioning through the actions of β-catenin. Finally, the HMGB, which when released by dying cells acts as a DAMP by binding to TLRs and other receptors [79], have been found to perform similar functions in the cytosol, where they are normally present—able to bind RNA and DNA (although HMGB2 binds only DNA) [80]. HMGB proteins appear to assist in the detection of these nucleic acid immunogens by improving presentation to other PRRs. Whether these promiscuous sensors are receptors in their own right or chaperones to sort out particular types of nucleic acid for other receptors to detect is still unknown, but further research may demonstrate that there are more nucleic acids sensors still to be found.
The understanding of the nature of the detection may provide clues to the unchanging features of these nucleic acids, useful for improving vaccine formulation. In addition, the understanding of the different mechanisms of nucleic acid sensing demonstrates the requirement for complementary detection of these different features and evolutionary-produced functional redundancy to circumvent pathogen antagonism.
Other pathogen receptors
Following from these relatively well-characterized groups of pathogen sensors are other newly described sensors, capable of detecting pathogens, independent of their nucleic acids.
The Mx GTPase family of proteins are dynamin-like, INF-inducible sensors [81], showing activity for viruses, such as influenza, hepatitis B virus, Thogoto virus, and HIV. The mechanism is not fully understood, although Mx GTPases are believed to be able to sense nucleocapsid structures for viruses, with a direct interaction described for the Thogoto virus [82]. How this interaction takes place is not fully understood, as it is not virus-specific, even with viruses from vastly different families—ortho-Mxs, lentiviruses, bunyaviruses, picornaviruses, and hepadnaviruses all thought to be able to be sensed by this mechanism. A lattice of Mx proteins is believed to be formed around the nucleocapsid by activation, with the outcome of sensing appearing to be a direct block to infection, which will be discussed later in this review.
Another nucelocapsid sensor, specific to lentiviruses, is TRIM5α. This member of the TRIM-containing family has a RING, B-box, coiled-coils, and PRYSPRY domains. The RING and B-box are important in producing polyubiquitin chains for the targeted neutralization of the virus and the activation of transduction cascades for transcription factor activation, with the PRYSPRY domain important for detection of nucleocapsid. As with the Mx GTPases, a lattice is believed to be formed around the offending nucleocapsid before its destruction, with many interactions via the PRYSPRY domain required for binding [83]. In addition, binding appears to be necessary for the restriction of lentiviruses, but as PRYSPRY mutants exist, which bind equally well to the capsid but have no restriction function, it suggests that other factors may be required [84].
Whereas the Mx GTPases and TRIM5α interact directly with lentiviral capsids, another indirect, but potentially more versatile, method of recognizing the surface of the pathogen has been described. Recently, antibodies were demonstrated to be able to be carried into the cytosol when bound to nonenveloped viruses and bacteria. These intracellular, pathogen-bound antibodies are sensed by the cytosolic antibody receptor TRIM21 [85–90]. This is a detection of pathogen surfaces in the cytosol, via the previous attachment of antibody as an intermediate. This antibody-dependent intracellular immunity has the added advantage of using an intermediate that can adapt to pathogen evolution (especially viruses, given the rapid rate of evolution), while having a defined, conserved binding site for TRIM21 to detect [87, 91]. However, this process is highly dependent on the pathogen carrying antibody into the cytosol, so any virus or bacteria that changes its surface as it enters the cell will probably shed this layer of detection (e.g., enveloped viruses). Like its paralogue TRIM5α, TRIM21 function appears to be dependent on the ability to form polyubiquitin chains. The discovery of the function of TRIM21 as a sensor for intracellular antibody opens up further questions (antibody was always thought to only function in the extracellular spaces, with its presence in the cytosol a flag for a breach in the membrane integrity of the cell), such as: could other serum proteins also function in a similar manner?
In addition to sensors that detect the surface of the pathogen, further intracellular sensors for infection have been described. Recently, endosomal fusion was found to be detected by cells in a STING-dependent manner [92] but unlike the previous description of the function of STING, was independent of nucleic acids or viral capsid. The actual mechanism of this detection, including the sensor itself, is still currently unknown, but it suggests that the cell might be able to sense small changes to its barrier integrity before there is the spilling of the classically described DAMPs of ATP and cellular forms of glucose [11].
For many pathogens, endosomal escape is required to move from the extracellular space to the cytosol. Recently, galectin 8 was found to act as a PRR for complex glycans that are present on the outer membrane leaflet of the plasma membrane [93]. Breakdown of the endosome integrity leads to the activation of autophagy via the ubiquitin-mediated activation of the adaptor molecule NDP52, and NDP52 activation on ubiquitin-coated Salmonella typhimurium enterica enterica has been described as leading to the recruitment of TBK1, via adaptors Nap1 and Sintbad (similar to Nap1 TBK1 adaptor) [94], suggesting that galectin 8 detection of cytosolically exposed surface glycans may also lead to immune activation.
In addition, intracellular detection of LPS, independent of its classical receptor TLR4, has been described in macrophages [95]. Whereas unstimulated Tlr4−/− mice were resistant to LPS challenge, Tlr4−/−mice primed with poly(I:C) were as susceptible to LPS-mediated sepsis as wild-type mice, suggesting that there was an additional pathway for detection of this endotoxin up-regulated by poly(I:C) stimulation. As with the fusion detection, the sensor and the signaling mechanisms are currently unknown.
Evidence suggests that innate immune activation in cells depends on at least two signals [2]; it is likely that all of these sensors function together to alert of a microbial presence. Having several layers of receptors, each of which is detecting a different aspect of the pathogen, enables not only a safety feature to prevent accidental activation of the immune system but also provides a method of recognizing the type of pathogen that is present and hence, modulating the subsequent immune response accordingly.
Whereas the sensors may have a different mechanism for detection, they all fit with Janeway's original idea: a conserved pattern is detected by the cell, leading to immune activation. In the case of TLRs and RLRs, the pattern can be DAMPs or PAMPs, detected through germline-encoded, LRR domains. Some of the nucleic acid sensors detect particular motifs, such as pan-handle RNA, hairpin loops, and A:T rich regions, whereas the capsid sensors seem to detect the repeating subunits of the viral nucleocapsid. TRIM21 detects pathogens via the repeated pattern on the antibody intermediate. The detection of burst endosomes, through the recognition of the exposure of complex glycans during endosomal rupture by galectin 8, is based on the detection of host ligands used as a danger signal to possible damage by a pathogen. Together, these patterns are present in the “lifestyle” of the pathogens, properties that are difficult for them to avoid completely.
Cross-species analysis of these receptors allows an evolutionary viewpoint of how these methods of detection arrived. The production of molecular scaffolds with domains with easily modifiable regions attached to activation domains is the repeated method of generating receptors (Fig. 2). TLRs have LRRs modified for binding their respective ligands [14], which are also found on the intracellular NLRs, albeit with a different activatory domain [24]. TLRs signal via the adaptor molecule MyD88, which contains a DD motif, whereas the NLRs use the evolutionarily similar CARD domain for their activated interactions, which is also shared by the RLRs [32], as well as the adaptor molecules for the ALRs [57]. Interestingly, fish NLRs possess PRYSPRY domains, which are traditionally associated with butyrophilins or the TRIM family [96]. Together, these modular assemblies suggest that the evolutionary process of creating these diverse receptors is through duplication and fusion of genes to create a new family of receptors, followed by selection for variants that are better at binding to PAMPs or DAMPs. This process is best exemplified by TRIMCyp, a lentiviral-restriction factor found in owl monkeys [97] and macaques [98], which is believed to have evolved independently twice. This gene was created by a fusion of the capsid-binding properties of CypA (an interaction required by the virus for evasion from TRIM5α) [99] and nucleic acid detection [70], with the activatory domains of TRIM5. This has provided these monkeys with strong resistance to infection by HIV-1 [97]. In essence, the simplicity of these sensors enables the greater ability of these genes to recombine into a useful receptor by attaching a binding domain of a protein to an action domain in a modular manner, should a selection pressure arise. Changing the activatory domain on the sensor can lead to a different response, based on the location of the receptor; for example, DNA-binding TLRs lead to transcription factor activation, whereas similarly binding NLRs would also lead to activation of the inflammasome and apoptosis—an important difference, as TLRs may pick up extracellular DNA, whereas cytosolic DNA is a sure sign that a pathogenic process is present. This versatility of the innate immune system to make use of several component domains and combine them to form functional receptors has led to the variety of different sensors that are present.
Figure 2. Domain architecture of intracellular sensors.

PRRs of the TLR, NLR, RLR, ALR, and TRIM families possess similar binding and signaling domains, suggesting that activation leads to similar innate programs of responses against pathogens. Note that the death fold is conserved in CARD, DD, and PYDs, with PYDs related to the PRYSPRY domains. NACHT, Nucleotide-binding and CARD.
The discovery of sensors for endosomal lysis, endosomal fusion, a novel intracellular LPS receptor, as well as the detection of cytosolic serum proteins shows that much more work is left to be carried out on these intracellular receptors and even suggests that further sensors may still be discovered.
INDUCTION OF AN ANTIMICROBIAL STATE
In response to the detection of the pathogen, the cell is able to make it more difficult for the organism to persist. The production of “antimicrobial states”, which make it more difficult for the pathogen to take control, has mostly been studied in the context of IFN stimulation, from its initial discovery as an inhibitory factor [10] to use as an antiviral drug [100, 101]. These studies all suggested that individual cells were either capable of preventing themselves from being infected or were able to limit the spread of the infection. Current work has been involved in finding and characterizing the mechanisms by which pathogens are restricted by these cellular changes.
It was thought for a long time that the only way to target an intracellular pathogen would be by destruction of the cell by cytotoxic cells, such as NK cells [102, 103] and T cells [104], or by monocyte/macrophage phagocytosis of infected cells [105, 106]. In addition, cell death by apoptosis [107] or pyroptosis [108, 109] after caspase induction is a well-known response to infection. However, in the context of this review, we will focus on more recently discovered noncytotoxic methods of dealing with a pathogen in the cell.
Whole pathogen destruction
The cytoplasm contains both “generic” and “pathogen-specific” mechanisms for pathogen destruction. Perhaps the best-studied pathogen-specific machinery is involved in the TRIM5α restriction of retroviruses that occurs in the primates. Human TRIM5α is capable of restricting equine infectious anemia virus (a lentivirus) and N-MLV (a γ-retrovirus) but not HIV-1. However, TRIM5α from certain species of Old World monkeys, such as the African green monkey and Tantalus monkey, are capable of restricting HIV-1 but not the equivalent primate lentivirus, SIV [110]. The observation that a virus must overcome TRIM5α-mediated virus restriction to establish infection reflects the potency of this strategy for denying the invading pathogen passage across the cytoplasm.
Despite differences in viral recognition among primate species, the effector mechanism of virus restriction appears to be conserved. During cell entry, retroviruses shed their envelope, and the viral capsid becomes exposed to the cytoplasm. During the normal course of infection, a carefully orchestrated and inter-related series of events must occur before the integration of the viral DNA into the host genome: reverse transcription, capsid uncoating, and (in the case of lentiviruses) nuclear entry. Whereas the mechanism of action of TRIM5α is not known conclusively, it is known that TRIM5α interferes with these processes. Studies with proteasome inhibitors have suggested that TRIM5α has two distinct mechanisms. Firstly, physical interaction of TRIM5α with the capsid protein results in a lattice of TRIM5α forming around the viral core [111], which interferes with the normal passage of the virus across the cytoplasm into the nucleus, whereas secondly, TRIM5α-meditated ubiquitination targets the virus for destruction by the proteasome [112].
Whereas TRIM5α represents a restriction factor dedicated to protection against retroviruses, TRIM21 provides defense against a broader range of invaders. Any event, be it infection or damage, that results in the presence of antibodies in the cytoplasm will activate TRIM21. In the case of invading, antibody-coated viruses, TRIM21, like TRIM5α, provides a potent, early postentry block to infection by using its E3 ubiquitin-ligase activity to target the incoming virions for proteasomal degradation [86]. Interestingly, the antibody-coated virus represents a challenging substrate for the proteasome and requires prior disassembly by the AAA ATPase p97/VCP (an enzyme with segregase and unfoldase activity) [85]. Whether TRIM5α also requires the activity of p97/VCP is unknown.
In addition to the TRIM E3 ligase-mediated degradation of the virus, the proteasome is involved in the direct destruction of pathogens from other sensors.
The Mx GTPases form around the viral capsid [113] and interfere with the transport of viruses to their sites of replication, such as the Golgi compartment or the nucleus, possibly by preventing host-cofactor recruitment. Viral capsids have been found by microscopy to be located in a perinuclear location, suggesting that their infectious cycle has been stalled. This implies that at some stage, the cell would dispose of these potentially infectious virions. Mx GTPases have been found to colocalize in promyelocytic leukemia protein bodies with SUMO-1 and SUMO-1-activating enzyme [114], providing a mechanism for proteasomal degradation of viral components.
Aside from proteasomal destruction, whole pathogens can also be cleared from the cell by autophagy, which is a catabolic process, in which the cell encloses substances to be degraded within an isolation membrane, which later fuses with a lysosome. Macroautophagy is the main pathway to be investigated and involves the degradation of large organelles or protein complexes in the cytosol [115]. The isolation membrane is produced when the mammalian target of rapamycin complex is inhibited, leading to the activation of the ULK1 complex (ULK1, ATG13, ATG17), along with ATG12 and LC3 proteins. Autophagy has been discovered to play important roles in immunity [116]. Detection of the pathogen was thought to be a result of engagement with cell-surface receptors, such as TLRs, or ubiquitination of the pathogen components themselves [116], but recently, it was discovered that broken endosomes can be sensed by galectin-8, activating autophagy through the adaptor NDP52 [93]. This phenomenon was demonstrated for Salmonella infection and sterile damage. In addition to Salmonella, autophagy has been shown to be important in the defense against Mycobacterium tuberculosis [117] and has been demonstrated to be antagonized by picornaviruses [118]. Interestingly, influenza infection requires autophagy for replication [119]. In addition to the direct destruction of viruses, autophagy is thought to improve antigen presentation.
Genome destruction
The cytosol possesses resting nuclease activity. IFN is known to create a state that leads to the increased degradation of long nucleotide sequences in the cytosol. This leads to efficient removal of the genetic material of the pathogen that has entered the cell. In addition, aside from the destruction of the nucleic acid polymers themselves, a loss of the integrity of the genetic code can be created, such that the produced pathogen is no longer viable.
One of the best described of these methods is the RNase L pathway [37]. The strands of 2′-5′ chains of adenosine produced by OAS lead to the activation of RNase L, binding via ankyrin repeats, causing dimerization of RNase L and enabling its nuclease activity. This nuclease activity particularly favors uracil-rich regions, preferentially cleaving after UU or UA sequences [120]. Both host and viral RNA are cleaved in this process, but whereas the host is able to transcribe mRNA from chromosomal DNA, an RNA virus would be destroyed in this process [38]. The nuclease activity is able to create the stretches of RNA that are detected by other PRRs, especially those of the RLR family [121]. A combination of the two roles allows RNase L to have antiviral and antibacterial roles [122, 123].
Whereas RNase L is involved in genome destruction, the APOBEC family of proteins is capable of compromising the integrity of the pathogen's genetic code through the deamination of cytosine to uracil [124]. This activity is usually used for processes, such as somatic hypermutation, gene conversion, and class-switch recombination. However, it was discovered that APOBEC3G is able to lead to immunity against retroviruses [124, 125]. APOBEC3G and APOBEC3F are packaged into produced virions by a process that is not understood [126], later acting to mutate the genome of these viruses after infection. This hypermutation does not occur on the viral RNA but only occurs after reverse transcription and occurs in the negative sense strand of the DNA [127], registering as guanosine-to-adenine transitions in the positive sense strand. These mutations would lead to recognition via DNA damage-response proteins or create nonviable genomes. In addition, APOBEC3G was able to cause interruption with the processing of the long-terminal repeat regions, disrupting integration into the host genome [127]. Along with this defect, an interruption to reverse transcription has been found, involving the aberrant processing of the primer tRNA [128], suggesting that the APOBEC family may have other functions in addition to simply editing and hypermutating [129].
Nutritional immunity
In addition to removing the pathogen directly, another method in which to restrict the persistence of infection is to prevent replication. One mechanism by which this takes place is by limiting the number of nutrients that the pathogen requires.
Whereas the cytosol is used as a store for many nutrients in cells, most of these are not free to be used by the pathogen. For instance, glucose is phosphorylated quickly when inside of the cell to prevent its egress from the cell [130], as well as any additional diffusion through bacterial membranes. In addition, it is increasingly being demonstrated that bacteria rely on calcium gradients for signaling [131]. In the resting cytosol, the concentration of calcium is 95 nM [132]. A bacterium would have to possess proteins of incredibly high affinity to produce these gradients when the cytosolic concentration is already so low. Iron availability is also poor intracellularly, as it is in the wrong redox state (3+ as opposed to 2+) for binding to bacterial proteins [133]. Intracellular bacteria have to find methods of being able to use the ferric protein-bound iron, and indeed, professional intracellular dwellers have been identified to modulate how the host cells handle iron [134].
In fact, the redox state of the intracellular space provides incredible difficulty for nonspecialized organisms. Bacteria carry out respiration at their periplasmic membrane, requiring the production of proton or sodium ion gradients across this lipid bilayer. However, the cytosol is already more acidic (pH 7.0 vs. 7.4) [130] and has a much lower sodium concentration (12 mM vs. 145 mM) [130], making the maintenance of these gradients difficult. In Escherichia coli and Propionigenium modestum [135], membrane potentials in the order of 100 mV are required for production of ATP. Eukaryotic organelles, which are adapted to these conditions, are at their maximal production of ATP with gradients of ∼50 mV. Prokaryotes in these same gradients are at only 15% of their maximal rate of ATP synthesis.
In addition, it has long been known that cells become quiescent when treated with IFN, arrested at the G1 stage of the cell cycle, with the mechanisms well-reviewed elsewhere [136]. This stall in the cell cycle limits nutrients, such as nucleotides, for viral replication, as the synthesis of such raw materials occurs predominantly in S-phase of cell division. The best studied of these is thymidine synthesis. Deoxythymidine is required to be phosphorylated to be used by replication machinery, a process carried out by thymidine kinase 1 in the cytosol [137]. This enzyme is under very specific timing regulation, only being expressed preceding replication [138] and rapidly degraded after [139]. Prevention of cells entering cell division limits the quantity of raw materials available for viral replication. In addition, the chromosomes will still be packaged in the nucleus, hence, enabling easier discrimination between self and incoming nucleic acids.
Similar to this method of nucleotide limitation, SAMHD1 is an IFN-induced restriction factor that has been found to possess triphosphohydrolase activity [140], converting the nucleoside triphosphate back into the nucleoside, as well as possible nuclease activity [141]. Together with tight transcriptional control over the phosphorylation enzymes, these enzymatic abilities allow SAMHD1 to restrict herpes simplex virus 1 [142] and HIV [143] by limiting the available dNTP pool.
Infected cells can also inhibit invading pathogens at the level of protein production. In addition to its role in sensing dsRNA, PKR is able to lead to stalling of ribosomes, so that protein synthesis is halted [33, 34]. Phosphorylation of eIF2α causes a sequestration of the guanine nucleotide exchange factor eIF2β, preventing the exchange of GDP for GTP on eIF2α, so that the ribosome preinitiation complex cannot be reformed [34]. This prevention of the recycling of ribosomes causes a decrease in total protein synthesis, which in the context of infection, is aimed at limiting the number of viral progeny produced [144]. This translational control appears to be somewhat selective, not only sparing the production of IFN but also being required for increased induction of this cytokine [145].
Pathogen egress prevention
If the pathogen cannot be cleared by the aforementioned mechanisms, in addition to signaling, the cell has another method to limit the spread of infection. If the cell makes the pathogen unable to leave, then the pathogen will not be able to infect a neighboring cell. This act of “cell altruism” (given that the cell has already been infected and the virus replicated) has been found to be the property of an IFN-inducible plasma membrane protein, tetherin [146], which is a disulfide-linked homodimer with a TM domain at the N-terminus and a GPI anchor at the C-terminus. When a virus attempts to bud from the infected cell, tetherin ties the virus to the cell by these two domains. This method of virus restriction has been found to be active against HIV-1 [146], dengue virus [147], Ebola virus [148], and Sendai virus [149]. It appears that budding HIV-1 virions have a preference for incorporating the GPI anchor into their envelopes over the TM domain [150], suggesting that this enables tetherin to engage in other functions, such as initiating immune signaling [151]. Notably, many methods of viral antagonism to this restriction mechanism have been found, such as Vpu for HIV-1 [146] and Sendai virus hemagglutinin–neuraminidase and fusion proteins [149], which target tetherin for ubiquitination and subsequent degradation, whereas Ebola virus glycoprotein disrupts tetherin homodimer formation [148].
Paracrine signaling
With all of these cell-autonomous mechanisms for removing a pathogen, it is worth noting that cells in a multicellular organism do not function alone. Even attachment to basement membranes, neighboring cells, and collagen leads to constant signaling about the nature of the cell. One of the main activities of the transcriptional changes in the cell is to lead to the production of cytokines, which can initiate some of these inhibitory effectors in neighboring cells before infection (e.g., IFN), act as a chemotactic attractant for professional cells (e.g., CCL4), or can lead to the production of acute-phase proteins (e.g., IL-6). The functions of these secreted proteins are beyond the scope of this review but are well-described elsewhere [7, 10, 152, 153]. Notably, most of the methods that act to remove the pathogen directly also lead to some level of signaling. For example, TRIM5α [154] and TRIM21 [88, 90] signal via the production of lysine-63-linked ubiquitin chains, APOBECs and RNase L can lead to sensing of viral nucleic acids from their edited products [37, 121, 129], and tetherin leads to transcription factor activation [151].
Increasing evidence has amassed that cells are capable of dealing with infections themselves in cell-autonomous immunity. Many of these features are highly conserved, with phylogeny studies demonstrating that the basic strategies are evolutionarily ancient [155] but with hallmarks of selection to counter different pathogens, as in the Red Queen hypothesis. In fact, the general principles are clear demonstrations of convergent evolution, such as the nucleic acid destruction of RNase L and APOBECs when compared with the bacterial endonuclease-restriction enzyme-defense system against bacteriophages [156].
Knowledge of the ability of single cells to deal with infection provides us with interesting insight into how the immune system functions as a whole. When a cell is infected by a pathogen, the cell is not merely a hijacked bystander but can sense infection, alerting other cells (neighboring cells as well as professional immune cells), as well as dealing with the pathogen by limiting its spread. The possible speed of these systems when compared with classical adaptive immunity is particularly beneficial, given the rate at which pathogens can replicate and the importance for the host organism to limit this replication for survival.
CONCLUDING REMARKS
The observation that much of intracellular immunity has been described only relatively recently may, in fact, reflect what a successful defensive strategy it is. In humans, many potential pathogens (e.g., N-MLV) that may be targeted consistently by intracellular immune mechanisms do not commonly cause disease for this very reason. Those pathogens that are able to infect cells and result in disease must necessarily subvert these systems to establish infection. Further understanding of intracellular immunity will lead to increased understanding of mechanisms of pathogenesis and inform the development of antimicrobial therapeutics aimed at revealing the pathogen to the immune system.
ACKNOWLEDGMENTS
J.C.H.T. was funded by the Frank Edward Elmore Fund of the University of Cambridge, School of Clinical Medicine. D.A.J. was supported by National Health and Medical Research Council Early Career Fellowship GNT1036521. We thank the Medical Research Council (UK; U105181010).
Footnotes
- AIM2
- absent in melanoma 2
- ALR
- absent in melanoma 2-like receptors
- APOBEC
- apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like
- ASC
- apoptosis-associated Speck-like protein containing a caspase activation and recruitment domain
- ATG
- autophagy-related
- CARD
- caspase recruitment domain
- cGAMP
- cyclic 2′-5′GMP-3′-5′AMP
- cGAS
- cyclic GMP-AMP synthase
- CypA
- cyclophilin A
- DAI
- DNA-dependent activator of IFN regulatory factors
- DAMP
- danger-associated molecular pattern
- DC
- dendritic cell
- DD
- death domain
- DDX
- DEAD box
- DEAD
- Asp-Glu-Ala-Asp
- DEXH
- Asp-Glu-X-His
- DHX
- DEXH box
- DNA PK
- DNA-dependent protein kinase
- eIF2α/β
- eukaryotic initiation factor 2 α/β
- HIN200
- hematopoietic IFN-inducible nuclear antigens with 200 aa repeats
- HMGB
- high-mobility group box family
- IFI16
- IFN-inducible protein 16
- IRF
- IFN regulatory factor transcription factor
- LPG2
- Laboratory of Genetics and Physiology 2
- LRR
- leucine-rich repeat
- LRRFIP1
- leucine-rich repeat flightless-interacting protein 1
- LSm14A
- like Sm proteins homolog 14A
- MAVS
- mitochondrial antiviral-signaling protein
- MDA5
- melanoma differentiation-associated protein 5
- MRE11
- meiotic recombination 11 homolog A
- Mx GTPase
- myxovirus-resistance GTPase
- N-MLV
- N-tropic murine leukemic virus
- Nap1
- NAK-associated protein 1
- NLR
- nucleotide-binding oligomerization-like receptor
- NLRP
- nucleotide-binding oligomerization-like receptor pyrin domain
- NOD1/2
- nucleotide-binding oligomerization 1/2
- OAS
- 2′-5′ oligoadenylate synthetase
- PAMP
- pattern-associated molecular pattern
- PKR
- dsRNA-activated protein kinase
- Pol III
- polymerase III
- poly(I:C)
- polyinosinic:polycytidylic acid
- PRR
- pattern recognition receptor
- PYD
- pyrin domain
- RD
- repressor domain
- RIG-I
- retinoic acid-inducible gene I
- RING
- really interesting new gene
- RLR
- retinoic acid-inducible gene-like receptor
- SAMHD1
- sterile α motif domain and HD domain-containing protein 1
- STING
- stimulator of IFN gene
- SUMO
- small ubiquitin-like modifier
- TBK1
- TNFR-associated factor family member-associated NF-κB activator-binding kinase 1
- TIR
- Toll-IL-1R
- TM
- transmembrane
- TRIF
- Toll-IL-1R domain-containing adaptor-inducing IFN-β
- TRIM
- tripartite motif-containing
- TRIMCyp
- tripartite motif-containing cyclophilin A domain
- VCP
- valosin-containing protein, p97
REFERENCES
- 1. Rajewsky K. (1996) Clonal selection and learning in the antibody system. Nature 381, 751–758 [DOI] [PubMed] [Google Scholar]
- 2. Fontana M. F., Vance R. E. (2011) Two signal models in innate immunity. Immunol. Rev. 243, 26–39 [DOI] [PubMed] [Google Scholar]
- 3. Janeway C. A., Jr., (2013) Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989. 54: 1–13. J. Immunol. 191, 4475–487 [PubMed] [Google Scholar]
- 4. Medzhitov R. (2009) Approaching the asymptote: 20 years later. Immunity 30, 766–775 [DOI] [PubMed] [Google Scholar]
- 5. Medzhitov R. (2013) Pattern recognition theory and the launch of modern innate immunity. J. Immunol. 191, 4473–4474 [DOI] [PubMed] [Google Scholar]
- 6. Topham N. J., Hewitt E. W. (2009) Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 128, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murphy K., Travers P., Walport M., Janeway C. (2012) Janeway's Immunobiology. Garland Science, New York, NY, USA [Google Scholar]
- 8. Barry M., Bleackley R. C. (2002) Cytotoxic T lymphocytes: all roads lead to death. Nat. Rev. Immunol. 2, 401–409 [DOI] [PubMed] [Google Scholar]
- 9. Randow F., MacMicking J. D., James L. C. (2013) Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science 340, 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watanabe Y. (2004) Fifty years of interference. Nat. Immunol. 5, 1193. [DOI] [PubMed] [Google Scholar]
- 11. Bianchi M. E. (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 [DOI] [PubMed] [Google Scholar]
- 12. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 13. Medzhitov R., Preston-Hurlburt P., Janeway C. A., Jr., (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 [DOI] [PubMed] [Google Scholar]
- 14. Uematsu S., Akira S. (2006) Toll-like receptors and innate immunity. J. Mol. Med. (Berl). 84, 712–725 [DOI] [PubMed] [Google Scholar]
- 15. Janssens S., Beyaert R. (2003) Role of Toll-like receptors in pathogen recognition. Clin. Microbiol. Rev. 16, 637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mogensen T. H. (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chuang T., Ulevitch R. J. (2001) Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta 1518, 157–161 [DOI] [PubMed] [Google Scholar]
- 18. Yarovinsky F., Zhang D., Andersen J. F., Bannenberg G. L., Serhan C. N., Hayden M. S., Hieny S., Sutterwala F. S., Flavell R. A., Ghosh S., Sher A. (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629 [DOI] [PubMed] [Google Scholar]
- 19. Koblansky A. A., Jankovic D., Oh H., Hieny S., Sungnak W., Mathur R., Hayden M. S., Akira S., Sher A., Ghosh S. (2013) Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi Z., Cai Z., Sanchez A., Zhang T., Wen S., Wang J., Yang J., Fu S., Zhang D. (2011) A novel Toll-like receptor that recognizes vesicular stomatitis virus. J. Biol. Chem. 286, 4517–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oldenburg M., Kruger A., Ferstl R., Kaufmann A., Nees G., Sigmund A., Bathke B., Lauterbach H., Suter M., Dreher S., Koedel U., Akira S., Kawai T., Buer J., Wagner H., Bauer S., Hochrein H., Kirschning C. J. (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337, 1111–1115 [DOI] [PubMed] [Google Scholar]
- 22. Kane M., Case L. K., Wang C., Yurkovetskiy L., Dikiy S., Golovkina T. V. (2011) Innate immune sensing of retroviral infection via Toll-like receptor 7 occurs upon viral entry. Immunity 35, 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen G., Shaw M. H., Kim Y. G., Nunez G. (2009) NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4, 365–398 [DOI] [PubMed] [Google Scholar]
- 24. Wen H., Miao E. A., Ting J. P. (2013) Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franchi L., Warner N., Viani K., Nunez G. (2009) Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sirard J. C., Vignal C., Dessein R., Chamaillard M. (2007) Nod-like receptors: cytosolic watchdogs for immunity against pathogens. PLoS Pathog. 3, e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixit E., Kagan J. C. (2013) Intracellular pathogen detection by RIG-I-like receptors. Adv. Immunol. 117, 99–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schlee M., Roth A., Hornung V., Hagmann C. A., Wimmenauer V., Barchet W., Coch C., Janke M., Mihailovic A., Wardle G., Juranek S., Kato H., Kawai T., Poeck H., Fitzgerald K. A., Takeuchi O., Akira S., Tuschl T., Latz E., Ludwig J., Hartmann G. (2009) Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T. S., Fujita T., Akira S. (2008) Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saito T., Hirai R., Loo Y. M., Owen D., Johnson C. L., Sinha S. C., Akira S., Fujita T., Gale M., Jr. (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 104, 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Satoh T., Kato H., Kumagai Y., Yoneyama M., Sato S., Matsushita K., Tsujimura T., Fujita T., Akira S., Takeuchi O. (2010) LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 107, 1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bowzard J. B., Davis W. G., Jeisy-Scott V., Ranjan P., Gangappa S., Fujita T., Sambhara S. (2011) PAMPer and tRIGer: ligand-induced activation of RIG-I. Trends Biochem. Sci. 36, 314–319 [DOI] [PubMed] [Google Scholar]
- 33. Garcia M. A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 70, 1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Haro C., Mendez R., Santoyo J. (1996) The eIF-2α kinases and the control of protein synthesis. FASEB J. 10, 1378–1387 [DOI] [PubMed] [Google Scholar]
- 35. Zheng X., Bevilacqua P. C. (2004) Activation of the protein kinase PKR by short double-stranded RNAs with single-stranded tails. RNA 10, 1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patel C. V., Handy I., Goldsmith T., Patel R. C. (2000) PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J. Biol. Chem. 275, 37993–37998 [DOI] [PubMed] [Google Scholar]
- 37. Chakrabarti A., Jha B. K., Silverman R. H. (2011) New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 31, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silverman R. H. (2007) Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81, 12720–12729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rusch L., Zhou A., Silverman R. H. (2000) Caspase-dependent apoptosis by 2′,5′-oligoadenylate activation of RNase L is enhanced by IFN-β. J. Interferon Cytokine Res. 20, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 40. Kristiansen H., Gad H. H., Eskildsen-Larsen S., Despres P., Hartmann R. (2011) The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. 31, 41–47 [DOI] [PubMed] [Google Scholar]
- 41. Slattery E., Ghosh N., Samanta H., Lengyel P. (1979) Interferon, double-stranded RNA, and RNA degradation: activation of an endonuclease by (2′-5′)An. Proc. Natl. Acad. Sci. USA 76, 4778–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou A., Paranjape J., Brown T. L., Nie H., Naik S., Dong B., Chang A., Trapp B., Fairchild R., Colmenares C., Silverman R. H. (1997) Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 16, 6355–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt A., Chernajovsky Y., Shulman L., Federman P., Berissi H., Revel M. (1979) An interferon-induced phosphodiesterase degrading (2′-5′) oligoisoadenylate and the C-C-A terminus of tRNA. Proc. Natl. Acad. Sci. USA 76, 4788–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kubota K., Nakahara K., Ohtsuka T., Yoshida S., Kawaguchi J., Fujita Y., Ozeki Y., Hara A., Yoshimura C., Furukawa H., Haruyama H., Ichikawa K., Yamashita M., Matsuoka T., Iijima Y. (2004) Identification of 2′-phosphodiesterase, which plays a role in the 2-5A system regulated by interferon. J. Biol. Chem. 279, 37832–37841 [DOI] [PubMed] [Google Scholar]
- 45. Paludan S. R., Bowie A. G. (2013) Immune sensing of DNA. Immunity 38, 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sharma S., Fitzgerald K. A. (2011) Innate immune sensing of DNA. PLoS Pathog. 7, e1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Unterholzner L. (2013) The interferon response to intracellular DNA: why so many receptors? Immunobiology 218, 1312–1321 [DOI] [PubMed] [Google Scholar]
- 48. Chiu Y. H., Macmillan J. B., Chen Z. J. (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Minamitani T., Iwakiri D., Takada K. (2011) Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J. Virol. 85, 4035–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ablasser A., Bauernfeind F., Hartmann G., Latz E., Fitzgerald K. A., Hornung V. (2009) RIG-I-dependent sensing of poly(dA: dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takaoka A., Wang Z., Choi M. K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., Ohba Y., Taniguchi T. (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 [DOI] [PubMed] [Google Scholar]
- 52. Wang Z., Choi M. K., Ban T., Yanai H., Negishi H., Lu Y., Tamura T., Takaoka A., Nishikura K., Taniguchi T. (2008) Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 105, 5477–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Upton J. W., Kaiser W. J., Mocarski E. S. (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ishii K. J., Kawagoe T., Koyama S., Matsui K., Kumar H., Kawai T., Uematsu S., Takeuchi O., Takeshita F., Coban C., Akira S. (2008) TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 [DOI] [PubMed] [Google Scholar]
- 55. Lippmann J., Rothenburg S., Deigendesch N., Eitel J., Meixenberger K., van Laak V., Slevogt H., N′guessan P. D., Hippenstiel S., Chakraborty T., Flieger A., Suttorp N., Opitz B. (2008) IFNβ responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell. Microbiol. 10, 2579–2588 [DOI] [PubMed] [Google Scholar]
- 56. Unterholzner L., Keating S. E., Baran M., Horan K. A., Jensen S. B., Sharma S., Sirois C. M., Jin T., Latz E., Xiao T. S., Fitzgerald K. A., Paludan S. R., Bowie A. G. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., Alnemri E. S. (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schroder K., Muruve D. A., Tschopp J. (2009) Innate immunity: cytoplasmic DNA sensing by the AIM2 inflammasome. Curr. Biol. 19, R262–R265 [DOI] [PubMed] [Google Scholar]
- 59. Yan H., Dalal K., Hon B. K., Youkharibache P., Lau D., Pio F. (2008) RPA nucleic acid-binding properties of IFI16-HIN200. Biochim. Biophys. Acta 1784, 1087–1097 [DOI] [PubMed] [Google Scholar]
- 60. Jin T., Perry A., Jiang J., Smith P., Curry J. A., Unterholzner L., Jiang Z., Horvath G., Rathinam V. A., Johnstone R. W., Hornung V., Latz E., Bowie A. G., Fitzgerald K. A., Xiao T. S. (2012) Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jakobsen M. R., Bak R. O., Andersen A., Berg R. K., Jensen S. B., Jin T., Laustsen A., Hansen K., Ostergaard L., Fitzgerald K. A., Xiao T. S., Mikkelsen J. G., Mogensen T. H., Paludan S. R. (2013) From the cover: IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 110, E4571–E4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Monroe K. M., Yang Z., Johnson J. R., Geng X., Doitsh G., Krogan N. J., Greene W. C. (2014) IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun L., Wu J., Du F., Chen X., Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li X., Shu C., Yi G., Chaton C. T., Shelton C. L., Diao J., Zuo X., Kao C. C., Herr A. B., Li P. (2013) Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gao P., Ascano M., Wu Y., Barchet W., Gaffney B. L., Zillinger T., Serganov A. A., Liu Y., Jones R. A., Hartmann G., Tuschl T., Patel D. J. (2013) Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang X., Shi H., Wu J., Zhang X., Sun L., Chen C., Chen Z. J. (2013) Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell. 51, 226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Davies B. W., Bogard R. W., Young T. S., Mekalanos J. J. (2012) Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lam E., Stein S., Falck-Pedersen E. (2014) Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol. 88, 974–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gao D., Wu J., Wu Y. T., Du F., Aroh C., Yan N., Sun L., Chen Z. J. (2013) Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rasaiyaah J., Tan C. P., Fletcher A. J., Price A. J., Blondeau C., Hilditch L., Jacques D. A., Selwood D. L., James L. C., Noursadeghi M., Towers G. J. (2013) HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kondo T., Kobayashi J., Saitoh T., Maruyama K., Ishii K. J., Barber G. N., Komatsu K., Akira S., Kawai T. (2013) DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 110, 2969–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ferguson B. J., Mansur D. S., Peters N. E., Ren H., Smith G. L. (2012) DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ishikawa H., Ma Z., Barber G. N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim T., Pazhoor S., Bao M., Zhang Z., Hanabuchi S., Facchinetti V., Bover L., Plumas J., Chaperot L., Qin J., Liu Y. J. (2010) Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 107, 15181–15186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Z., Yuan B., Bao M., Lu N., Kim T., Liu Y. J. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Miyashita M., Oshiumi H., Matsumoto M., Seya T. (2011) DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 31, 3802–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li Y., Chen R., Zhou Q., Xu Z., Li C., Wang S., Mao A., Zhang X., He W., Shu H. B. (2012) LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc. Natl. Acad. Sci. USA 109, 11770–11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yang P., An H., Liu X., Wen M., Zheng Y., Rui Y., Cao X. (2010) The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nat. Immunol. 11, 487–494 [DOI] [PubMed] [Google Scholar]
- 79. Yanai H., Ban T., Taniguchi T. (2012) High-mobility group box family of proteins: ligand and sensor for innate immunity. Trends Immunol. 33, 633–640 [DOI] [PubMed] [Google Scholar]
- 80. Yanai H., Ban T., Wang Z., Choi M. K., Kawamura T., Negishi H., Nakasato M., Lu Y., Hangai S., Koshiba R., Savitsky D., Ronfani L., Akira S., Bianchi M. E., Honda K., Tamura T., Kodama T., Taniguchi T. (2009) HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103 [DOI] [PubMed] [Google Scholar]
- 81. Haller O., Stertz S., Kochs G. (2007) The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect. 9, 1636–1643 [DOI] [PubMed] [Google Scholar]
- 82. Kochs G., Haller O. (1999) Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc. Natl. Acad. Sci. USA 96, 2082–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ohkura S., Goldstone D. C., Yap M. W., Holden-Dye K., Taylor I. A., Stoye J. P. (2011) Novel escape mutants suggest an extensive TRIM5α binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog. 7, e1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yang Y., Brandariz-Nuñez A., Fricke T., Ivanov D. N., Sarnak Z., Diaz-Griffero F. (2014) Binding of the rhesus TRIM5α PRYSPRY domain to capsid is necessary but not sufficient for HIV-1 restriction. Virology 448, 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hauler F., Mallery D. L., McEwan W. A., Bidgood S. R., James L. C. (2012) AAA ATPase p97/VCP is essential for TRIM21-mediated virus neutralization. Proc. Natl. Acad. Sci. USA 109, 19733–19738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mallery D. L., McEwan W. A., Bidgood S. R., Towers G. J., Johnson C. M., James L. C. (2010) Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 107, 19985–19990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McEwan W. A., Hauler F., Williams C. R., Bidgood S. R., Mallery D. L., Crowther R. A., James L. C. (2012) Regulation of virus neutralization and the persistent fraction by TRIM21. J. Virol. 86, 8482–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McEwan W. A., Tam J. C., Watkinson R. E., Bidgood S. R., Mallery D. L., James L. C. (2013) Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 14, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vaysburd M., Watkinson R. E., Cooper H., Reed M., O'Connell K., Smith J., Cruickshanks J., James L. C. (2013) Intracellular antibody receptor TRIM21 prevents fatal viral infection. Proc. Natl. Acad. Sci. USA 110, 12397–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Watkinson R. E., Tam J. C. H., Vaysburd M. J., James L. C. (2013) Simultaneous neutralization and innate immune detection of a replicating virus by TRIM21. J. Virol. 87, 7309–7313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Keeble A. H., Khan Z., Forster A., James L. C. (2008) TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc. Natl. Acad. Sci. USA 105, 6045–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Holm C. K., Jensen S. B., Jakobsen M. R., Cheshenko N., Horan K. A., Moeller H. B., Gonzalez-Dosal R., Rasmussen S. B., Christensen M. H., Yarovinsky T. O., Rixon F. J., Herold B. C., Fitzgerald K. A., Paludan S. R. (2012) Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 13, 737–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Thurston T. L., Wandel M. P., von Muhlinen N., Foeglein A., Randow F. (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Thurston T. L., Ryzhakov G., Bloor S., von Muhlinen N., Randow F. (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221 [DOI] [PubMed] [Google Scholar]
- 95. Kayagaki N., Wong M. T., Stowe I. B., Ramani S. R., Gonzalez L. C., Akashi-Takamura S., Miyake K., Zhang J., Lee W. P., Muszynski A., Forsberg L. S., Carlson R. W., Dixit V. M. (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 [DOI] [PubMed] [Google Scholar]
- 96. Laing K. J., Purcell M. K., Winton J. R., Hansen J. D. (2008) A genomic view of the NOD-like receptor family in teleost fish: identification of a novel NLR subfamily in zebrafish. BMC Evol. Biol. 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ribeiro I. P., Menezes A. N., Moreira M. A., Bonvicino C. R., Seuanez H. N., Soares M. A. (2005) Evolution of cyclophilin A and TRIMCyp retrotransposition in New World primates. J. Virol. 79, 14998–15003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wilson S. J., Webb B. L., Ylinen L. M., Verschoor E., Heeney J. L., Towers G. J. (2008) Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc. Natl. Acad. Sci. USA 105, 3557–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Berthoux L., Sebastian S., Sokolskaja E., Luban J. (2005) Cyclophilin A is required for TRIM5{α}-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. USA 102, 14849–14853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Brok J., Gluud L. L., Gluud C. (2005) Ribavirin plus interferon versus interferon for chronic hepatitis C. Cochrane Database Syst. Rev. 2005, CD005445. [DOI] [PubMed] [Google Scholar]
- 101. Castet V., Fournier C., Soulier A., Brillet R., Coste J., Larrey D., Dhumeaux D., Maurel P., Pawlotsky J. M. (2002) α Interferon inhibits hepatitis C virus replication in primary human hepatocytes infected in vitro. J. Virol. 76, 8189–8199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Biron C. A., Nguyen K. B., Pien G. C., Cousens L. P., Salazar-Mather T. P. (1999) Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17, 189–220 [DOI] [PubMed] [Google Scholar]
- 103. Bancroft G. J. (1993) The role of natural killer cells in innate resistance to infection. Curr. Opin. Immunol. 5, 503–510 [DOI] [PubMed] [Google Scholar]
- 104. Stenger S., Modlin R. L. (1998) Cytotoxic T cell responses to intracellular pathogens. Curr. Opin. Immunol. 10, 471–477 [DOI] [PubMed] [Google Scholar]
- 105. Turrini F., Ginsburg H., Bussolino F., Pescarmona G. P., Serra M. V., Arese P. (1992) Phagocytosis of Plasmodium falciparum-infected human red blood cells by human monocytes: involvement of immune and nonimmune determinants and dependence on parasite developmental stage. Blood 80, 801–808 [PubMed] [Google Scholar]
- 106. Shiratsuchi A., Kaido M., Takizawa T., Nakanishi Y. (2000) Phosphatidylserine-mediated phagocytosis of influenza A virus-infected cells by mouse peritoneal macrophages. J. Virol. 74, 9240–9244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Barber G. N. (2001) Host defense, viruses and apoptosis. Cell Death Differ. 8, 113–126 [DOI] [PubMed] [Google Scholar]
- 108. Doitsh G., Galloway N. L., Geng X., Yang Z., Monroe K. M., Zepeda O., Hunt P. W., Hatano H., Sowinski S., Muñoz-Arias I., Greene W. C. (2014) Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Masters S. L., Gerlic M., Metcalf D., Preston S., Pellegrini M., O'Donnell J. A., McArthur K., Baldwin T. M., Chevrier S., Nowell C. J., Cengia L. H., Henley K. J., Collinge J. E., Kastner D. L., Feigenbaum L., Hilton D. J., Alexander W. S., Kile B. T., Croker B. A. (2012) NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37, 1009–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Song B., Javanbakht H., Perron M., Park D. H., Stremlau M., Sodroski J. (2005) Retrovirus restriction by TRIM5α variants from Old World and New World primates. J. Virol. 79, 3930–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Grutter M. G., Luban J. (2012) TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2, 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu X., Parast A. B., Richardson B. A., Nduati R., John-Stewart G., Mbori-Ngacha D., Rainwater S. M., Overbaugh J. (2006) Neutralization escape variants of human immunodeficiency virus type 1 are transmitted from mother to infant. J. Virol. 80, 835–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Haller O., Kochs G. (2002) Interferon-induced Mx proteins: dynamin-like GTPases with antiviral activity. Traffic 3, 710–717 [DOI] [PubMed] [Google Scholar]
- 114. Engelhardt O. G., Ullrich E., Kochs G., Haller O. (2001) Interferon-induced antiviral Mx1 GTPase is associated with components of the SUMO-1 system and promyelocytic leukemia protein nuclear bodies. Exp. Cell. Res. 271, 286–295 [DOI] [PubMed] [Google Scholar]
- 115. Mizushima N., Ohsumi Y., Yoshimori T. (2002) Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429 [DOI] [PubMed] [Google Scholar]
- 116. Deretic V., Saitoh T., Akira S. (2013) Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Deretic V., Delgado M., Vergne I., Master S., De Haro S., Ponpuak M., Singh S. (2009) Autophagy in immunity against Mycobacterium tuberculosis: a model system to dissect immunological roles of autophagy. Curr. Top. Microbiol. Immunol. 335, 169–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jackson W. T., Giddings T. H., Jr., Taylor M. P., Mulinyawe S., Rabinovitch M., Kopito R. R., Kirkegaard K. (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhou Z., Jiang X., Liu D., Fan Z., Hu X., Yan J., Wang M., Gao G. F. (2009) Autophagy is involved in influenza A virus replication. Autophagy 5, 321–328 [DOI] [PubMed] [Google Scholar]
- 120. Floyd-Smith G., Slattery E., Lengyel P. (1981) Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate-dependent endonuclease. Science 212, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 121. Malathi K., Dong B., Gale M., Jr., Silverman R. H. (2007) Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ireland D. D., Stohlman S. A., Hinton D. R., Kapil P., Silverman R. H., Atkinson R. A., Bergmann C. C. (2009) RNase L mediated protection from virus induced demyelination. PLoS Pathog. 5, e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Li X. L., Ezelle H. J., Kang T. J., Zhang L., Shirey K. A., Harro J., Hasday J. D., Mohapatra S. K., Crasta O. R., Vogel S. N., Cross A. S., Hassel B. A. (2008) An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc. Natl. Acad. Sci. USA 105, 20816–20821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Neuberger M. S., Harris R. S., Di Noia J., Petersen-Mahrt S. K. (2003) Immunity through DNA deamination. Trends Biochem. Sci. 28, 305–312 [DOI] [PubMed] [Google Scholar]
- 125. Sheehy A. M., Gaddis N. C., Choi J. D., Malim M. H. (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650 [DOI] [PubMed] [Google Scholar]
- 126. Strebel K., Khan M. A. (2008) APOBEC3G encapsidation into HIV-1 virions: which RNA is it? Retrovirology 5, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Mbisa J. L., Barr R., Thomas J. A., Vandegraaff N., Dorweiler I. J., Svarovskaia E. S., Brown W. L., Mansky L. M., Gorelick R. J., Harris R. S., Engelman A., Pathak V. K. (2007) Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 81, 7099–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Guo F., Cen S., Niu M., Saadatmand J., Kleiman L. (2006) Inhibition of tRNA3Lys-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 80, 11710–11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Holmes R. K., Malim M. H., Bishop K. N. (2007) APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 32, 118–128 [DOI] [PubMed] [Google Scholar]
- 130. Alberts B. (1998) Essential Cell Biology: An Introduction to the Molecular Biology of the Cell. New York, NY, USA, Garland Science [Google Scholar]
- 131. Dominguez D. C. (2004) Calcium signalling in bacteria. Mol. Microbiol. 54, 291–297 [DOI] [PubMed] [Google Scholar]
- 132. Usachev Y. M., Marchenko S. M., Sage S. O. (1995) Cytosolic calcium concentration in resting and stimulated endothelium of excised intact rat aorta. J. Physiol. 489, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Andrews S. C., Robinson A. K., Rodriguez-Quinones F. (2003) Bacterial iron homeostasis. FEMS Microbiol. Rev. 27, 215–237 [DOI] [PubMed] [Google Scholar]
- 134. Pan X., Tamilselvam B., Hansen E. J., Daefler S. (2010) Modulation of iron homeostasis in macrophages by bacterial intracellular pathogens. BMC Microbiol. 10, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kaim G., Dimroth P. (1999) ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J. 18, 4118–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sangfelt O., Erickson S., Grander D. (2000) Mechanisms of interferon-induced cell cycle arrest. Front. Biosci. 5, D479–D487 [DOI] [PubMed] [Google Scholar]
- 137. Munch-Petersen B., Cloos L., Jensen H. K., Tyrsted G. (1995) Human thymidine kinase 1. Regulation in normal and malignant cells. Adv. Enzyme. Regul. 35, 69–89 [DOI] [PubMed] [Google Scholar]
- 138. Bello L. J. (1974) Regulation of thymidine kinase synthesis in human cells. Exp. Cell. Res. 89, 263–274 [DOI] [PubMed] [Google Scholar]
- 139. Zhu C., Harlow L. S., Berenstein D., Munch-Petersen S., Munch-Petersen B. (2006) Effect of C-terminal of human cytosolic thymidine kinase (TK1) on in vitro stability and enzymatic properties. Nucleosides Nucleotides Nucleic Acids 25, 1185–1188 [DOI] [PubMed] [Google Scholar]
- 140. Goldstone D. C., Ennis-Adeniran V., Hedden J. J., Groom H. C., Rice G. I., Christodoulou E., Walker P. A., Kelly G., Haire L. F., Yap M. W., de Carvalho L. P., Stoye J. P., Crow Y. J., Taylor I. A., Webb M. (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 [DOI] [PubMed] [Google Scholar]
- 141. Beloglazova N., Flick R., Tchigvintsev A., Brown G., Popovic A., Nocek B., Yakunin A. F. (2013) Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 288, 8101–8110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kim E. T., White T. E., Brandariz-Nunez A., Diaz-Griffero F., Weitzman M. D. (2013) SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J. Virol. 87, 12949–12956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E. C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T., Pancino G., Priet S., Canard B., Laguette N., Benkirane M., Transy C., Landau N. R., Kim B., Margottin-Goguet F. (2012) SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Balachandran S., Roberts P. C., Brown L. E., Truong H., Pattnaik A. K., Archer D. R., Barber G. N. (2000) Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13, 129–141 [DOI] [PubMed] [Google Scholar]
- 145. McAllister C. S., Taghavi N., Samuel C. E. (2012) Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2α-mediated translational control. J. Biol. Chem. 287, 36384–36392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Neil S. J., Zang T., Bieniasz P. D. (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430 [DOI] [PubMed] [Google Scholar]
- 147. Pan X. B., Han J. C., Cong X., Wei L. (2012) BST2/tetherin inhibits dengue virus release from human hepatoma cells. PloS One 7, e51033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kuhl A., Pohlmann S. (2012) How Ebola virus counters the interferon system. Zoonoses Public Health 59 (Suppl. 2), 116–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Bampi C., Rasga L., Roux L. (2013) Antagonism to human BST-2/tetherin by Sendai virus glycoproteins. J. Gen. Virol. 94, 1211–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Venkatesh S., Bieniasz P. D. (2013) Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 9, e1003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Galao R. P., Le Tortorec A., Pickering S., Kueck T., Neil S. J. (2012) Innate sensing of HIV-1 assembly by tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 12, 633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Dinarello C. A. (2007) Historical insights into cytokines. Eur. J. Immunol. 37 (Suppl. 1), S34–S45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Miyajima A., Kitamura T., Harada N., Yokota T., Arai K. (1992) Cytokine receptors and signal transduction. Annu. Rev. Immunol. 10, 295–331 [DOI] [PubMed] [Google Scholar]
- 154. Pertel T., Hausmann S., Morger D., Zuger S., Guerra J., Lascano J., Reinhard C., Santoni F. A., Uchil P. D., Chatel L., Bisiaux A., Albert M. L., Strambio-De-Castillia C., Mothes W., Pizzato M., Grutter M. G., Luban J. (2011) TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kimbrell D. A., Beutler B. (2001) The evolution and genetics of innate immunity. Nat. Rev. Genet. 2, 256–267 [DOI] [PubMed] [Google Scholar]
- 156. Arber W., Linn S. (1969) DNA modification and restriction. Annu. Rev. Biochem. 38, 467–500 [DOI] [PubMed] [Google Scholar]