

Abstract.
Antibiotic resistance (AR) is increasingly prevalent in low and middle income countries (LMICs), but the extent of the problem is poorly understood. This lack of knowledge is a critical deficiency, leaving local health authorities essentially blind to AR outbreaks and crippling their ability to provide effective treatment guidelines. The crux of the problem is the lack of microbiology laboratory capacity available in LMICs. To address this unmet need, we demonstrate a rapid and simple test of -lactamase resistance (the most common form of AR) that uses a modified -lactam structure decorated with two fluorophores quenched due to their close proximity. When the -lactam core is cleaved by -lactamase, the fluorophores dequench, allowing assay speeds of 20 min to be obtained with a simple, streamlined protocol. Furthermore, by testing in competition with antibiotics, the -lactamase-associated antibiotic susceptibility can also be extracted. This assay can be easily implemented into standard lab work flows to provide near real-time information of -lactamase resistance, both for epidemiological purposes as well as individualized patient care.
Keywords: rapid diagnostic, β-lactam, bodipy, quenching, fluorescence, point-of-care
1. Introduction
The rising use of antibiotics worldwide has led to increasing rates of antibiotic resistance (AR), with low and middle income countries (LMICs) disproportionately affected.1 Monitoring the spread of AR is critical, as failure to treat with an effective antibiotic is correlated with a lack of response, increased mortality, and increased healthcare costs.2,3 In developed nations, broad-spectrum antibiotics are often prescribed as a first-line treatment in an attempt to prevent this scenario. However, determining the most appropriate antibiotics requires detailed knowledge of the local pathogen antibiotic susceptibilities; information that is typically unavailable in LMICs due to the inadequate microbiological laboratory capacity available for AR surveillance.4 This can be critical in remote or low-resource areas where only a limited set of antibiotics is available for use and/or empirically made incorrect antibiotic decisions may cost a patient’s life, particularly in bacteremia cases. This diagnostic deficiency is so severe that the measured effects of AR on the health and economies of LMICs are widely acknowledged to be largely underestimated and poorly understood.5 The single most common cause of AR is often stated to be due to the bacterial β-lactamase enzyme,6 and a rapid test characterizing the antibiotic susceptibility of this resistance mechanism appropriate for low-resource environments would go a long way in addressing these needs.
β-Lactamase enzymes confer resistance to the -lactam class of antibiotics (e.g., penicillins and cephalosporins; Fig. 1), which are commonly prescribed due to their minimal side effects and good physiological compatibility. These antibiotics specifically inhibit bacterial cell wall synthesis by binding with peptidoglycan transpeptidases and blocking their cross-linking activity.7 Essential to this function is the -lactam ring, the common core structure shared by all -lactam antibiotics (Fig. 1). -Lactamase hydrolyzes this -lactam ring,7 splitting the antibiotic into two pieces, and neutralizing its efficacy. First generation cephalosporins and penicillin were especially susceptible to this form of attack, with later generation cephalosporins proving more resistant. However, extended-spectrum -lactamases, which can neutralize many of the higher generation cephalosporins, have become more prevalent in recent years and often require an entirely different class of antibiotic treatment (e.g., carbapenem, Fig. 1).8–10 Alarmingly, -lactamases capable of hydrolyzing even the carbapenems (i.e., carbapenemases) are now an emerging problem.10,11 Thus, beyond simply detecting -lactamase existence, characterizing the local spectrum of -lactamase expression is extremely valuable, as it provides critical information for guiding initial treatment as well as local outbreak control, particularly in LMICs where antibiotic use is poorly regulated.
Fig. 1.

General structures of commonly used -lactam class of antibiotics: cephalosporin, penicillin, and carbapenem.
Existing methodologies for determining antibiotic susceptibility include conventional broth/agar dilution and disk-diffusion methods. All these methods involve culturing bacteria in the presence of antimicrobials, observing the resultant growth pattern, and interpreting the antibiotic susceptibility by using interpretive charts from the Clinical and Laboratory Standards Institute (CLSI) or other regulatory bodies.12–16 Broth or agar dilution involves culturing bacteria with differing concentrations of antibiotic. Following incubation, the lowest antibiotic concentration inhibiting bacterial growth is taken as the minimum inhibitory concentration (MIC) and is used to determine the resistance. Strips with antibiotic gradients may also be used to facilitate the MIC readout from a plate (E-test).17 The disk diffusion method uses an antibiotic impregnated disk which is placed on the surface of an agar plate plated with a known quantity of bacteria. The antibiotic diffuses out from the disk to create an antibiotic gradient. Bacteria grow around the disk with a clear “zone of inhibition” surrounding the disk, the size (diameter) of which is used to assess the susceptibility. Notably, variations of the disk-diffusion method can be employed to determine -lactamase activity.18 These include the three-dimensional disk tests, double-disk tests, and cloverleaf method. The latter two of these are used to demonstrate the loss of activity of -lactam antibiotics.19,20 In particular, the zone edge test, which is derived from disk diffusion and involves observing the sharpness of the edge of the zone of inhibition following disk diffusion, is one of only two currently recommended CLSI tests for -lactamase detection.15,21
Even though these tests are relatively inexpensive, they are laborious and require at least 1 working day to perform due to an overnight incubation step, substantially reducing the diagnostic throughput and laboratory capacity. While use of automated testing equipment can improve the throughput, the initial equipment investment significantly increases the cost for diagnostics.
Rapid detection of -lactamase status can be achieved by using other nongrowth based methods. The most established of these is the nitrocefin test, which is the second CLSI recommended method for -lactamase detection.15 In the presence of β-lactamase positive bacteria, nitrocefin is rapidly degraded and produces a visible color change in less than 1 h. While inexpensive and rapid, this method is mainly used to detect -lactamase and does not provide detailed information on the antibiotic susceptibility (e.g., a -lactamase producing bacterium may be resistant to penicillin but sensitive to a fourth-generation cephalosporin). Furthermore, the standard CLSI-approved nitrocefin test is used as a screening method for -lactamase only15 and does not provide enough information for an informed -lactam antibiotic choice.
More sophisticated approaches for rapid -lactamase detection include polymerase chain reaction (PCR)-based testing and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF).22 PCR tests are becoming increasingly powerful with the ability to determine a wide variety of AR genes,23 as well as other characteristics.24 MALDI-TOF has also made significant advances in recent years, allowing rapid microbial identification and -lactamase characterization.22,25 Both testing modalities require roughly a few hours to perform and will likely play prominent roles in the future. However, the cost for both of these methods is currently higher than the traditional methods mentioned above, limiting their use in LMICs.
Recently, our group has developed a unique photochemical technology designed to provide both diagnosis and treatment against -lactamase positive bacteria.26–29 A probe, termed -lactamase activated photosensitizer/fluorophore (referred to as the first generation probe throughout the manuscript) was designed by taking advantage of the static quenching phenomenon, which occurs when two fluorophores (or photosensitizers) are in close proximity (Fig. 2). Consequently, they are not readily excited due to a change in their ground state properties. In order to create this scenario, the two photosensitizers are anchored to opposite ends of a modified -lactam core so as to be quenched and inactive. However, after probe cleavage by -lactamase, the photosensitizers diffuse away from each other, at which time these photosensitizers can then be activated by light, thereby producing toxic species that destroy nearby pathogens26 (i.e., photodynamic therapy30). Also, following the cleavage, photosensitizers produce weak fluorescence that can be used for diagnostic purposes. Our group has previously published evidence that this generated fluorescence can be used to characterize the β-lactamase status of bacteria through the analysis of the probe cleavage kinetics.27,28 For diagnostic application, the fluorescence is exploited, and the probe is defined as a -lactamase enzyme-activated fluorophore (-LEAF). The first generation prototype probe published by our group uses 5-(4′-carboxybutylamino)-9-diethylaminobenzo[a]phenothiazinium chloride (EtNBS) as the fluorophore/photosensitizer. Even though rapid () diagnostics were obtained with the first generation probe using detailed enzyme kinetics studies,27 the probe was initially designed as a phototherapeutic, and therefore, was composed of photosensitizer EtNBS, not optimal for diagnostics.
Fig. 2.

Principle of -lactamase enzyme-activated fluorophore (-LEAF). Before the introduction of the enzyme (the system is in “off” state), two fluorophores are in close proximity to each other and as a result of static quenching, the system does not produce fluorescence. When the enzyme cleaves the β-lactam ring (the system is in “on” state), the two fluorophores are separated from each other and consequently the system produces fluorescence.
For LMIC implementation, a combination of cost, speed, ease of use, and antibiotic susceptibility characterization is required. Here, we report on an improved methodology based on our original -LEAF technology27 involving: (1) a new second generation -LEAF probe which has been optimized for high fluorescence to increase the diagnostic speed and (2) a streamlined protocol which uses a single competitive reaction between the -LEAF probe and a -lactam antibiotic to determine the -lactamase-related antibiotic susceptibility. To validate the clinical applicability of the methodology, the new protocol is demonstrated with -lactamase positive and negative bacteria isolated from patient samples.
2. Methods
2.1. Reagents and Instruments for Synthesis of -LEAF Probe
ACLE hydrochloride was a generous gift from Otsuka Chemical Co., Ltd., Tokyo, Japan. Bodipy-FL was purchased from Invitrogen (Grand Island, New York). Other chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, Missouri) and used without further purification. UV–visible and fluorescence spectra were recorded on a Hewlett Packard 8453 UV-Visible spectrophotometer (Palo Alto, California) and Horiba JobinYvon FluoroMax-3 spectrofluorometer (Kyoto, Japan), respectively. Mass spectrometry was recorded on MALDI-TOF using 2,5-dihydroxybenzoic acid as a matrix. Liquid chromatography mass spectrometry (LC-MS) was obtained using Agilent 6430 Triple Quad LC/MS system (Santa Clara, California). High-performance liquid chromatography (HPLC) was performed with reverse phase Alltima C18 column using a Shimadzu SCL-10AVP controller with a SPD-M10AVP photodiode array detector (Kyoto, Japan).
2.2. Reagents for -LEAF Assay, Bacterial Strains, and Culture Conditions
Penicillinase enzyme (-lactamase from Bacillus cereus) and cefazolin-sodium were purchased from Sigma-Aldrich. Staphylococcus aureus strains used in this study were purchased from ATCC (Manassas, Virginia), and clinical isolates were provided by Dr. M.J. Ferraro (Microbiology Labs, Massachusetts General Hospital, Boston, Massachusetts) (Table 1). Brain heart infusion (BHI) broth and BHI agar were obtained from BD Difco (BD: Becton, Dickinson and Company, Franklin Lakes, New Jersey). Penicillin disks (10 U) were purchased from BD BBL. All strains were routinely cultured in BHI agar or broth at 37°C. The isolates were grown in the presence of penicillin disks to induce and enhance -lactamase production as required.
Table 1.
Staphylococcus aureus strains and isolates used in this study.
| # | S. aureus isolatea | Sourceb |
|---|---|---|
| 1 | 29213 [-lactamase (+)] | ATCC |
| 2 | 25923 [-lactamase (-)] | ATCC |
| 3 | 75391-09 | Clinical isolate |
| 4 | W5337 | Clinical isolate |
| 5 | W53156 | Clinical isolate |
| 6 | AI5070237 | Clinical isolate |
The S. aureus clinical isolates (#3 to #6) were provided by Dr. Mary Jane Ferraro (Microbiology Labs, Massachusetts General Hospital, Boston, Massachusetts, USA).
ATCC=American Type Culture Collection; Clinical Isolate=bacteria isolated from a clinical infection.
2.3. -LEAF Synthesis
The first generation probe with the photosensitizer EtNBS-COOH was synthesized as previously published.26
The synthesis of the second generation probe -LEAF Bodipy-FL was as follows:
-
•
7-Amino-3-(4-aminophenylthio)methyl-3-cephem-4-carboxylic acid p-methoxybenzyl ester (Compound 1, Fig. 3). The compound was synthesized and characterized by previously published methods.26
-
•
-LEAF with p-methoxybenzyl protection group (Compound 2, Fig. 3). The mixture of carboxylic acid-modified Bodipy-FL (5 mg, ), compound 1 (1.5 mg, ), and -(7-azabenzotriazole-1-yl)-N,N,N,N′-tetramethyluroniumhexafluorophosphate (HATU, 26 mg, ) in dry N,N-dimethylformamide (DMF, 500 μl) was stirred for 30 min. Diisopropylethylamine (DIPEA, 2.5 μl, ) was added to the stirring solution. The resulting reaction mixture was protected from light and stirred overnight. The solvent was removed under vacuum, and the residue was reconstituted in dichloromethane (DCM). The organic layer was washed with brine. After removing the solvent under vacuum, the crude product was purified by HPLC using the method described below. Retention time of the product is 60.2 min (50% yield). MALDI-MS (): calculated [] for is 1028.32; found: 1028.20
-
•
-LEAF (Compound 3, Fig. 3). Compound 2 (2 mg, 0.0022 mmol) was dissolved in a solvent mixture of trifluoroacetic acid:anisole:DCM (1.5 ml, 1:1:5) and stirred at 0°C for 1 h. The solvent was removed under vacuum, and the residue was purified by HPLC to yield 3 in an 85% yield. Retention time of the product is 55.4 min. MALDI-MS () calculated [] for is 908.55; found: 908.10, LC-MS () calculated [] for is 465.60; found: 465.00; . The analytical HPLC chromatogram of the product is shown in Fig. 4. The LC-MS chromatogram of the product is shown in Fig. 5.
-
•
HPLC Condition. HPLC was performed on Alltima C18 column (reverse phase) at flow rate using a Shimadzu SCL-10AVP controller with an SPD-M10AVP photodiode array detector. The eluents were TFA in water and TFA in acetonitrile. Initially, the column was equilibrated with 75% A and 25% B solution. After equilibration, the gradient started with 75% of A and 25% of B and then changed to 55% of A and 45% of B in 4 min at which time the ratio of B increased to 85% in 35 min. The concentration of B was ramped to 100% in 5 min, and the column was held at that condition for an additional 30 min.
-
•
LC-MS Condition. LC-MS was performed on Agilent 6430 Triple Quad LC/MS system. The eluents were and . The column was equilibrated with 20% B and 80% A solution. Following the injection, the concentration of B was ramped from 20% to 100% in 5 min. The retention time of the desired product is 1.5 min.
-
•Fluorescence Quantum Yield Calculations. The quantum yields were calculated using a secondary standard method.31,32 Fluorescein, a dye with excitation/emission wavelengths similar to Bodipy-FL, was used as a reference compound. Optically dilute solutions () of sample and analyte were prepared and used throughout the experiment. In this approach, the integrated fluorescence intensity of the analyte () and standard (), the optical density of the analyte (OD) and the standard (), and the refractive index of the analyte solvent () and standard solvent () are related to the quantum yield of the analyte () as
where is a quantum yield of the reference standard (0.94 for fluorescein33).(1)
Fig. 3.
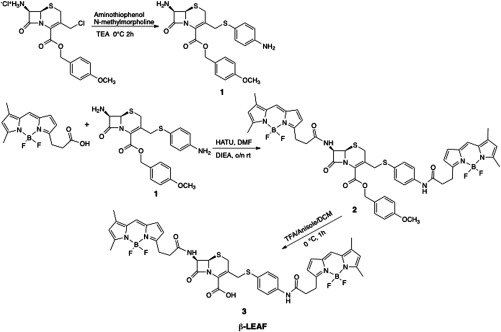
Synthesis of -LEAF probe.
Fig. 4.

Analytical high-performance liquid chromatography (HPLC) chromatogram of the -LEAF Bodipy-FL probe.
Fig. 5.

Liquid chromatography mass spectrometry (LC-MS) analysis of the -LEAF Bodipy-FL probe.
2.4. -LEAF Assays
2.4.1. -LEAF- -lactamase enzyme assays
Penicillinase enzyme was prepared in solution form by dissolving the purchased powder in 10% glycerol in phosphate buffered saline (PBS) and storing in the long term at . Required stock solutions were prepared prior to experiments in PBS. A 20 μM -LEAF probe solution ( stock) was prepared in 40% dimethyl sulfoxide (DMSO) in PBS. The assays were performed in 96-well white clear-bottom plates in a total volume of , respectively, to include probe stock solution and enzyme solution, with a resultant buffer concentration of 20% DMSO in PBS in each reaction. Time course assays were carried out, monitoring -LEAF cleavage by measuring fluorescence for 60 min, at 1-min intervals (Spectramax M5 Plate Reader, Molecular Devices, Sunnyvale, California). Instrument settings were kept as excitation at 640 nm and emission at 700 nm for the first generation probe (-LEAF EtNBS). For the second generation probe (-LEAF Bodipy-FL), settings were excitation at 450 nm and emission at 510 nm. Temperature was maintained at 37°C throughout. The -LEAF cleavage rate in each case was determined as the slope, i.e., fluorescence change as a function of time (obtained from instrument software—SoftMax Pro5).
Determination of kinetic parameters. To determine the kinetic constants with respect to first and second generation probes for penicillinase enzyme, reactions were carried out in volumes containing penicillinase and varying concentrations of the respective probes (1.25 to ). The reactions were performed for 20 min while monitoring fluorescence. The product formed was calculated as
| (2) |
where is a constant calculated as an average of values of [] divided by (final relative fluorescence units i.e., RFUs, when the probe completely cleaved at respective substrate concentration), [] is product, i.e., cleaved probe formed by the enzyme action, concentration, and [] is the starting substrate, i.e., intact probe, concentration. and values were determined by nonlinear regression analyses carried out with GraphPad Prism software.
Data from three independent experiments were plotted, and the results are presented in tabular form as average deviation.
2.4.2. -LEAF—antibiotic bacteria fluorescence assays
Bacterial strains were cultured overnight on BHI agar plates in the presence of a penicillin disk (10 U) respectively. For each bacterial isolate, colonies closest to the penicillin disk were transferred to PBS to make a homogenous suspension [ colony forming units (CFU)/ml]. Bacterial OD was measured at 600 nm. Serial dilutions were also prepared with tenfold lower bacterial concentrations in PBS. A 20 μM β-LEAF Bodipy-FL probe solution ( stock) was prepared in 40% DMSO in PBS, and a 100-mM cefazolin solution ( stock) was prepared by dissolving the antibiotic powder in PBS. The assays were performed in 96-well white clear-bottom plates in a total volume of , respectively, to include bacteria and the -LEAF probe, with or without 25-mM cefazolin. Each reaction was set up as follows: bacterial suspension, antibiotic stock solution or PBS only, and probe stock solution, with the resultant buffer concentration as 20% DMSO in PBS in each reaction. For each isolate, reactions were performed in triplicate in the absence and presence of the test antibiotic, respectively. Time course assays were carried out, monitoring -LEAF cleavage by measuring fluorescence for 60 min, at 1-min intervals (Spectramax M5 Plate Reader, Molecular Devices). Instrument settings were kept as excitation at 450 nm and emission at 510 nm. Temperature was maintained at 37°C throughout. The -LEAF cleavage rate in each case was determined as the slope, i.e., fluorescence change as a function of time (obtained from instrument software—SoftMax Pro5), normalized by bacterial OD.
3. Results
The new -LEAF construct was synthesized by modifying previously published procedures from our group.26 Briefly, following the generation of two primary amines on the opposite sides of the cephalosphorin ring (compound 1 as shown in Fig. 3), carboxylic acid functionalized Bodipy-FL was coupled to the cephalosporin core in the presence of HATU and DIPEA via amidation, while it was protected from light. Following the purification of the compound 2 (Fig. 3), a -methoxybenzyl protection group was hydrolyzed under acidic conditions to yield a crude mixture of -LEAF probe. The timing of the reaction was carefully monitored, as longer reaction times () resulted in rapid decomposition of the product. The final product 3 (Fig. 3) (-LEAF) was purified by HPLC, and the desired second generation -LEAF probe was obtained with a 43% overall yield.
Figure 6(a) shows the degree of fluorescent quenching achieved in the -LEAF construct. Free Bodipy-FL dye and the -LEAF Bodipy-FL probe were prepared at equimolar concentrations (). The concentration of each solution was determined based on the extinction coefficient of Bodipy-FL to ensure that there was an equal amount of fluorescent dye in both samples. Following the excitation at 450 nm, emission profiles of the solutions were recorded from 455 to 655 nm. As shown in Fig. 6(a), a fluorescence quenching factor of 7.5 was achieved with the -LEAF Bodipy-FL probe, whereas the previously published first generation -LEAF probe (-LEAF EtNBS) had a fluorescence quenching factor of 5. Besides having a high degree of quenching, close proximity of the two fluorophores was further verified by the newly generated shoulder at 475 nm in the -LEAF absorbance profile [Fig. 6(b)], which is due to homotransfer between the two Bodipy-FL molecules (homotransfer can only occur for molecules in close proximity). The homotransfer generates a new ground state which differs from the Bodipy-FL ground state, and this change in electronic structure manifests as an altered probe absorption spectrum (i.e., the 475-nm shoulder). The absorption spectrum of compound 1 is also provided in Fig. 7.
Fig. 6.

(a) Fluorescence spectra of Bodipy-FL and -LEAF Bodipy-FL probe. Fluorescence spectra of Bodipy-FL and -LEAF Bodipy-FL probe were taken at equimolar concentrations () in 20% DMSO in PBS solution. Fluorescence quenching factor of 7.5 was achieved with the Bodipy-FL -LEAF probe. (b) Absorbance spectra of Bodipy-FL and -LEAF Bodipy-FL probe. Absorbance spectra of Bodipy-FL and -LEAF Bodipy-FL probe were taken at equimolar concentrations () in 20% DMSO in PBS solution. The concentration of the solution was determined based on the extinction coefficient of Bodipy-FL. Generation of shoulder peak at 475 nm in the -LEAF absorbance profile further verifies the close proximity of the fluorophores in the -LEAF construct.
Fig. 7.

Absorbance spectra of modified cephalosporin compound 1 in DMSO.
3.1. Second Generation Probe (-LEAF Bodipy-FL) is Sensitive to -Lactamase
We then performed studies to characterize the ability of the second generation probe to measure -lactamase activity. To ascertain the efficiency and benchmark the total fluorescence output of the second generation probe, of the first and second generation probes were assayed with penicillinase enzyme (a common -lactamase) (Fig. 8). A nearly 30-fold higher rate of fluorescence change (RFU/min) was noted with the second versus the first generation probe with enzymatic cleavage. Furthermore, assaying with different enzyme concentrations showed an appreciable fluorescence increase with concentrations as low as penicillinase enzyme with the second generation probe (data not shown). To more systematically compare the kinetic properties of the two probes as substrates for -lactamase, enzyme kinetics assays with penicillinase enzyme were performed. Data were taken at penicillinase enzyme concentration for 20 min and analyzed with Michealis–Menten kinetics (Table 2). Both first and second generation probes showed a linear increase in fluorescence at this enzyme concentration (for different probe concentrations) over 20 min. Though affinity (ascertained through ) of the second generation probe only shows marginal improvement, the is more than two-fold higher than the first generation probe. This presents the possibility of the reducing assay time to half or less.
Fig. 8.

Fluorescence change due to the action of penicillinase (-lactamase) on first and second generation probes. 10 μM first and second generation probes were incubated with penicillinase enzyme (final concentration in each reaction) or plain PBS, respectively. The fluorescence change over time was monitored for 60 min and is presented as bar graphs. The -axis shows the measured fluorescence change rate in relative fluorescence units (RFU)/min, which reflects the probe cleavage in respective reactions.
Table 2.
Kinetic parameters for the first and second generation probes, respectively, as substrates of -lactamase (penicillinase) enzyme. Values presented are averages from three independent experiments and presented as . deviation.
| Probe | ( penicillinase) | () |
|---|---|---|
| First generation (-LEAF EtNBS) | ||
| Second generation (-LEAF Bodipy-FL) |
3.2. Optical Assay and Probe Validation with Bacteria
To validate the second generation probe in bacterial assays, known -lactamase producer and nonproducer strains were tested with the commonly used β-lactam antibiotic cefazolin to provide a realistic test of this methodology. When assayed with the probe alone, an increase in fluorescence indicates bacterial -lactamase activity, which is a phenotypic measure of -lactamase production. For assessing antibiotic susceptibility, the assay is also designed to include the probe and an excess antibiotic concentration in a competition reaction. The fluorescence increase observed in this case is compared to the fluorescence increase with the probe alone. As antibiotic is in excess compared with the probe, the antibiotic will be preferentially cleaved by lactamase, if the lactamase can target the antibiotic. Thus, a substantially decreased fluorescence output in the presence of antibiotic predicts that the antibiotic may be easily targeted and destroyed by the lactamase. On the other hand, if the antibiotic cannot be efficiently targeted by the lactamase despite its excess concentration, only the probe will get cleaved to show a fluorescent increase comparable to that observed with the probe only.
Fluorescence over 60 min with the known lactamase producer and nonproducer strains was monitored with the second generation probe to obtain fluorescent trend lines, both in the absence and presence of cefazolin (Fig. 9). These data are graphically presented to show the fluorescence change over time in each sample [Fig. 10(a)]. Results show a good range of detection with the second generation probe, with clearly distinguishable profiles for lactamase producing and nonproducing bacterial strains. Due to improved probe properties, only 20 min of data were necessary for analysis [Fig. 10(b)]. These 20-min results were consistent with results found at 60 min.
Fig. 9.

Time course to represent the fluorescence change observed during -LEAF assays using second generation probe with S. aureus ATCC strains with known production of -lactamase. -LEAF assays were performed with the two ATCC S. aureus control strains, known -lactamase producer #1 – B-lac(+) and nonproducer #2 – B-lac(-). The bacteria were incubated with probe alone and probe+cefazolin as a test antibiotic, respectively, and fluorescence was monitored over 60 min, at 1-min intervals. The -axis shows the fluorescence as RFUs and -axis shows the time in minutes.
Fig. 10.

-LEAF assays with second generation probe to determine -lactamase production and cefazolin susceptibility in S. aureus ATCC strains with known production of -lactamase. -LEAF assays were performed with the two ATCC S. aureus control strains [known -lactamase producer #1 – B-lac(+) and nonproducer #2 – B-lac(-)], with cefazolin as a test antibiotic. The bacteria were incubated with probe alone and probe+cefazolin, respectively, and fluorescence monitored over 60 min. (a) The data from 60 min are graphically presented. (b) Data from only the first 20 min are graphically shown. For both panels (a) and (b), the -axis represents the cleavage rate of -LEAF (measured as RFU change rate—RFU/min) normalized by bacterial OD (optical density) at 600 nm. The white patterned bars depict the fluorescence change in probe alone reaction to show -lactamase production. The black bars depict the fluorescence change when both the probe and cefazolin are included in the reactions. Where the two bars are significantly different, the antibiotic is predicted to be less active. Results are presented as the average of three independent experiments (each experiment contained samples in triplicates), and error bars represent the standard error.
Furthermore, four clinical isolates of S. aureus were tested in addition to the control bacterial strains (Fig. 11). These clinical isolates could be readily classified as -lactamase producing (isolate #6) or nonproducing (isolates #3, #4, #5) as per observed profiles and compared to the control bacteria (the -lactamase status of the clinical isolates was verified with the gold standard nitrocefin and zone edge tests). Of note, strain #2 displays a mild negative fluorescence change. This is likely due to the uncleaved probe associating with the pathogen, thereby changing the microenvironment of the probe and reducing the fluorescence.
Fig. 11.

-LEAF assays with second generation probe to determine -lactamase production and cefazolin susceptibility in S. aureus clinical isolates. -LEAF assays were performed with the two ATCC S. aureus control strains (known -lactamase producer #1 and nonproducer #2) and four S. aureus clinical isolates (#3 to #6), with cefazolin as a test antibiotic. The different bacterial isolates were incubated with probe alone and probe+cefazolin, respectively, and fluorescence was monitored over 60 min. The -axis represents the cleavage rate of -LEAF (measured as fluorescence change rate—RFU/min) normalized by bacterial OD (optical density) at 600 nm. The white patterned bars depict the fluorescence change in probe alone reaction, to show -lactamase production. The black bars depict the fluorescence change when both the probe and cefazolin are included in the reactions. Where the two bars are significantly different, the antibiotic is predicted to be less active. Results are presented as the average of two independent experiments (each experiment contained samples in triplicates), and error bars represent the standard error. The -lactamase status of the clinical isolates was verified by using the nitrocefin and zone edge tests.
4. Discussion
This study demonstrates the utility of a new Bodipy-FL based probe in a rapid assay of -lactamase presence and related antibiotic susceptibility for use in LMIC. This required overcoming a number of issues including: (1) reducing the measurement background by improving the degree of static quenching; (2) enhancing the -lactamase limit of detection by choosing fluorophores with enhanced photophysical properties; and (3) compressing the measurement time by increasing the rate of -LEAF cleavage by -lactamase.
This work is a continuation of previous studies where we reported on a phenothiazine-based probe designed primarily as a phototherapeutic agent,26 which was also fluorescent, but poorly so, as expected. This study had a diagnostic focus, so an increase in sensitivity and fluorescent signal was desirable. Among other parameters, the quenching factor is an important component of this technology and plays a central role in determining the overall signal-to-noise of the measurement. The choice of the fluorophore has an important role in determining the degree of quenching, as well as the speed of -lactamase cleavage. With the aim of achieving high sensitivity, several different fluorophore–fluorophore and fluorophore–quencher pairs with promising photophysical properties were explored. Commercially available Cyanine 5.5 dye, Bodipy-FL-Black Hole Quencher-1 (BHQ1) pairs, and Cyanine 5.5-BHQ3 fluorescence–quencher pairs were employed to synthesize different -LEAF probes (data not shown). Even though these new probes showed high degrees of fluorescence quenching (Table 3), they proved to be extremely poor substrates for -lactamase cleavage. Among several probes, only Bodipy-FL constructs provided a rapid fluorescence increase. Commercially available green fluorescent dye, Bodipy-FL, has an exceptionally high-fluorescence quantum yield (0.9) in addition to a respectable extinction coefficient (). These photophysical properties play an important role in the enhanced performance of this new second generation probe.
Table 3.
Photophysical properties of number of -LEAF probes consisting of different fluorophore–fluorophore and/or fluorophore–quencher pairs.
| Fluorophore–fluorophore and fluorophore–quencher pairs | Emission maxima (nm) | Fluorescence quantum yield of the fluorophore | Quenching factor | |
|---|---|---|---|---|
| 1 | EtNBS-EtNBS | 681a | 0.2 | 4.5 |
| 2 | Cy5.5-Cy5.5 | 699a | 0.2 | 1.6 |
| 3 | Bodipy-FL-Bodipy-FL | 509b | 0.9 | 7.5 |
| 4 | Cy5.5-BHQ3 | 699a | 0.2 | 18.8 |
| 5 | Bodipy-FL-BHQ1 | 509b | 0.9 | 118 |
Spectra were recorded in methanol.
Spectra were recorded in 20% DMSO in PBS solution.
The fluorophore–fluorophore proximity is another key determinant of the quenching factor and is determined by the length of the linkers connecting the fluorophores to the -lactam core. Short linkers improve the quenching efficiency; however, they may also interfere with probe cleavage by impeding access of the probe to the -lactamase catalytic site (i.e., steric hindrance). With these factors in mind, we explored several chemistries and honed in on a three-carbon length linker with the Bodipy-FL fluorophore with which we were able to obtain a quenching factor nearly 70% greater than the previous probe. In addition, as seen in Fig. 6(b), while free Bodipy-FL and the -LEAF probe showed the same OD at the equimolar concentration, Bodipy-FL had a shoulder peak at as a result of the two fluorophores being in close proximity to each other. Surprisingly, this excellent degree of quenching resulted in no penalty in terms of the -lactamase catalytic efficiency. In fact, detailed kinetic analysis with penicillinase enzyme revealed that the new probe has a maximum catalytic velocity () more than two times larger than that of the first generation probe (Table 2).
A compelling aspect of these results is the simplicity of the resulting data interpretation. In the basic protocol (bacteria plus probe with and without antibiotic; Figs. 10 and 11), we found only two outcomes: (1) No fluorescence change under either conditions, indicating a -lactamase negative strain and (2) The fluorescence increases without the antibiotic, but does not increase with the antibiotic. This indicates that the -lactamase is preferentially cleaving the antibiotic over -LEAF. Thus, the antibiotic is sensitive to -lactamase-mediated destruction and is not a good treatment choice. This is seen in Fig. 10, where the -lactamase positive control strain only displays a large fluorescence increase when incubated without antibiotic (and negligible change with antibiotic), confirming the strain to have -lactamase-mediated resistance to cefazolin. This behavior was also seen in a clinical isolate (Fig. 11, strain #6). In contrast, the -lactamase negative control strain and three of the clinical isolates show little fluorescence change, indicating no -lactamase production (Fig. 11). The assay’s -lactamase predictions were all confirmed with nitrocefin and penicillin zone edge tests, which are the gold-standard tests performed in clinical microbiology laboratories for -lactamase production (data not shown).
Beyond the two outcomes listed above, a third possibility exists. If the -lactam antibiotic used is resistant to -lactamase-mediated destruction, the fluorescence will increase equally for both conditions, thereby indicating that -lactamase is cleaving the probe and not the antibiotic. We did not observe this condition in any of the isolates tested here. However, we have studied this phenomenon in a previous work29 using the first generation probe, where clinical isolates were found to be susceptible to a cephalosporin antibiotic (cefepime) despite producing -lactamase. In these cases, the fluorescence did increase independently of the antibiotic presence. As the -lactamase is agnostic to the antibiotic in this situation, we expect similar fluorescence behavior to the second generation probe (i.e., the fluorescence will also increase with and without antibiotic), since the probe chemistry has no influence on the lack of -lactamase interaction.
Further opportunities to characterize the subtype of -lactamase are available by studying the hydrolysis kinetics with varying concentrations of -LEAF and antibiotic.27 From these curves, antibiotic association constants and -LEAF Michaelis–Menten parameters can be extracted. While the catalytic rate of antibiotic destruction is only indirectly probed by these parameters, they can provide a rapid “fingerprint” of the particular -lactamase, which can allow laboratory personnel to report other catalytic properties based on the regional -lactamase spectrum.
The method is simple and requires only two conditions: (1) and (2) . Furthermore, the improved catalytic and fluorescent properties of this second generation probe allow a reduction in the assay time. This is a key distinction from a nitrocefin-based assay which can provide similar information but, in practice, requires a significantly longer assay time (due to the limited dynamic range of the colorimetric readout, requiring tuning of the pathogen concentration).34 These improvements represent a significant advance in both speed and simplicity and are essential requirements for LMIC implementation, where laboratory capacity is often a limiting factor. As the fluorescence readout only uses single excitation and emission wavelengths, battery-powered hand-held fluorescent readers, utilizing simple LED sources, dichroics, and emission filters, can be easily designed around this probe technology [Fig. 12(a)]. By incorporating two different wavelength LEDs, it is also possible to incorporate a simultaneous absorbance measurement into the system while sharing the same photodetector, further reducing the cost. Critically, these readers require almost no maintenance, as they are comprised of only a few, low cost but robust components. A simple workflow providing a binary output is described in Fig. 12(b). This is in contrast to other, more complicated methodologies (e.g., automated blood testing), which require considerable maintenance even in developed countries. Furthermore, the readers proposed here can be made portable, allowing deployment in remote areas. This, coupled with the probe’s exceptionally stability, having a room temperature shelf life of at least 1 year in the dark, may markedly extend the coverage of antibiotic -lactamase testing. Ideally, this would allow local health authorities to both provide guidelines on optimum antibiotic prescriptions, as well as chart the spread of AR. One potential point-of-care application of the assay is depicted in schematic form in Fig. 13. Beyond this, the development of rapid testing, such as the -LEAF assay, allows the possibility of personalized medicine, with its concomitant health benefits, to be extended to the LMIC. The system reported here is promising for low-income settings and merits further validation with a large number of clinical samples.
Fig. 12.
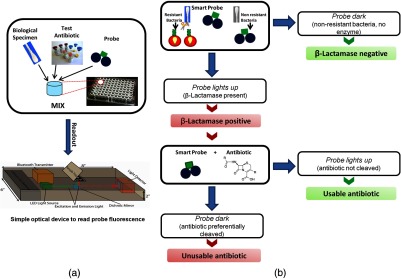
-LEAF assay setup and clinical interpretation. (a) A simple assay setup in a microtiter plate is depicted. Multiple reactions can be set up simultaneously. Each reaction would include the biological specimen (containing bacteria), probe, and respective test antibiotic. Several antibiotics could be tested concurrently. These assays can be read using standard plate readers or with simple, low-cost readers designed for low-resource settings (bottom figure panel). (b) Flowchart of simple binary output results obtainable from the -LEAF assay. The “probe alone” assay gives a yes/no result of whether -lactamase is produced. Further, when the probe is assayed along with a test antibiotic, a yes/no answer for whether the antibiotic is usable or not is provided.
Fig. 13.
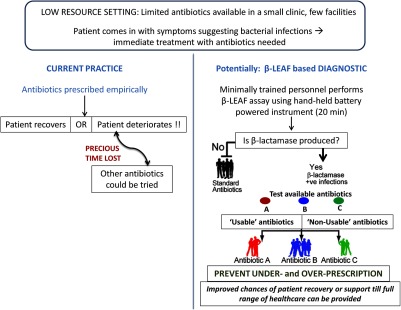
Envisioned application of the -LEAF assay (in a low-resource setting in particular). One scenario for application of the -LEAF assay as a rapid diagnostic is presented, comparing the current practice and potential usage of the assay.
Acknowledgments
We thank Dr. Mary Jane Ferraro (Microbiology Labs, Massachusetts General Hospital, Boston, Massachusetts, USA) for providing the S. aureus clinical isolates. We also thank Drs. Sarika Verma and Ulysses W. Sallum for useful early discussions. This research was funded by the Department of Defense/Air Force Office of Research (DOD/AFOSR) (Grant No. FA9550-11-1-0331) and NIH/NIBIB (National Institute of Biomedical Imaging and Bioengineering) (Point of Care Technology in Primary Care) through CIMIT (Centre for Integration of Medicine and Innovation Technology) (Grant No. U54 EB015408). SSE and SK contributed to the design, conduct and analyses of experiments, and the writing and preparation of the manuscript. AP contributed to the design and analyses of experiments and the writing and preparation of the manuscript. TH contributed to the study conception and design, writing of the manuscript, and overall supervision. All authors read and approved the final manuscript.
Biography
Biographies of the authors are not available.
References
- 1.Winokur P. L., et al. , “Variations in the prevalence of strains expressing an extended-spectrum beta-lactamase phenotype and characterization of isolates from Europe, the Americas, and the Western Pacific region,” Clin. Inf. Dis. 32(Suppl 2), S94–S103 (2001). 10.1086/cid.2001.32.issue-s2 [DOI] [PubMed] [Google Scholar]
- 2.Paterson D. L., et al. , “Outcome of cephalosporin treatment for serious infections due to apparently susceptible organisms producing extended-spectrum beta-lactamases: implications for the clinical microbiology laboratory,” J. Clin. Microbiol. 39(6), 2206–2212 (2001). 10.1128/JCM.39.6.2206-2212.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibrahim E. H., et al. , “The influence of inadequate antimicrobial treatment of bloodstream infections on patient outcomes in the ICU setting,” Chest 118(1), 146–155 (2000). 10.1378/chest.118.1.146 [DOI] [PubMed] [Google Scholar]
- 4.Okeke I. N., Divining Without Seeds: A Case for Strengthening Laboratory Medicine in Africa, Cornell University Press, Ithaca, New York: (2011). [Google Scholar]
- 5.Laxminarayan R., et al. , “Antibiotic resistance-the need for global solutions,” Lancet Inf. Dis. 13(12), 1057–1098 (2013). 10.1016/S1473-3099(13)70318-9 [DOI] [PubMed] [Google Scholar]
- 6.Livermore D. M., “beta-Lactamases in laboratory and clinical resistance,” Clin. Microbiol. Rev. 8(4), 557–584 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch A. L., “Penicillin binding proteins, beta-lactams, and lactamases: offensives, attacks, and defensive countermeasures,” Crit. Rev. Microbiol. 26(4), 205–220 (2000). 10.1080/10408410091154228 [DOI] [PubMed] [Google Scholar]
- 8.Bush K., Jacoby G. A., Medeiros A. A., “A functional classification scheme for beta-lactamases and its correlation with molecular structure,” Antimicrob. Agents Chemother. 39(6), 1211–1233 (1995). 10.1128/AAC.39.6.1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez-Bano J., Pascual A., “Clinical significance of extended-spectrum beta-lactamases,” Expert Rev. Antiinf. Ther. 6(5), 671–683 (2008). 10.1586/14787210.6.5.671 [DOI] [PubMed] [Google Scholar]
- 10.Jacoby G. A., Munoz-Price L. S., “The new beta-lactamases,” N. Engl. J. Med. 352(4), 380–391 (2005). 10.1056/NEJMra041359 [DOI] [PubMed] [Google Scholar]
- 11.Nordmann P., Poirel L., “Emerging carbapenemases in Gram-negative aerobes,” Clin. Microbiol. Inf. 8(6), 321–331 (2002). 10.1046/j.1469-0691.2002.00401.x [DOI] [PubMed] [Google Scholar]
- 12.Bauer A. W., et al. , “Antibiotic susceptibility testing by a standardized single disk method,” Am. J. Clin. Pathol. 45(4), 493–496 (1966). [PubMed] [Google Scholar]
- 13.Bauer A. W., et al. , “Antibiotic susceptibility testing by a standardized single disk method,” Tech. Bull. Regist. Med. Technol. 36(3), 49–52 (1966). [PubMed] [Google Scholar]
- 14.Ericsson H. M., Sherris J. C., “Antibiotic sensitivity testing. Report of an international collaborative study,” Acta Pathol. Microbiol. Scand. B Microbiol. Immunol. 217(Suppl 217), 1–90 (1971). [PubMed] [Google Scholar]
- 15.CLSI, Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Second Informational Supplement; CLSI Document M100-S22, Clinical and Laboratory Standards Institute, Wayne, Pennsylvania: (2012). [Google Scholar]
- 16.CLSI, Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard—Eleventh Edition. CLSI Document M02-A11, Clinical and Laboratory Standards Institute Wayne, Pennsylvania: (2012). [Google Scholar]
- 17.Brown D. F., Brown L., “Evaluation of the E test, a novel method of quantifying antimicrobial activity,” J. Antimicrob. Chemother. 27(2), 185–190 (1991). 10.1093/jac/27.2.185 [DOI] [PubMed] [Google Scholar]
- 18.Livermore D. M., Brown D. F., “Detection of beta-lactamase-mediated resistance,” J. Antimicrob. Chemother. 48(Suppl 1), 59–64 (2001). 10.1093/jac/48.suppl_1.59 [DOI] [PubMed] [Google Scholar]
- 19.Thomson K. S., Sanders C. C., “Detection of extended-spectrum beta-lactamases in members of the family Enterobacteriaceae: comparison of the double-disk and three-dimensional tests,” Antimicrob. Agents Chemother. 36(9), 1877–1882 (1992). 10.1128/AAC.36.9.1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livermore D. M., Williams J. D., “Beta-lactams mode of action and mechanisms of bacterial resistance,” in Antibiotics in Laboratory Medicine, Lorian V., Ed., pp. 502–578, Williams and Wilkins, Baltimore, Maryland: (1996). [Google Scholar]
- 21.Gill V. J., Manning C. B., Ingalls C. M., “Correlation of penicillin minimum inhibitory concentrations and penicillin zone edge appearance with staphylococcal beta-lactamase production,” J. Clin. Microbiol. 14(4), 437–440 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sparbier K., et al. , “Matrix-assisted laser desorption ionization-time of flight mass spectrometry-based functional assay for rapid detection of resistance against beta-lactam antibiotics,” J. Clin. Microbiol. 50(3), 927–937 (2012). 10.1128/JCM.05737-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodford N., “Rapid characterization of beta-lactamases by multiplex PCR,” Antibiotic Resistance Protocols, Methods in Molecular Biology Gillespie S. H., McHugh T. D., Eds., Vol. 642, pp. 181–192, Humana Press; (2010). [DOI] [PubMed] [Google Scholar]
- 24.Espy M. J., et al. , “Real-time PCR in clinical microbiology: applications for routine laboratory testing,” Clin. Microbiol. Rev. 19(1), 165–256 (2006). 10.1128/CMR.19.1.165-256.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wieser A., et al. , “MALDI-TOF MS in microbiological diagnostics—identification of microorganisms and beyond (mini review),” Appl. Microbiol. Biotechnol. 93(3), 965–974 (2012). 10.1007/s00253-011-3783-4 [DOI] [PubMed] [Google Scholar]
- 26.Zheng X., et al. , “Exploiting a bacterial drug‐resistance mechanism: a light‐activated construct for the destruction of MRSA,” Angew. Chem. 121(12), 2182–2185 (2009). 10.1002/ange.v121:12 [DOI] [PubMed] [Google Scholar]
- 27.Sallum U. W., et al. , “Rapid functional definition of extended spectrum beta-lactamase activity in bacterial cultures via competitive inhibition of fluorescent substrate cleavage,” Photochem. Photobiol. 86(6), 1267–1271 (2010). 10.1111/php.2010.86.issue-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan S., Hasan T., “Functional targeting of bacteria: a multimodal construct for PDT and Diagnostics of drug-resistant bacteria,” in Photodynamic Therapy: From Theory to Application, Abdelkader M., Ed., pp. 237–253, Springer-Verlag, Berlin, Heidelberg: (2014). [Google Scholar]
- 29.Khan S., et al. , “Rapid optical determination of beta-lactamase and antibiotic activity,” BMC Microbiol. 14, 84 (2014). 10.1186/1471-2180-14-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jori G., et al. , “Photodynamic therapy in the treatment of microbial infections: basic principles and perspective applications,” Lasers Surg. Med. 38(5), 468–481 (2006). 10.1002/(ISSN)1096-9101 [DOI] [PubMed] [Google Scholar]
- 31.Fery-Forgues S., Lavabre D., “Are fluorescence quantum yields so tricky to measure? A demonstration using familiar stationery products,” J. Chem. Educ. 76(9), 1260–1264 (1999). 10.1021/ed076p1260 [DOI] [Google Scholar]
- 32.Lakowicz J. R., Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publisher, New York: (1999). [Google Scholar]
- 33.Fluorescein, http://www.lifetechnologies.com/us/en/home/references/molecular-probes-the-handbook/fluorophores-and-their-amine-reactive-derivatives/fluorescein-oregon-green-and-rhodamine-green-dyes.html#head1 (2014).
- 34.Papanicolaou G. A., Medeiros A. A., “Discrimination of extended-spectrum beta-lactamases by a novel nitrocefin competition assay,” Antimicrob. Agents Chemother. 34(11), 2184–2192 (1990). 10.1128/AAC.34.11.2184 [DOI] [PMC free article] [PubMed] [Google Scholar]