

Abstract
Mutations of VPS35, a component of the retromer complex have been associated with late onset familial Parkinson’s disease. The D620N mutation in VPS35 appears to be most prevalent, however, P316S was found in two cases within the same family and a control, whereas L774M was identified in 6 cases and 1 control. In vivo evidence of their pathogenicity is lacking. Here we investigated the in vivo effects of P316S, D620N and L774M using Drosophila as a model. We generated transgenic human VPS35-expressing mutations and demonstrated that VPS35 D620N transgenic flies led to late-onset loss of TH-positive DA neurons, poor mobility, shortened lifespans and increased sensitivity to rotenone, a PD-linked environmental toxin, with some of these phenotypes observed for P316S but not in L774M transgenic flies. We conclude that D620N and to a smaller extent P316S are associated with pathogenicity in PD.
Electronic supplementary material
The online version of this article (doi:10.1186/s13041-014-0073-y) contains supplementary material, which is available to authorized users.
Keywords: Parkinson’s disease, Drosophila, VPS35, Retromer complex
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder that affects 1% of the aging population. It is characterized by the loss of midbrain dopaminergic (DA) neurons in the substantia nigra region as well as accumulation of Lewy body aggregates. Although PD has been studied extensively, the molecular etiology is still unknown, with many trying to unravel the genetic determinants of PD [1]. VPS35 encodes for a subunit of the retromer, a coat-like protein complex that is involved in the recycling of membrane proteins. The retromer complex helps to guide protein sorting from the endosome-lysosome degradation pathway retrogradely to the Golgi apparatus and VPS35 functions as a platform for binding to other subunits (VPS26 and VPS29) with its distal ends in retromer assembly [2,3].
Recently, a number of reports determined a link between mutations in VPS35 and familial late-onset PD [4,5]. Successive studies showed that D620N mutation in VPS35 is the most prevalent and found to be associated with autosomal dominant PD in Caucasian and Japanese families but not in the Han Chinese population [6-9]. There are reports on other single mutations such as L774M and P316S but their pathogenicity remains unclear. P316S was found in two cases within the same family however, with a control subject also identified with the same mutation, genetic evidence for the pathogenicity of P316S remains unconvincing. L774M was identified in 6 cases and 1 control, hence the inconclusive evidence [5,10].
In flies, VPS35 was found to be involved in a number of physiological functions. Knockdown of VPS35 led to the inhibition of scavenger receptor endocytosis and VPS35 mutation resulted in the overproliferation and excessive differentiation of haemocytes, suggesting that it might have a tumor suppressor function. Also, there is likely a conserved function of VPS35 playing a role in endocytic trafficking [11]. In addition, VPS35 was shown to act in the same pathway with Rab9 to control tracheal length by positively regulating the luminal deposition of Serp, a specific cargo for the retromer-dependent endocytic trafficking [12]. Macleod et al., demonstrated that overexpression of VPS35 rescues dopaminergic neuronal loss and reduced lifespan caused by expression of LRRK2 G2019S in dopaminergic neurons, showing interaction between VPS35 and LRRK2 [13].
To date, there is limited in vivo evidence of pathogenicity of VPS35 mutations reported in single patients. To address these gaps in knowledge, utilizing Drosophila as a model, we investigated the pathogenicity of three reported VPS35 mutations, P316S, D620N and L774M in vivo. We showed evidence that D620N is the most pathogenic mutation in VPS35 followed by P316S, and that these mutations in VPS35 are linked to PD.
Materials and methods
Experiments carried out adhered to ethical standards of the Research Center with regards to non-human experiments.
Fly stocks
The following flies were used in this study: dopa decarboxylase (ddc)-GAL4 and yellow white (yw) (Bloomington Drosophila Stock Center). Human VPS35-expressing flies were created by generating transgenic human VPS35 wild type or mutant (P316S, D620N and L774M) cDNA containing a Hemagglutinin (HA) tag at the C-terminus and inserted into the pUAST-attB plasmid, which will allow the UAS constructs to land into a chosen attP site in the fly genome during microinjection. Constructs were then sent for microinjection into Drosophila embryos (BestGene). Flies were raised on standard yeast-cornmeal-agar medium at 25°C with 12-hour light and dark cycle.
Western blot, immunofluorescence and confocal microscopy
Protein was extracted from fly head homogenates and equal amounts of protein from the various genotypes were resolved by SDS-PAGE. HA-tagged VPS35 was detected using mouse anti-HA (1:1000, Santa Cruz). Flies were aged to day 20 and 60 after eclosion, before fly brains were dissected, fixed and stained according to published protocols [14]. Brains were probed with rabbit anti-tyrosine hydroxylase (1:500, Sigma-Aldrich) and mouse anti-HA (1:200, Santa Cruz).
Climbing, lifespan assays and rotenone treatment
Climbing assay was performed as described previously [15]. To determine adult lifespan, 100 flies from each genotype under the direction of ddc-GAL4 were maintained on standard media. Newly eclosed adult flies were transferred into vials containing fresh media every 3 days and mortality was scored daily. Age-matched ddc-GAL4/+flies used as controls. In rotenone-treated flies, flies were fed with cornmeal-agar medium containing 500 μM rotenone (Sigma), which was first dissolved in DMSO, immediately after eclosion and during entire experimental period.
Statistical analysis
Quantitative data are expressed as mean ± SEM, unless otherwise stated. Statistical significance for climbing assay and differences in the number of TH-positive DA neurons were analyzed using one-way Anova with Bonferroni’s post hoc test, unless otherwise stated. Lifespan assay was analyzed with Log-rank test.
Results
VSP35 D620N promotes DA neurodegeneration and loss of locomotor activity
To study the pathogenic effects of VPS35 mutants in vivo, we generated the following HA tagged transgenic flies: wild type VPS35 as control, and mutant VPS35 flies, P316S, D620N and L774M. They were verified via immunostaining and immunoblot that the VPS35 from the different variants were expressed and in comparable amounts (Additional file 1: Figure S1).
ddc-GAL4 flies, commonly used in drosophila PD research was used here for the overexpression of the various VPS35 transgenes in TH-positive DA neurons and to examine the gain-of-function effects of mutant VPS35 [16]. We first studied if DA neuronal integrity was affected, since this is a characteristic of PD patients, by comparing the number of TH-positive DA neurons (six clusters in each brain hemisphere). There was no apparent difference at 20 days after eclosion across all genotypes (Figure 1A) but in aged 60 days old flies, we saw a significant loss of TH-positive DA neurons in the PPL1 clusters of VPS35 D620N mutant flies (p < 0.05) compared to age matched wild type and control (ddc-GAL4/+) flies (Figure 1B, C). Interestingly, there was also significant difference between PPL1 clusters of control and wild type VPS35 flies (p < 0.05) (Figure 1B).
Figure 1.
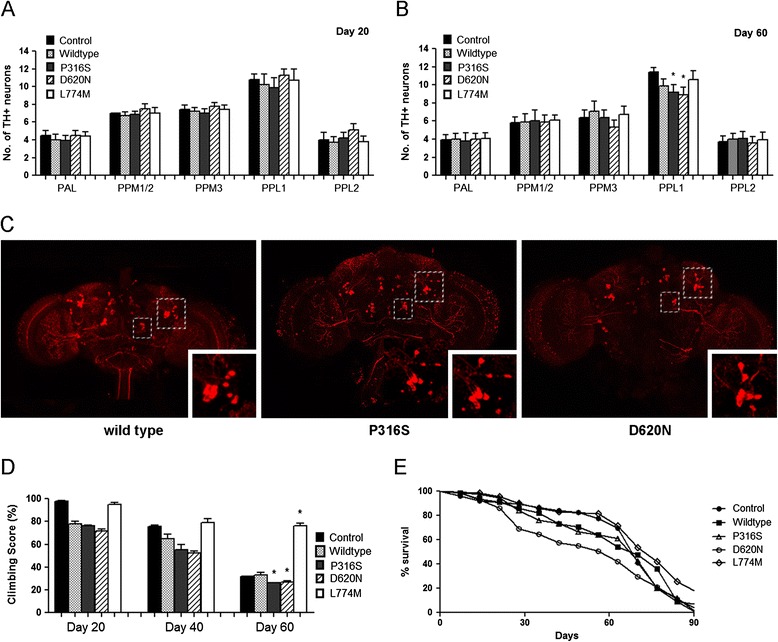
Expression of VPS35 pathogenic variant D620N in flies result in DA neuronal degeneration and locomotive dysfunction. (A, B) Bar graphs show number of TH-positive DA neurons in flies at 20 and 60 days after eclosion (n = 3, cohort of 10). (C) Representative confocal images of whole mount brains 60 days after eclosion of VPS35 wild type, P316S and D620N flies with magnified views of PPL1 cluster (boxed on the right). (D) Bar graph shows age-dependent climbing scores of female flies at different days after eclosion. Percentage of flies that reached the top of the column after 1 min were counted (n = 3, cohort of 20). (E) Survival curves were plotted as percentage of living flies. (n = 3, cohort of 100).
As DA neuronal loss has been linked to locomotor dysfunction, we compared the mobility of the various genotypes. At 20 and 40 days post eclosion, we observed a significant difference in climbing ability when comparing control flies to wildtype, P316S and D620N flies (p < 0.05) (Figure 1D). At 60 days post eclosion, there was a marked reduction in climbing abilities for all genotypes except L774M transgenic flies, with the climbing scores for VPS35 P316S and D620N flies significantly lower (p < 0.05) compared to age matched control and VPS35 wild type flies (Figure 1D). This correlated with the significant loss of DA neurons in VPS35 D620N mutant flies at 60 days. Interestingly, L774M transgenic flies displayed robust climbing even after 60 days of aging. There was a significant decrease (p < 0.05) in the median lifespan of D620N flies (63 days) compared to wild type and control flies (70 days) (Figure 1E).
Rotenone treatment further exacerbated the pathogenicity of D620N VPS35 variant in DA neuron degeneration
We challenged the various transgenic VPS35-expressing flies with rotenone to examine their relative susceptibility of their TH-positive DA neurons to degeneration in response to a PD-linked environmental toxin. We observed a faster loss (15 days) of DA neurons in the PPL1 clusters of rotenone-treated flies expressing VPS35 P316S or D620N flies compared to age-matched control flies. Accelerated loss of TH-positive DA neurons was also observed in the PPL2 cluster of VPS35 P316S and D620N flies (p < 0.05) (Figure 2). Again, we did not find any loss of TH-positive DA neurons in flies expressing VPS35 L774M as rotenone treatment failed to enhance any loss of TH-positive DA neurons. These results suggest that D620N VPS35 flies are highly susceptible to rotenone DA neurodegeneration.
Figure 2.
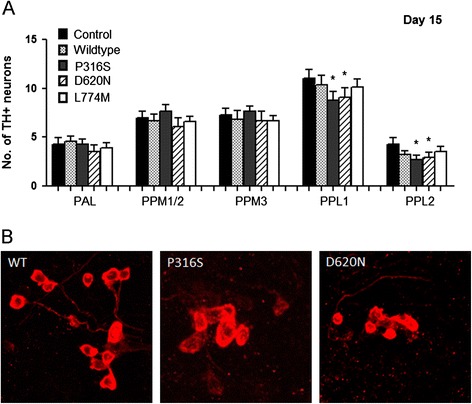
Exposure of VPS35 P316S and D620N mutant flies to rotenone accelerates DA neuronal loss. (A) Bar graph shows the quantification of the number of DA neurons in different clusters of various fly species 15 days after rotenone treatment (n = 3, cohort of 10). (B) Representative confocal images showing TH-positive DA neurons in the PPL1 cluster of rotenone treated fly species in VPS35 wild type, P316S and D620N flies.
Discussion
VPS35 is a component of the retromer complex recently linked to autosomal dominant late onset PD and in vivo evidence of pathogenicity of VPS35 mutations in single patient is lacking. Here, we characterized three reported variants of VPS35, P316S, D620N and L774M, in flies and provided in vivo evidence of the pathogenicity of VPS35 D620N mutation. We demonstrated that the expression of VPS35 D620N and to a smaller extent P316S, lead to the loss of TH-positive DA neurons, locomotor dysfunction and reduced lifespan.
The C-terminal amino acid residues 307 – 796 form an α-solenoid fold that fits into the metal-binding site of VPS29, suggesting that the known mutations of VPS35 might interfere with the interaction and assembly with VPS29, and disrupt retromer function in trafficking proteins intracellularly [17,18]. D620N is at the region where VPS35 binds not only to VPS29, but also accessory proteins such as cation-independent mannose-6-phosphate receptor (CI-MPR) and SNX1, whereby the former is essential for efficient anterograde transport of soluble hydrolases and also retrieval of mislocalized resident Golgi proteins, and the latter implicated in sorting of activated signaling receptors for lysosomal degradation [19]. Zavodszky et al. found that unlike a complete knockdown of VPS35 that eliminates retromer function, the D620N mutation affects retromer function linked to the WASH complex. They recently published findings showing that D620N mutation in VPS35 destabilizes the retromer-WASH complex interaction, resulting in reduced endosomal localization of the WASH complex, and that cells expressing the D620N VPS35 mutation exhibited defects in autophagy [20]. Defects in autophagy could be a contributor to PD pathogenesis as it may aggravate pathologies associated with PD, such as mitochondrial abnormalities, protein aggregation, increased reactive oxygen species and higher sensitivity to cell death. These may have contributed to the more severe phenotype observed in D620N flies. Although the precise mechanism of retromer dysfunction in PD is unclear, one report observed that the expression of VPS35 D620N led to endosomal alterations and defects in trafficking, which may partially explain its action in PD [21].
An interesting observation was the robust climbing of L774M mutation that appeared to have a protective effect during the climbing assay after flies were aged. As the mutation is at the C-terminus of VPS35 and is part of the binding region of VPS35 to VPS29, the mutation may have modulated structural changes in VPS35 such that it binds to VPS29 more efficiently. Another interesting observation was the toxic effect of over-expressing wild type VPS35, as we observed significant differences in the number of TH-positive neurons between age matched control and wild type flies as well as climbing abilities of age matched control and wild type flies at 20 and 40 days old. These results suggest that over-expressing wild type VPS35 alone causes the loss of DA neurons, and impairs locomotion even though Macleod and colleagues observed that the over-expression of wild type VPS35 rescued DA neuronal loss and reduced life span caused by LRRK2 G2019S expression in DA neurons [13]. In addition, the over-expression of D620N and P316S mutations aggravate this effect, which is consistent with the dominant nature of these mutations.
In a recent study by Tsika and colleagues, the common VPS35 D620N mutation did not compromise the protein stability, localization to endosomal and lysosomal vesicles, or the vesicular sorting of retromer cargo in rodent primary neurons or patient-derived human fibroblasts, suggesting that there is no loss of function. Instead, the overexpression of D620N VPS35 induced marked degeneration of substantia nigra dopaminergic neurons and axonal pathology in a rat model, implying that dominant VPS35 mutations lead to neurodegeneration in PD, which is similar to our results in flies [22].
We observed that the pathogenic process caused by single mutations of D620N and P316S can be exacerbated by rotenone, an environmental mitochondrial complex I inhibitor that has been linked to PD. Other findings have shown that the retromer complex regulates the transport of mitochondrial-anchored protein ligase to peroxisomes and that enhanced VPS35 expression protects dopaminergic cells against mitochondrial stress, suggesting that there might be a connection between VPS35 and pathogenesis of PD via mitochondrial stress [23,24].
Our study in characterizing the various VPS35 mutations will contribute to the understanding of VPS35-induced PD, with in vivo confirmation of the pathogenicity VPS35 D620N mutation. Further functional and genetic studies can be carried out to resolve the underlying mechanisms of VPS35 in PD, in addition to studying the interactions between VPS35 and other PD associated genes, which might provide important insights in the identification of more effective drug or molecular therapeutic targets for PD patients with VPS35 mutations.
Acknowledgements
National Medical Research Council, Singapore Millennium Foundation, Singapore General Hospital, and Duke NUS Graduate School of Medicine. Supported by the Singapore National Research Foundation under its Translational and Clinical Research Flagship Programme and administered by the Singapore Ministry of Health’s National Medical Research Council.
Additional file
HA-tagged VPS35 transgenes tested for expression and protein levels. (A) Immunostaining of whole-mount adult fly brains expressing wild type or mutant human VPS35 in TH + neurons by overexpressing HA-tagged VPS35 variants in TH + neurons using ddc-GAL4. Top row: anti-HA. Middle row: anti-TH. Bottom row: Merged images of anti-HA (green) and anti-TH (red). (B) An anti-HA immunoblot of adult brain lysates prepared from control (elav-GAL4/+) or transgenic flies expressing the various VPS35 variants (elav-GAL4 > VPS35 variants).
Footnotes
Hua-shan Wang, and Joanne Toh contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WHS conceived and designed the study. WHS, JT and PH participated in carrying out the experiments. WHS and JT participated in analyzing the data. JT and ET drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Hua-shan Wang, Email: mitsuvolvo@gmail.com.
Joanne Toh, Email: joanne.toh.y.e@singhealth.com.sg.
Patrick Ho, Email: patrick.ho.g.h@sgh.com.sg.
Murni Tio, Email: murnittio@yahoo.co.uk.
Yi Zhao, Email: murnittio@yahoo.co.uk.
Eng-King Tan, Email: gnrtek@sgh.com.sg.
References
- 1.Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat. 2007;28(7):641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- 2.Skinner C, Seaman M, Skinner C, Seaman M. The role of retromer in neurodegenerative disease. In: St. George-Hyslop PH, Mobley WCC, Yves C, editors. Research and Perspectives in Alzheimer’s Disease. Heidelberg: Springer Berlin; 2009. pp. 125–140. [Google Scholar]
- 3.Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20(4):427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brücke T, Poewe W, Auff E, Trenkwalder C, Rost B, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ando M, Funayama M, Li Y, Kashihara K, Murakami Y, Ishizu N, Toyoda C, Noguchi K, Hashimoto T, Nakano N, Sasaki R, Kokubo Y, Kuzuhara S, Ogaki K, Yamashita C, Yoshino H, Hatano T, Tomiyama H, Hattori N. VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov Disord. 2012;27(11):1413–1417. doi: 10.1002/mds.25145. [DOI] [PubMed] [Google Scholar]
- 7.Guo JF, Sun QY, Lv ZY, Yu RL, Li K, Zhang YH, Tian JY, Xia K, Yan XX, Tang BS. VPS35 gene variants are not associated with Parkinson’s disease in the mainland Chinese population. Parkinsonism Relat Disord. 2012;18(8):983–985. doi: 10.1016/j.parkreldis.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Lesage S, Condroyer C, Klebe S, Honoré A, Tison F, Brefel-Courbon C, Dürr A, Brice A, French Parkinson’s Disease Genetics Study Group Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology. 2012;78(18):1449–1450. doi: 10.1212/WNL.0b013e318253d5f2. [DOI] [PubMed] [Google Scholar]
- 9.Sheerin UM, Charlesworth G, Bras J, Guerreiro R, Bhatia K, Foltynie T, Limousin P, Silveira-Moriyama L, Lees A, Wood N: Screening for VPS35 mutations in Parkinson’s disease.Neurobiol Aging 2012, 33(4):838. e1-838.e5. [DOI] [PMC free article] [PubMed]
- 10.Sharma M, Ioannidis JPA, Aasly JO, Annesi G, Brice A, Bertram L, Bozi M, Barcikowska M, Crosiers D, Clarke CE, Facheris MF, Farrer M, Garraux G, Gispert S, Auburger G, Vilariño-Güell C, Hadjigeorgiou GM, Hicks AA, Hattori N, Jeon BS, Jamrozik Z, Krygowska-Wajs A, Lesage S, Lill CM, Lin JJ, Lynch T, Lichtner P, Lang AE, Libioulle C, Murata M, et al. A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet. 2012;49(11):721–726. doi: 10.1136/jmedgenet-2012-101155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korolchuk VI, Schütz MM, Gómez-Llorente C, Rocha J, Lansu NR, Collins SM, Wairkar YP, Robinson IM, O’Kane CJ. Drosophila Vps35 function is necessary for normal endocytic trafficking and actin cytoskeleton organisation. J Cell Sci. 2007;120(24):4367–4376. doi: 10.1242/jcs.012336. [DOI] [PubMed] [Google Scholar]
- 12.Dong B, Kakihara K, Otani T, Wada H, Hayashi S: Rab9 and retromer regulate retrograde trafficking of luminal protein required for epithelial tube length control.Nat Commun 2013, 4:1358. [DOI] [PMC free article] [PubMed]
- 13.MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, MacCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitworth AJ, Theodore DA, Greene JC, Beneš H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci. 2005;102(22):8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Lu R, Ouyang X, Ho MWL, Chia W, Yu F, Lim KL. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27(32):8563–8857. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng CH, Mok SZS, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29(36):11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hierro A, Rojas AL, Rojas R, Murthy N, Effantin G, Kajava AV, Steven AC, Bonifacino JS, Hurley JH. Functional architecture of the retromer cargo-recognition complex. Nature. 2007;449(7165):1063–1067. doi: 10.1038/nature06216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins BM, Skinner CF, Watson PJ, Seaman MNJ, Owen DJ. Vps29 has a phosphoesterase fold that acts as a protein interaction scaffold for retromer assembly. Nat Struct Mol Biol. 2005;12(7):594–602. doi: 10.1038/nsmb954. [DOI] [PubMed] [Google Scholar]
- 19.Collins BM. The structure and function of the retromer protein complex. Traffic. 2008;9(11):1811–1822. doi: 10.1111/j.1600-0854.2008.00777.x. [DOI] [PubMed] [Google Scholar]
- 20.Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, Rubinsztein DC: Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy.Nat Commun 2014, 5:3828. [DOI] [PMC free article] [PubMed]
- 21.Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, Collins BM, Bugarcic A, Teasdale RD. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic. 2014;15(2):230–244. doi: 10.1111/tra.12136. [DOI] [PubMed] [Google Scholar]
- 22.Tsika E, Glauser L, Moser R, Fiser A, Daniel G, Sheerin UM, Lees A, Troncoso JC, Lewis PA, Bandopadhyay R, Schneider BL, Moore DJ. Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum Mol Genet. 2014;23(17):4621–4638. doi: 10.1093/hmg/ddu178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol. 2010;20(14):1310–1315. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 24.Bi F, Li F, Huang C, Zhou H. Pathogenic mutation in VPS35 impairs its protection against MPP(+) cytotoxicity. Int J Biol Sci. 2013;9(2):149–155. doi: 10.7150/ijbs.5617. [DOI] [PMC free article] [PubMed] [Google Scholar]