

Abstract
Background
We hypothesized that poor control of Epstein-Barr virus (EBV) infection, leading to reactivation of the virus, increases the risk of non-Hodgkin lymphoma (NHL) in the general population of primarily immunocompetent persons.
Methods
We conducted a case-control study nested within the Women’s Health Initiative Observational Study cohort in which we measured antibodies to EBV antigens (IgG to viral capsid antigen [VCA], nuclear antigen [EBNA1], and early antigen [EA-D]) and EBV DNA load in prediagnostic samples of 491 B-cell NHL cases and 491 controls.
Results
We found no association with established EBV infection, based on seropositivity for VCA. Seropositivity for EBNA1 was associated with decreased risk of B-cell NHL, overall (odds ratio [OR]=0.5, 95% confidence interval [CI]: 0.3-0.8) and for each of the histologic subtypes examined. Increased risk of chronic lymphocytic leukemia (CLL) and related subtypes was observed with higher levels of EBV DNA and antibody to EA-D, both markers reflective of reactivation. These associations were strongest for cases with the shortest time interval between blood draw and diagnosis.
Conclusions
In balance, these results do not provide strong evidence of EBV playing a causal role in B-cell NHL in general population women. The associations we observed may reflect increased risk of NHL with underlying immune impairment or could be due to reverse causation.
Impact
Further characterization of the subtype-specific association with CLL is warranted. Exclusion of cases with preclinical disease markers (such as monoclonal B-lymphocytosis for CLL) may help rule out reverse causation in future studies.
Keywords: non-Hodgkin lymphoma, Epstein-Barr virus, epidemiology, cohort studies
INTRODUCTION
Epstein-Barr virus (EBV) is a γ-herpesvirus that infects the great majority of adults worldwide, usually since childhood, resulting in life-long infection. Following primary infection, EBV typically enters a latent phase, in which EBV DNA persists as a closed circular nuclear genome (episome) in a subpopulation of resting memory B cells (1). Regulation of the established EBV infection in a healthy host involves a cellular immune response featuring EBV-specific cytotoxic T-lymphocytes that keep the number of infected B cells in check (2). Poorly-controlled EBV infection, such as that observed in clinical states of immune dysregulation, is characterized by B cell proliferation and lytic replication of the EBV genome (3). Even in an immunocompetent host, spontaneous activation occurs in a small proportion of infected B cells (4). EBV infection can also be transiently reactivated in response to physical or psychological stressors (5, 6).
There is compelling evidence for an etiologic role of EBV in certain B-cell lymphoid diseases including endemic Burkitt lymphoma (4, 7), aggressive lymphoproliferative disease seen in people who have severe immunodeficiencies (4, 7), HIV-related lymphomas (8), and lymphomas in the elderly (9). Many of the NHL tumor cells in these patients carry a monoclonal form of the EBV genome (i.e., are EBV-positive tumors), indicating that the tumor arose from a single infected cell and suggesting a direct effect of EBV on lymphomagenesis (3, 7). EBV has also been postulated as a risk factor for NHL in the general (primarily immunocompetent) population (10). EBV is not detected in the majority of these tumors (11); therefore, a direct role of EBV in an immortalizing transformation of lymphocytes is less likely in these cases. However, alternative pathways have been hypothesized for a causal role of poorly-controlled EBV infection in NHL occurring in the general population, such as EBV-induced stimulation of B-cell proliferation (10), which increases the pool of B cells in which transforming mutations can occur or can promote clonal expansion in already-transformed B cells. Another proposed alternative pathway is a “hit-and-run” mechanism in which EBV antigens or other infection-induced molecules cause mutations that initiate or promote lymphomagenesis, followed by loss of EBV episomes from the clonal tumor cell line (1, 12, 13).
The hypothesis that EBV may be associated with NHL in the general population has been investigated in several prospective cohort studies, in which antibodies to EBV antigens measured in prediagnostic serum samples were used to characterize control of established EBV infection (10, 14-17). Persons who have established infection with EBV typically have IgG antibodies specific to the viral capsid antigen (VCA) and nuclear antigen type 1 (EBNA1); therefore, most adults test seropositive for these antibodies. However, elevated titers of anti-VCA and anti-EBNA may imply poor of control of EBV-transformed cells (18, 19). Antibodies to early antigen (EA) are seen less frequently and reflect viral replication (i.e., reactivation) (14, 20). The previous epidemiologic studies provide some evidence of an abnormal pattern of EBV serology preceding NHL diagnosis, including several studies finding an association with elevated anti-EA titers (10, 14, 15). However, inconsistencies in the methods and results of the previous studies leave the role of EBV in NHL open to interpretation.
We investigated the association between EBV and risk of NHL in a case-control study nested within the Women’s Health Initiative Observational Study (WHI OS) prospective cohort. Our hypothesis was that poor control of established EBV infection, as reflected by relatively high levels of antibodies to EBV antigens (IgG to VCA, EBNA1, and EA-D), is a causal factor for NHL in the general population of primarily immunocompetent persons. We limited our study to B-cell NHL based on a hypothesized mechanism of EBV-induced B-cell proliferation leading to increased rates of critical mutations. Our study is the largest to date on this topic, allowing well-powered estimation of EBV-related risks by major NHL subtypes – an important contribution given that the one previous study to examine subtypes found differences in risk (16). In addition to serology, we evaluated risk of NHL according to EBV DNA load, a measure used frequently in patient groups (EBV DNA load is a strong predictor for the development of lymphoproliferative disorders among transplant patients (21-23)), but not yet examined as a quantitative risk factor in general population studies of NHL.
MATERIALS AND METHODS
The study was designed as a nested case-control study in the WHI OS. All study activities were approved by Human Subjects review by the FHCRC IRB (IR #6897).
Study Population
The design of the WHI OS and clinical trials has been published previously (24). The OS includes 93,676 community-dwelling, postmenopausal women enrolled between 1994 and 1998 at 40 clinical centers distributed widely throughout the US, including targeted enrollment of minority women.
We included 491 B-cell NHL cases diagnosed before April 2009. Women were mailed questionnaires annually to report a wide variety of outcomes including cancers of any type. Patient charts were reviewed by coders trained in Surveillance, Epidemiology, and End-Results (SEER) program guidelines to classify the case morphology according to the ICD-O-3 coding (25). Excluded were women with cancer history (except non-melanoma skin cancer) at enrollment (n=88) or incident cancer during follow-up but before the NHL diagnosis (n=34), and cases without a control match (n=2; see below). B-cell NHL cases were further grouped based on the World Health Organization and the International Lymphoma Epidemiology Consortium (InterLymph) guidelines (26, 27), including 142 chronic lymphocytic leukemia/small lymphocytic lymphoma/ prolymphocytic leukemia (CLL/SLL/PLL; 28.8% of cases), 138 diffuse large B cell lymphoma (DLBCL; 28.2%), and 102 follicular lymphoma (FL; 20.9%).
We selected one control for each of the cases from WHI OS participants who had never been diagnosed with cancer (except non-melanoma skin cancer) at the time of case diagnosis. Each control was selected from the pool of eligible non-cases living at the time of a case diagnosis, and was individually matched to the corresponding case by age (±1 year), US region (Northeast, South, Midwest, West), and date of enrollment blood draw (±3 months).
EBV Biomarker Measurement
We conducted multiplexed assays for the determination of antibodies to EBV in serum samples from the WHI study baseline, collected an average of 6 years before NHL diagnosis (or control reference date). IgG antibodies to several EBV-encoded antigens, including VCA, EBNA1, and EA-diffuse (EA-D) were measured simultaneously using a multiplexed fluorescent bead-based Luminex platform assay, the Plexus EBV IgG Multi-Analyte Diagnostics (Focus Diagnostics, Cypress, CA), according to the manufacturer’s instructions, using a Bio-Plex 200 Luminex dual laser, flow-based reader. This assay is designed to qualitatively assess the presence or absence of human IgG class antibodies to these EBV-encoded molecules. For each specific antibody, a standardized median fluorescence index (MFI) was calculated by subtracting the background MFI from the sample MFI and dividing by the standard cutoff value, giving a unit-less continuous measure of the amount of the antibody. The standardized MFI (‘index value’) was used to categorize samples for presence of antibody to the specific EBV antigen as positive (>1.10, greater than the standard cutoff MFI value), negative (<0.90), or borderline (≥0.90 and ≤1.10); we combined positive and borderline results as ‘positive’ for the purpose of statistical analyses. Case and control serum samples were placed in random order, with matched case/control pairs located next to each other. We also included 50 blind duplicate pairs (100 samples), interspersed randomly among the study samples. Coefficients of variation (CVs) based on blind duplicate samples were 5.0% (IgG to VCA), 5.8% (IgG to EBNA1), and 10.5% (IgG to EA-D).
EBV DNA load was measured at the University of Washington Diagnostic Molecular Virology Laboratory. The DNA samples were from the WHI baseline or later follow-up visits (i.e., the DNA samples were not necessarily from the same blood draw as the serum samples from the study baseline used for antibody measurements), but for all subjects, were collected before the case diagnosis date (or control reference date). 464 cases and 469 controls had available 1-μg DNA samples with appropriate collection timing. The final selected samples were collected, on average, 3.9 years before diagnosis. A detailed description of the methods for EBV DNA load determination is presented as an Appendix. In brief, the EBV PCR assay used primers specifically designed for this study, which amplified a repetitive area in the IR-1 (internal repeat) region of the EBV genome (forward: CCCACGCGCGCATAAT; reverse: GCTTATTCCTCTTTTCCCCTCTAAA; probe: 6FAM-TAGGCCTAAAACCCCCAGGAAGCGG-TAMRA); for the purposes of this study, we estimated 10 IR-1 repeats per one EBV genome. High and low positive controls consisted of the EBV-containing cell line Namalwa (28). Each EBV load determination was performed in parallel with determination of betaglobin copy number to allow conversion of EBV results to cellular viral load, calculated as EBV genome copies/106 cells (1 EBV genome=10 IR-1 copies). We transformed EBV DNA values for statistical analyses using the base 2 logarithm, in order to reduce the influence of extreme values in this right-skewed variable. The CV between blind duplicate samples for the log-transformed values was 16.5%.
Statistical Analysis
All analyses were performed using the SAS system, version 9.2 (SAS Institute, Cary, NC). We generated odds ratios (ORs) and 95% confidence intervals (CIs) as estimates of relative risks of the association between B-cell NHL and each EBV measure, using conditional logistic regression (29). In addition to adjustment for the study matching factors, we evaluated a set of potential confounders selected a priori, including race/ethnicity (coded as ‘Black or African American’, ‘White, not Hispanic’, and ‘Other’), education (grouped into 7 categories and evaluated as a continuous variable), body mass index (BMI, continuous), and smoking status (coded as ‘Former’, ‘Current’, and ‘Never’). Potential confounding was evaluated by the change in the effect estimate for a specific EBV measure when including the covariate in the model compared to the model without the covariate.
All of the EBV measures were analyzed first as dichotomous variables, estimating risks for positive versus negative in order to determine risk associated with mere presence of the marker. We limited all analyses of EBNA1, EA-D, and EBV DNA to participants who were seropositive for VCA, indicating an established EBV infection. We also evaluated the EBV measures as categorical variables (with cutpoints defined according to the distribution of each measure among control subjects) in order to evaluate whether NHL risk differed by quantified value of the EBV measure, without any a priori assumption about the fit of the relationship. Tests for trend across categories (including the reference group and all categories above it) were performed by testing the statistical significance of a continuous variable with assigned values equal to the intra-category median analyte level among controls. Finally, EBV measures were modeled as continuous variables for an examination of linear fit with NHL risk (analyses for VCA and EBNA1 were restricted to women with detected values for those analytes; all observations were included in analyses of continuous EA-D and EBV DNA). Because EBV DNA values were transformed based on the base 2 logarithm, relative risks for the continuous measure are estimated per a doubling of the untransformed analyte level.
We evaluated etiologic heterogeneity by fitting unconditional polytomous logistic regression models for major subtypes of B-cell NHL (including CLL/SLL/PLL, DLBCL, and FL) and testing for homogeneity of risk estimates among the subtypes. If the p-value in the test for homogeneity among subtypes was <0.05, we then conducted pairwise comparisons to test for heterogeneity in the magnitude of risk estimates between specific subtypes. To describe the patterns of association between EBV measures and NHL according to the number of years between blood sample collection and NHL diagnosis (‘prediagnostic interval’), we conducted polytomous logistic regression analyses with NHL cases classified according to time intervals of <4, 4-7, ≥8 years.
We performed several subanalyses to evaluate consistency of our findings. We estimated risks separately by ages <65 and ≥65, and tested for interaction by age group on the multiplicative scale. To further evaluate the magnitude of the main associations of interest among otherwise apparently immunocompetent subjects, we conducted subanalyses in which we excluded subjects who reported at baseline that they had ever been diagnosed with certain conditions or taken medications known to be characterized by/cause altered immune status. Specifically, exclusions were based on a list of conditions queried in a baseline medical history questionnaire, which included physician diagnoses of systemic lupus erythematosus, rheumatoid arthritis, colitis, diverticulitis, pancreatitis, kidney stones, gallbladder disease, stomach ulcer, goiter, and asthma (but did not include other conditions of interest such as allergy). In addition, we excluded women who reported having taken glucocorticosteroid medications or disease-modifying antirheumatic drugs. Analyses with these exclusions included 346 cases and 349 controls.
RESULTS
Our study population of postmenopausal women (Table 1) was mostly 60 years and older at the study baseline (>80% of controls), of white, not Hispanic race/ethnicity (85%), and educated beyond high school (>80%). Approximately half of the women never smoked, and more than half were classified as overweight or obese (BMI ≥25; >60%). The EBV measure values did not differ significantly by the matching factors: age, US region, or blood draw date (not shown).
Table 1.
Characteristics of study population (N [%])
| Cases (N=491) | Controls (N=491) | |
|---|---|---|
| Age (years)a | ||
| 50-59 | 95 (19.4) | 95 (19.4) |
| 60-69 | 247 (50.3) | 244 (49.7) |
| 70-79 | 149 (30.4) | 152 (31.0) |
| Study regiona | ||
| Northeast | 124 (25.3) | 124 (25.3) |
| South | 109 (22.2) | 109 (22.2) |
| Midwest | 119 (24.2) | 119 (24.2) |
| West | 139 (28.3) | 139 (28.3) |
| Race/ethnicity | ||
| White, not Hispanic | 449 (91.4) | 416 (84.7) |
| Black or African American | 19 (3.9) | 32 (6.5) |
| Other | 22 (4.5) | 38 (7.8) |
| Missing | 1 (0.2) | 5 (1.0) |
| Education | ||
| Less than high school diploma | 16 (3.3) | 29 (5.9) |
| High school diploma or GED | 87 (17.7) | 66 (13.5) |
| Vocational or training school | 50 (10.2) | 51 (10.4) |
| Some college or associate degree | 141 (28.7) | 142 (28.9) |
| Baccalaureate degree | 58 (11.8) | 64 (13.0) |
| Some postgraduate or professional | 59 (12.0) | 50 (10.2) |
| Master’s or doctoral degree | 78 (15.9) | 86 (17.5) |
| Missing | 2 (0.4) | 3 (0.6) |
| Smoking status | ||
| Never | 255 (51.9) | 246 (50.1) |
| Former | 206 (42.0) | 215 (43.8) |
| Current | 26 (5.3) | 24 (4.9) |
| Missing | 4 (0.8) | 6 (1.2) |
| Body mass index (kg/m2) | ||
| Underweight (<18.5) | 8 (1.6) | 10 (2.0) |
| Normal weight (18.5-24.9) | 184 (37.5) | 182 (37.1) |
| Overweight (25-29.9) | 187 (38.1) | 170 (34.6) |
| Obesity (30-34.9) | 71 (14.5) | 79 (16.1) |
| Extreme obesity (≥35) | 32 (6.5) | 44 (9.0) |
| Missing | 9 (1.8) | 6 (1.2) |
| Histologic subtype | ||
| CLL/SLL/PLL | 142 (28.8) | |
| Diffuse large B-cell (DLBCL) | 138 (28.2) | |
| Follicular lymphoma (FL) | 102 (20.9) | |
| Marginal zone lymphoma (MZL) | 45 (9.1) | |
| B-NHL, not otherwise specified (NOS) | 30 (6.1) | |
| Lymphoplasmacytic lymphoma (LPL)/ Waldenstrom macroglobulinemia (WM) |
16 (3.3) | |
| Mantle cell lymphoma (MCL) | 9 (1.8) | |
| Burkitt lymphoma (BL) | 5 (1.0) | |
| Hairy cell leukemia (HCL) | 4 (0.8) |
Controls were matched to cases on age, US region, and blood draw date
Associations between the EBV measures and B-cell NHL are shown in Table 2. There was no evidence of confounding by the covariates selected a priori (race/ethnicity, education, smoking, BMI); therefore, we present risk estimates adjusted for the matching factors only. There was no apparent association between seropositivity for the VCA antigen (i.e., women with evidence of established EBV infection; 92.5% of controls) and risk of B-cell NHL or any NHL subtype; likewise, risk did not vary by level of antibody to VCA.
Table 2.
EBV measures at study baseline in relation to incidence of B-cell NHLa
| Controls | All B-cell NHL | CLL/SLL/PLL | DLBCL | FL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | N | OR (95% CI) | p-trend | N | OR (95% CI) | p-trend | N | OR (95% CI) | p-trend | N | OR (95% CI) | p-trend | |
| IgG to VCA | |||||||||||||
| Detected (vs. not) | 454 (37) | 446 (43) | 0.9 (0.5-1.3) | 130 (10) | 1.0 (0.5-2.2) | 124 (14) | 0.7 (0.4-1.4) | 89 (13) | 0.6 (0.3-1.1) | ||||
| Not detected | 37 | 43 | 1.3 (0.8-2.1) | 10 | 1.1 (0.5-2.5) | 14 | 1.6 (0.8-3.5) | 13 | 1.7 (0.8-3.8) | ||||
| <4.07 | 113 | 101 | Referent | 28 | Referent | 26 | Referent | 23 | Referent | ||||
| 4.07 to <5.45 | 114 | 113 | 1.1 (0.8-1.6) | 34 | 1.2 (0.7-2.0) | 30 | 1.1 (0.6-2.0) | 19 | 0.8 (0.4-1.6) | ||||
| 5.45 to <6.50 | 113 | 135 | 1.3 (0.9-2.0) | 49 | 1.7 (1.0-2.9) | 33 | 1.3 (0.7-2.3) | 24 | 1.0 (0.5-1.9) | ||||
| ≥6.50 | 114 | 97 | 0.9 (0.6-1.4) | 0.15 | 19 | 0.7 (0.4-1.3) | 0.22 | 35 | 1.3 (0.7-2.3) | 0.43 | 23 | 1.0 (0.5-1.9) | 0.90 |
| Association per 1-unit increaseb |
454 | 446 | 1.03 (0.94-1.12) | 0.51 | 130 | 1.01 (0.90-1.14) | 0.88 | 124 | 1.04 (0.92-1.17) | 0.53 | 89 | 1.01 (0.88-1.15) | 0.94 |
| IgG to EBNA1b | |||||||||||||
| Detected (vs. not) | 426 (28) | 389 (57) | 0.5 (0.3-0.8) | 111 (19) | 0.4 (0.2-0.7) | 110 (14) | 0.5 (0.3-1.0) | 76 (13) | 0.4 (0.2-0.7) | ||||
| Not detected | 28 | 57 | 2.1 (1.2-3.7) | 19 | 2.7 (1.3-5.6) | 14 | 1.8 (0.8-3.9) | 13 | 4.0 (1.6-9.6) | ||||
| <2.55 | 106 | 94 | Referent | 27 | Referent | 29 | Referent | 13 | Referent | ||||
| 2.55 to <3.23 | 107 | 99 | 1.0 (0.6-1.5) | 29 | 1.0 (0.6-1.9) | 25 | 0.8 (0.5-1.5) | 24 | 1.8 (0.9-3.8) | ||||
| 3.23 to <3.64 | 108 | 90 | 0.8 (0.6-1.2) | 29 | 1.0 (0.6-1.9) | 22 | 0.7 (0.4-1.3) | 20 | 1.5 (0.7-3.2) | ||||
| ≥3.64 | 105 | 106 | 1.1 (0.7-1.6) | 0.33 | 26 | 1.0 (0.5-1.8) | 0.82 | 34 | 1.2 (0.7-2.0) | 0.18 | 19 | 1.5 (0.7-3.2) | 0.18 |
| Association per 1-unit increasec |
426 | 389 | 1.05 (0.87-1.26) | 0.61 | 111 | 1.05 (0.81-1.36) | 0.70 | 110 | 1.13 (0.87-1.46) | 0.37 | 76 | 1.14 (0.84-1.55) | 0.39 |
| IgG to EAb | |||||||||||||
| Detected (vs. not) | 154 (300) | 173 (273) | 1.2 (0.9-1.5) | 61 (69) | 1.8 (1.2-2.6) | 50 (74) | 1.3 (0.9-2.0) | 25 (64) | 0.8 (0.5-1.2) | ||||
| Not detected | 300 | 273 | Referent | 69 | Referent | 74 | Referent | 64 | Referent | ||||
| <1.65 | 52 | 54 | 1.1 (0.7-1.7) | 18 | 1.6 (0.9-2.9) | 16 | 1.3 (0.7-2.4) | 7 | 0.6 (0.3-1.4) | ||||
| 1.65 to <3.11 | 49 | 50 | 1.0 (0.7-1.6) | 16 | 1.4 (0.7-2.6) | 18 | 1.5 (0.8-2.6) | 6 | 0.6 (0.2-1.4) | ||||
| ≥3.11 | 53 | 69 | 1.4 (0.9-2.1) | 0.61 | 27 | 2.3 (1.3-3.9) | 0.004 | 16 | 1.3 (0.7-2.4) | 0.49 | 12 | 1.0 (0.5-2.0) | 0.89 |
| Association per 1-unit increase |
454 | 446 | 1.06 (0.99-1.12) | 0.09 | 130 | 1.12 (1.04-1.22) | 0.005 | 124 | 1.04 (0.95-1.15) | 0.37 | 89 | 0.95 (0.84-1.08) | 0.47 |
| EBV DNAb | |||||||||||||
| Detected (vs. not) | 221 (211) | 198 (225) | 0.8 (0.6-1.1) | 58 (68) | 0.8 (0.5-1.2) | 60 (55) | 1.0 (0.7-1.6) | 40 (41) | 1.0 (0.6-1.5) | ||||
| Not detected | 211 | 225 | Referent | 68 | Referent | 55 | Referent | 41 | Referent | ||||
| ≤2000 | 184 | 149 | 0.7 (0.5-1.0) | 38 | 0.6 (0.4-1.0) | 43 | 0.9 (0.6-1.4) | 34 | 1.0 (0.6-1.6) | ||||
| >2000 | 37 | 49 | 1.6 (0.9-2.7) | 0.13 | 20 | 1.7 (0.9-3.1) | 0.06 | 17 | 1.8 (0.9-3.4) | 0.07 | 6 | 0.9 (0.3-2.2) | 0.75 |
| Association per doubling of level |
432 | 423 | 1.00 (0.97-1.02) | 0.65 | 126 | 1.00 (0.97-1.03) | 0.77 | 115 | 1.01 (0.98-1.05) | 0.41 | 81 | 1.00 (0.96-1.04) | 0.96 |
Adjusted for matching factors (age, US region, and blood draw date)
Restricted to subjects testing seropositive for VCA
Limited to those with detected values
Seropositivity for antibody to the EBV EBNA1 antigen (with a frequency of 93.4% in controls with established infection) was associated with decreased risk of NHL, with ORs of 0.4-0.6 for all B-cell NHL and each of the subtypes (or conversely, risk increases with seronegativity, with ORs of 1.7-2.5). There was no evidence of heterogeneity in the magnitude these risk estimates for the three major subtypes, CLL/SLL/PLL, DLBCL, or FL. The risk decrease with anti-EBNA1 seropositivity was similar or strengthened with exclusion of women reporting immune-related medical conditions or medications (not shown). There was no association between the level of antibody to EBNA1 antigen and risk of NHL.
There was a weak suggestion of increased B-cell NHL risk associated with seropositivity for the EBV EA-D antigen (Table 2), with non-significant 1.4-fold increased risk for antibody level in the highest tertile versus not detected. Of the subtypes, CLL/SLL/PLL was most strongly associated with anti-EA-D, with significantly increased risks observed for seropositivity (OR=1.8 vs. seronegative), antibody level in the highest tertile (OR=2.3 vs. seronegative; p-trend across tertiles=0.004), and the continuous measure (OR=1.12 per unit increase in EA-D index value; p-value for linear fit=0.005). There was significant heterogeneity in the magnitude of risk estimates among the major NHL subtypes for anti-EA-D positivity (p=0.02) and the anti-EA-D continuous measure (p=0.047), and further pairwise comparisons revealed important differences in risk estimates between CLL/SLL/PLL and FL for anti-EA-D positivity (p=0.004) and continuous EA-D (p=0.02). The associations of anti-EA-D with CLL/SLL/PLL were robust to exclusion of women with immune-related medical conditions and medications (not shown), and were only present in older women. For example, we observed increased risk associated with the highest tertile anti-EA-D level in women ≥65 years of age (OR=3.5; 95% CI: 1.8-6.9) and a significant trend of increasing risk across tertiles (p-trend=0.0003), but found no association in women <65 years old (OR=1.0; p-trend=0.97).
EBV DNA load was weakly associated with all B-cell NHL (Table 2), with 1.6-fold non-significantly increased risk associated with greater than 2000 genome copies/106 cells compared to no detection. CLL/SLL/PLL and DLBCL were the subtypes with the highest risk estimates for this comparison, with ORs of 1.7 and 1.8 and borderline significant trends across EBV DNA level categories (p-trend=0.06 and 0.07, respectively); nevertheless, there was not significant heterogeneity in risk among the major subtypes. Upon exclusion of study subjects reporting immune-related medical conditions or medications, the associations of EBV DNA measures with DLBCL were somewhat strengthened and exhibited a trend of increasing risk across EBV DNA load categories (p=0.05), whereas those for CLL/SLL/PLL were diminished (not shown).
The associations we observed between CLL/SLL/PLL and EBV measures were strongest for cases with the shortest time interval between blood draw and diagnosis. For example, we observed associations for the shortest (<4 years) and longest (≥8 years) intervals, respectively, of an OR of 0.3 versus 0.7 with anti-EBNA1 seropositivity (Figure 1), 3.5 versus 1.7-fold increased risk with anti-EA-D level in the highest tertile (Figure 2), and 2.0 versus 1.5-fold increased risk with EBV DNA >2000 copies/106 cells (Figure 3). As noted, the only EBV measure associated with FL was anti-EBNA1 seropositivity. This association was present for cases in all three prediagnostic intervals (Figure 1), but was strongest for cases with ≥8 years since blood draw, with an OR of 0.3 (95% CI: 0.1-1.0).
Figure 1.

Associations between EBNA1 seropositivity and risk of NHL subtypes (OR and 95% CI), with cases categorized by decreasing time interval between blood draw and diagnosis (circle: ≥8 years; triangle: 4-7 years; square: <4 years)
Figure 2.
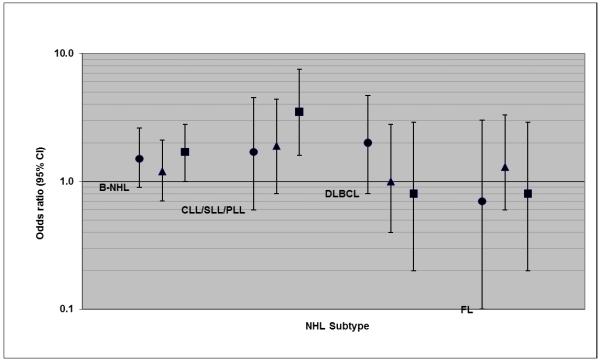
Associations between IgG antibody to EA-D level (highest tertile versus seronegative) and risk of NHL subtypes (OR and 95% CI), with cases categorized by decreasing time interval between blood draw and diagnosis (circle: ≥8 years; triangle: 4-7 years; square: <4 years)
Figure 3.
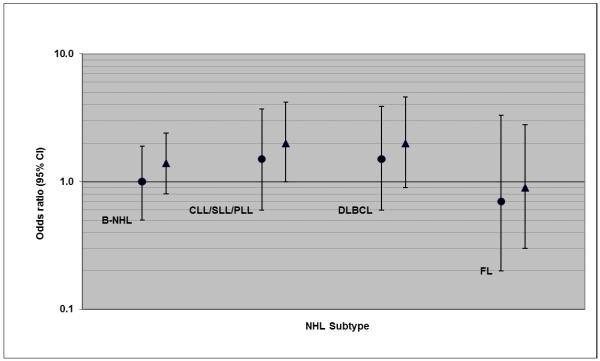
Associations between EBV DNA level (>2000 genome copies per 106 cells versus none detected) and risk of NHL subtypes (OR and 95% CI), with cases categorized by decreasing time interval between blood draw and diagnosis (circle: ≥4 years; triangle: <4 years)
DISCUSSION
We hypothesized that poorly-controlled EBV infection, leading to reactivation, increases the risk of B-cell NHL in the general population. The prospective design of the WHI OS and the banked samples offered a valuable resource to measure EBV markers in prediagnostic samples in a sizable study of postmenopausal women. We found that seropositivity to EBNA1 was associated with decreased risk of B-cell NHL, overall and for each of the histologic subtypes examined. However, we found no association between the levels of antibody to EBNA1 or VCA and NHL risk. Increased risks of NHL subtypes were observed with higher levels of antibody to EA-D (CLL/SLL/PLL) and EBV DNA (CLL/SLL/PLL and DLBCL), both markers reflective of active EBV infection. There are several possible scenarios that could have produced the associations we observed, including: 1) Poor control of EBV infection is a cause of certain NHL subtypes in the general population, in particular, CLL/SLL/PLL; 2) Underlying immune dysfunction, which is correlated with (and may cause) loss of control of EBV infection, is a cause of NHL in apparently immune competent women (as these results were robust to exclusion of women reporting immune-related conditions or medications); in this scenario, an association with EBV measures is due to confounding by subclinical, underlying immune dysfunction; and, 3) The associations with EBV measures are due to reverse causation, in that immune dysfunction from undiagnosed NHL caused EBV reactivation rather than EBV reactivation playing a causal role in lymphomagenesis; this scenario is supported by our finding that the associations with CLL/SLL/PLL were strongest for cases with the shortest time interval between blood draw and diagnosis. Although none of these scenarios can be proven from the data at hand, we provide further interpretation, below.
We found that women who were seropositive for IgG to EBNA1 (93.4% of control women with evidence of established EBV infection) were at decreased risk of developing B-cell NHL or, conversely, seronegativity to EBNA1 was associated with increased risk. Two previous cohort studies found non-significant reduced risks associated with EBNA seropositivity (10, 14). EBNA1 is expressed in all latently infected cells and is required for maintenance of EBV genome in the B cell as a circular DNA episome (4). Although anti-EBNA1 levels have been shown to be elevated in chronic EBV reactivation and in certain types of cancer (19), there is also evidence that EBNA1 expression may be lost when a lytic (replicative) cycle ensues. For example, transplant recipients undergoing immunosuppressive drug therapies (a scenario in which EBV reactivation is common) can fail to produce detectable antibodies to EBV antigens (22). Therefore, it is possible that the increased risk of NHL associated with EBNA1 seronegative status relates to a shift to EBV activation. Alternatively, it is possible that the presence of antibody to EBNA1 may simply reflect a more robust immune system, resulting in more efficient regulation of EBV infection and also improved means to prevent lymphomagenesis (whether caused by EBV or other factors). This alternative explanation is aligned with our results for the FL subtype, for which we observed a strongly reduced risk associated with EBNA1 seropositivity, but no association with any of the other measures of active EBV infection.
Similar to other studies on this topic conducted in general population samples, we found the most consistent EBV-NHL associations for the CLL/SLL/PLL subtype. A large study nested in the Physicians’ and Nurses’ Health Study cohorts (N=340 cases) found no evidence that antibodies to EBV antigens were associated with NHL overall, although there were some risk increases observed for CLL/SLL in relation to elevated anti-EBNA2 titer (typically seen in persistently active EBV infection) and lower EBNA1/EBNA2 ratio (16). A history of infectious mononucleosis (caused by delayed primary EBV infection) was associated with increased risk of NHL in an analysis of InterLymph consortium studies, and the strongest association was observed for the CLL/SLL/PLL/MCL subtype (30). In a study that used immunoblot analysis to characterize antibody diversity patterns, an abnormal EBV serologic pattern was found among CLL/SLL cases (40% of cases vs. 18% of controls), but not in other NHL subtypes (31). The predominance of CLL/SLL in these general population findings differs from the subtype spectrum of EBV-related lymphomas observed in immune-compromised populations – such as DLBCL and T-cell lymphomas (but not CLL or FL) in solid-organ transplant recipients (32) and Burkitt lymphoma, DLBCL, and T-cell lymphomas in HIV/AIDS (8). Because most CLL/SLL/PLL are EBV-negative neoplasms, a causal mechanism for EBV is hypothesized to involve stimulation of B-cell proliferation and/or induced expression of molecules with known potential for causing oncogenic mutations (i.e., a ‘hit-and-run’ mechanism), such as AID and pol-η (33-35). In our study, associations of CLL/SLL/PLL with anti-EA-D and EBV DNA were stronger for cases with a shorter time interval between blood draw and diagnosis, suggesting either that EBV acts as a late stage carcinogen for CLL/SLL/PLL or that the association may be due to reverse causastion, from EBV reactivation in response to underlying immune dysregulation that typically precedes CLL/SLL diagnosis. Features of immune dysregulation in CLL patients include decreased immunoglobulin production and antibody response, cytokine production abnormalities, abnormal immunoregulatory T-cell function, and diminished natural killer cell function (36); however, the length of time that such immune impairment precedes diagnosis is not well described. CLL/SLL is preceded in virtually all instances by small, preclinical clonal lymphocyte expansions termed “monoclonal B-cell lymphocytosis” (MBL) (37, 38), indicating an early pathogenic process, and use of such a biomarker to exclude persons with early signs of disease may be useful in future studies to help tease out a causal role of EBV.
The associations we observed for anti-EBNA1 with all B-NHL and anti-EA-D with CLL/SLL/PLL persisted upon exclusion of women with various immune-related medical conditions and medications (i.e., when limited to apparently immune-competent women). This subanalysis decreases our concern about false positive results from confounding by immune-related conditions/medications in which EBV reactivation is frequent and which are independently associated with increased NHL risk. Nevertheless, more subtle, subclinical immune dysregulation may still be a potential, underlying risk factor for NHL. The association we observed between anti-EA-D and CLL/SLL/PLL was limited to older women (≥65 years), and was strong among this subgroup. This may imply a synergistic effect between EBV and age-related biologic changes, or it may suggest confounding by other features of subclinical immune senescence in older women.
Our study was the largest to date on EBV infection in relation to NHL in a general population setting. The sample size enabled evaluation of risk for major subtypes of NHL, in addition to further stratification of case subtypes according to the length of time between blood draw and NHL diagnosis. In balance, the associations we observed do not provide strong evidence of EBV playing a causal role in B-cell NHL in our study population of general population, postmenopausal women. Further research within this cohort and in other studies may guide interpretation of findings for CLL/SLL/PLL. Characterization of cases by preclinical disease markers (such as MBL) to exclude persons with early disease may help rule out reverse causation in future studies. In addition, investigation of prediagnostic features of immune impairment and their timing in correspondence with EBV measures may provide evidence for EBV as a causal agent rather than a response to a dysregulated immune state.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A listing of WHI investigators can be found at https://cleo.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf.
Financial support: This WHI ancillary study was funded through a contract between the Fred Hutchinson Cancer Research Center and the National Heart, Lung, and Blood Institute, under the Broad Agency Announcement mechanism (contract HHSN268200900008C, Project: “Markers of B-Cell Stimulation as Potential Predictors of Non-Hodgkin Lymphoma”). This work was carried out, in part, in the facilities of the UCLA AIDS Institute, which were supported, in part, by funds from the James B. Pendleton Charitable Trust and the McCarthy Family Foundation. The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C.
Footnotes
Conflicts of interest: The authors have no conflicts of interest to report.
REFERENCES
- 1.Ambinder R. Infection and lymphoma. N Engl J Med. 2003;349(14):1309–11. doi: 10.1056/NEJMp030083. [DOI] [PubMed] [Google Scholar]
- 2.Khanna R, Burrows SR. Role of cytotoxic T lymphocytes in Epstein-Barr virus-associated diseases. Annu Rev Microbiol. 2000;54:19–48. doi: 10.1146/annurev.micro.54.1.19. [DOI] [PubMed] [Google Scholar]
- 3.Hardie DR. Human gamma-herpesviruses: a review of 2 divergent paths to oncogenesis. Transfus Apher Sci. 2010;42(2):177–83. doi: 10.1016/j.transci.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343(7):481–92. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- 5.Glaser R, Friedman SB, Smyth J, Ader R, Bijur P, Brunell P, et al. The differential impact of training stress and final examination stress on herpesvirus latency at the United States Military Academy at West Point. Brain Behav Immun. 1999;13(3):240–51. doi: 10.1006/brbi.1999.0566. [DOI] [PubMed] [Google Scholar]
- 6.Glaser R, Kiecolt-Glaser JK, Speicher CE, Holliday JE. Stress, loneliness, and changes in herpesvirus latency. J Behav Med. 1985;8(3):249–60. doi: 10.1007/BF00870312. [DOI] [PubMed] [Google Scholar]
- 7.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350(13):1328–37. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 8.Ambinder RF. Epstein-Barr virus associated lymphoproliferations in the AIDS setting. Eur J Cancer. 2001;37(10):1209–16. doi: 10.1016/s0959-8049(01)00123-x. [DOI] [PubMed] [Google Scholar]
- 9.Adam P, Bonzheim I, Fend F, Quintanilla-Martinez L. Epstein-Barr virus-positive diffuse large B-cell lymphomas of the elderly. Adv Anat Pathol. 2011;18(5):349–55. doi: 10.1097/PAP.0b013e318229bf08. [DOI] [PubMed] [Google Scholar]
- 10.Mueller NE, Mohar A, Evans A. Viruses other than HIV and non-Hodgkin’s lymphoma. Cancer Res. 1992;52(19 Suppl):5479s–81s. [PubMed] [Google Scholar]
- 11.Kuppers R. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol. 2003;3(10):801–12. doi: 10.1038/nri1201. [DOI] [PubMed] [Google Scholar]
- 12.Helmut NH, Salamon D, Ilg K, Koroknai A, Banati F, Schwarzmann F, et al. EBV-associated neoplasms: alternative pathogenetic pathways. Med Hypotheses. 2004;62(3):387–91. doi: 10.1016/j.mehy.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Ambinder RF. Gammaherpesviruses and “Hit-and-Run” oncogenesis. Am J Pathol. 2000;156(1):1–3. doi: 10.1016/S0002-9440(10)64697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehtinen T, Lumio J, Dillner J, Hakama M, Knekt P, Lehtinen M, et al. Increased risk of malignant lymphoma indicated by elevated Epstein-Barr virus antibodies--a prospective study. Cancer Causes Control. 1993;4(3):187–93. doi: 10.1007/BF00051312. [DOI] [PubMed] [Google Scholar]
- 15.Rothman N, Cantor KP, Blair A, Bush D, Brock JW, Helzlsouer K, et al. A nested case-control study of non-Hodgkin lymphoma and serum organochlorine residues. Lancet. 1997;350(9073):240–4. doi: 10.1016/S0140-6736(97)02088-6. [DOI] [PubMed] [Google Scholar]
- 16.Bertrand KA, Birmann BM, Chang ET, Spiegelman D, Aster JC, Zhang SM, et al. A prospective study of Epstein-Barr virus antibodies and risk of non-Hodgkin lymphoma. Blood. 2010;116(18):3547–53. doi: 10.1182/blood-2010-05-282715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richiardi L, De ML, Gillio-Tos A, Merletti F, Fiano V, Palli D, et al. Persistent infection by HCV and EBV in peripheral blood mononuclear cells and risk of non-Hodgkin’s lymphoma. Cancer Epidemiol. 2010;34(6):709–12. doi: 10.1016/j.canep.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Linde A. Diagnosis of Epstein-Barr virus-related diseases. Scand J Infect Dis Suppl. 1996;100:83–8. [PubMed] [Google Scholar]
- 19.Lennette ET, Rymo L, Yadav M, Masucci G, Merk K, Timar L, et al. Disease-related differences in antibody patterns against EBV-encoded nuclear antigens EBNA 1, EBNA 2 and EBNA 6. Eur J Cancer. 1993;29A(11):1584–9. doi: 10.1016/0959-8049(93)90299-u. [DOI] [PubMed] [Google Scholar]
- 20.Henle W, Henle G. Epstein-Barr virus-specific serology in immunologically compromised individuals. Cancer Res. 1981;41(11 Pt 1):4222–5. [PubMed] [Google Scholar]
- 21.Aalto SM, Juvonen E, Tarkkanen J, Volin L, Haario H, Ruutu T, et al. Epstein-Barr viral load and disease prediction in a large cohort of allogeneic stem cell transplant recipients. Clin Infect Dis. 2007;45(10):1305–9. doi: 10.1086/522531. [DOI] [PubMed] [Google Scholar]
- 22.Andreone P, Gramenzi A, Lorenzini S, Biselli M, Cursaro C, Pileri S, et al. Posttransplantation lymphoproliferative disorders. Arch Intern Med. 2003;163(17):1997–2004. doi: 10.1001/archinte.163.17.1997. [DOI] [PubMed] [Google Scholar]
- 23.Baldanti F, Grossi P, Furione M, Simoncini L, Sarasini A, Comoli P, et al. High levels of Epstein-Barr virus DNA in blood of solid-organ transplant recipients and their value in predicting posttransplant lymphoproliferative disorders. J Clin Microbiol. 2000;38(2):613–9. doi: 10.1128/jcm.38.2.613-619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Women’s Health Initiative Study Group Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials. 1998;19(1):61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 25.International Classification of Diseases for Oncology. 3rd Edition World Health Organization; Geneva: 2000. [Google Scholar]
- 26.Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107(1):265–76. doi: 10.1182/blood-2005-06-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morton LM, Turner JJ, Cerhan JR, Linet MS, Treseler PA, Clarke CA, et al. Proposed classification of lymphoid neoplasms for epidemiologic research from the Pathology Working Group of the International Lymphoma Epidemiology Consortium (InterLymph) Blood. 2007;110(2):695–708. doi: 10.1182/blood-2006-11-051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawrence JB, Villnave CA, Singer RH. Sensitive, high-resolution chromatin and chromosome mapping in situ: presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell. 1988;52(1):51–61. doi: 10.1016/0092-8674(88)90530-2. [DOI] [PubMed] [Google Scholar]
- 29.Breslow NE, Day NE. The Analysis of Case-Control Studies. Vol. 1. Lyon; France: 1980. Statistical Methods in Cancer Research. [PubMed] [Google Scholar]
- 30.Becker N, Falster MO, Vajdic CM, de SS, Martinez-Maza O, Bracci PM, et al. Self-reported history of infections and the risk of non-Hodgkin lymphoma: An InterLymph pooled analysis. Int J Cancer. 2012 doi: 10.1002/ijc.27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Sanjose S, Bosch R, Schouten T, Verkuijlen S, Nieters A, Foretova L, et al. Epstein-Barr virus infection and risk of lymphoma: immunoblot analysis of antibody responses against EBV-related proteins in a large series of lymphoma subjects and matched controls. Int J Cancer. 2007;121(8):1806–12. doi: 10.1002/ijc.22857. [DOI] [PubMed] [Google Scholar]
- 32.Clarke CA, Morton LM, Lynch C, Pfeiffer RM, Hall EC, Gibson TM, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer. 2013 doi: 10.1038/bjc.2013.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epeldegui M, Hung YP, McQuay A, Ambinder RF, Martinez-Maza O. Infection of human B cells with Epstein-Barr virus results in the expression of somatic hypermutation-inducing molecules and in the accrual of oncogene mutations. Mol Immunol. 2007;44(5):934–42. doi: 10.1016/j.molimm.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Epeldegui M, Vendrame E, Martinez-Maza O. HIV-associated immune dysfunction and viral infection: role in the pathogenesis of AIDS-related lymphoma. Immunol Res. 2010;48(1-3):72–83. doi: 10.1007/s12026-010-8168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He B, Raab-Traub N, Casali P, Cerutti A. EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J Immunol. 2003;171(10):5215–24. doi: 10.4049/jimmunol.171.10.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dasanu CA. Intrinsic and treatment-related immune alterations in chronic lymphocytic leukaemia and their impact for clinical practice. Expert Opin Pharmacother. 2008;9(9):1481–94. doi: 10.1517/14656566.9.9.1481. [DOI] [PubMed] [Google Scholar]
- 37.Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360(7):659–67. doi: 10.1056/NEJMoa0806122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rawstron AC, Bennett FL, O’Connor SJ, Kwok M, Fenton JA, Plummer M, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359(6):575–83. doi: 10.1056/NEJMoa075290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.