

Abstract
Introduction
Cowden syndrome (CS) is a hereditary cancer syndrome associated with a germline mutation in PTEN. Patients are predisposed to multiple malignancies including renal cell carcinoma (RCC).
Methods
Patients with CS were evaluated as part of a clinical protocol. Those with a history of RCC underwent review of clinical features, tumor characteristics, and family history. Renal tumors were evaluated for loss of heterozygosity (LOH).
Results
Among 24 CS patients, 4 were identified with RCC (16.7%). Three patients had solitary tumors, two with papillary type I histology and one with clear cell histology. The fourth patient had bilateral, synchronous chromophobe tumors. No patients had a prior family history of RCC. All RCC patients had dermatologic manifestations of CS and had macrocephaly. LOH at the PTEN mutation was identified in 4 tumors (80%). No genotype-phenotype association was found, as the same mutation was identified in different RCC histologies.
Conclusion
RCC is an underappreciated feature of CS. As most patients lack a prior family history or a distinctive RCC histology, recognition of the associated non-renal features should target referral for genetic counseling. PTEN LOH is common in CS renal tumors. Because loss of PTEN can activate mTOR and mTOR inhibitors are FDA-approved to treat RCC, these agents have clinical potential in RCC associated with CS.
Keywords: PTEN, RCC, Cowden syndrome, hereditary, mTOR
Introduction
The genetic basis of renal cell carcinoma (RCC) continues to be elucidated, with over a dozen genes implicated in the development of renal tumors.1,2 Approximately 4-6% of RCC is considered to have a hereditary component, which can often be attributed to a single germline alteration. Several of these conditions, including Von-Hippel Lindau (VHL), Hereditary Papillary RCC (HPRC), Birt-Hogg-Dube (BHD), and Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC), have improved our understanding of sporadic kidney tumors. The genetic basis of many patients with familial renal cancer (FRC), bilateral, multifocal or early-onset RCC has yet to be determined. With increasing availability of next generation sequencing technologies (whole genome/exome sequencing) our understanding of the genetic basis of RCC is expanding, including a class of kidney cancers associated with the succinate dehydrogenase (SDH)3,4 and the MITF transcription factor.5
One rare hereditary cancer syndrome, Cowden syndrome (CS), or PTEN hamartoma tumor syndrome, has been recognized for the past half century.6 This syndrome is inherited in an autosomal dominant pattern with an estimated incidence of 1 in 200,000 individuals.7 Over a decade ago patients with this syndrome were found to have germline alterations in the PTEN gene.8 Over 70% of CS patients have germline mutations in PTEN. Patients affected with CS develop multiple hamartomas and are at increased risk for breast, endometrial, and thyroid cancers. (Table 1) Dermatologic manifestations such as acral keratosis and facial trichilemmomas are common in CS patients. A clinical diagnosis is made based on a combination of pathognomonic, major, and minor criteria (Table 1).9 Genitourinary tumors such as kidney and prostate cancer are believed associated with CS, with kidney cancer considered one of the diagnostic criteria. Mester and colleagues reported the first cohort of patients with CS associated RCC (CS-RCC) and estimated these patients had a >30 fold increased risk of developing kidney cancer.10 However, the overall incidence of RCC in CS was low (4.1%), and no patients had a prior history of RCC. Also unlike many of the other hereditary cancer syndromes such as VHL and HPRC, CS-RCC tumors in this series had variable histology, specifically both papillary and chromophobe types.
Table 1.
| Pathognomonic Characteristics | Major Criteria | Minor Criteria |
|---|---|---|
|
| ||
| Mucocutaneous Lesions | Breast Cancer | Benign Thyroid Conditions |
| Mucosal Lesions | Thyroid Cancer | Mental Retardation |
| Acral Keratoses | Endometrial Cancer | GI Harmatomas |
| Facial Trichilemmomas | Macrocephaly | Lipomas |
| Papillomatous Papules | Lhermitte-Dulcos Disease (LDD) | Fibromas |
| Renal Cell Carcinoma (RCC) | ||
| Breast Fibrocystic Disease | ||
| Diagnostic Criteria |
|
| Pathognomonic Characteristics | Major Criteria | Minor Criteria | |
|---|---|---|---|
| Patient 1 | Mucosal Lesions, Papillomatous Papules, Acral Keratoses | Macrocephaly | GI Harmatomas, RCC, Breast Fibrocystic Disease, Benign Thyroid |
| Patient 2 | Trichilemmomas, Mucosal Lesions, Papillomatous Papules, Acral Keratoses | Macrocephaly, Breast Cancer, Uterine Cancer | GI Harmatomas, RCC, Lipomas, Benign Thyroid |
| Patient 3 | Mucosal Lesions, Papillomatous Papules, Acral Keratoses | Macrocephaly, Lhermitte-Duclos Disease, Breast Cancer, Uterine Cancer | GI Harmatomas, RCC, Benign Thyroid |
| Patient 4 | Mucosal Lesions, Papillomatous Papules, Acral Keratoses | Macrocephaly, Lhermitte-Duclos Disease | GI Harmatomas, RCC, Benign Thyroid |
To improve the understanding of this hereditary kidney cancer syndrome; we review the clinical features, family history, tumor characteristics, and mechanism of tumorigenesis for CS-RCC.
Methods
Patients affected with CS were evaluated at the National Cancer Institute's (NCI) Center for Clinical Research (CCR) as part of enrollment in a therapeutic trial in the NCI Medical Oncology Branch (NCT00971789). On evaluation, patients' medical and family medical history, clinical manifestations, imaging and germline mutation testing were evaluated. All CS patients met clinical diagnostic criteria and had PTEN germline mutations confirmed using a Clinical Laboratory Improvement Amendments (CLIA) certified laboratory.9
Patients with a history of kidney cancer were further investigated to characterize the features of CS associated RCC. Family pedigrees were created to ascertain the pattern of inheritance and penetrance of kidney cancer. Renal tumor histologic slides were reviewed by a single genitourinary pathologist (MJM). To evaluate for PTEN loss of heterozygosity (LOH), microdissection was performed from unstained formalin fixed, paraffin embedded (FFPE) slides of the primary tumor. DNA was extracted from the FFPE tissue with the assistance of an Applied Biosciences RecoverAll kit (Austin, TX). For normal control, peripheral blood samples or normal kidney parenchyma were evaluated when available. For germline mutation analysis, peripheral blood DNA extraction was performed using a Promega/Maxwell 16 purification kit (Madison, WI). Quantification of extracted DNA was performed using a ThermoScientific Nanodrop 1000 (Wilmington, DE). A Qiagen PCR Kit (Germantown, MD) was used to amplify the sequence at site of the PTEN germline mutation using standard primers, and purified DNA products were sequenced bidirectionally using the Big Dye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and run on an ABI 3130xl Genetic Analyzer. For clear cell RCC cases, VHL mutation status was analyzed by sequencing all three exons (primer pairs for PTEN and VHL available on request). Amplified forward and reverse sequences were evaluated using Sequencher 4.10.01 (Ann Arbor, MI). LOH of PTEN at the mutation site was determined by comparing the tumor and normal control to the reference germline sequence.
Data from this cohort was combined with a recently published cohort to gain insight on distribution of histologic subtypes and genotype-phenotype correlations.10 For each patient, the PTEN mutation type (missense, nonsense, deletion, or insertion), exon location, and proximity to known regulatory site were reviewed in relationship to histology.
Results
During the period from 2008 to 2011, a total of 24 patients with CS were evaluated. Four patients (16.7%), 2 men and 2 women, were found to have a history of RCC. Of these four patients, no other family members had a history of RCC, despite other relatives having other types of cancer as well as features suggestive of CS (Figure 1). Patient 4 had previously presented with bilateral renal tumors, as described by Mester, et al.10, while the remaining three had solitary renal lesions (Figure 2). All patients had renal tumors treated surgically.
Figure 1. Cowden/PTEN Syndrome Family Pedigrees.

Cowden/PTEN Syndrome families (patients 1-4) are shown with blue segments representing the occurrence of any Cowden Syndrome diagnostic features (described with age of diagnosis) and red segments highlighting the occurrence of Renal Cell Carcinoma (RCC). Ca. – cancer; Pap. – papillary.
Figure 2. CT Imaging of Cowden/PTEN Syndrome Associated RCC (CS-RCC).
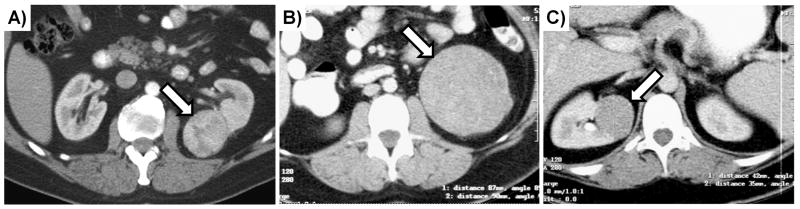
A) A contrast-enhanced CT scan of Patient 3 demonstrating a left-sided T1a clear cell RCC. B) & C) Contrast-enhanced CT scans of Patient 4 demonstrated bilateral, multifocal chromophobe RCC, a left-sided T2 and a right-sided T1b respectively.
All patients had renal tumors indistinguishable from established histologic subtypes.11 In our series, patients 1 and 2 had papillary type 1 RCC, patient 3 had clear cell RCC and patient 4 had bilateral chromophobe RCC (Figure 3a-d). Combining this series with the recent series reported by Mester, the first 11 CS associated RCC had papillary type I in 6 (54.5%), papillary type II in 2 (18.2%), chromophobe in 2 (18.2%), and clear cell in 1 (9.1%).10
Figure 3. Histology of Cowden/PTEN Syndrome Associated RCC (CS-RCC).

Patients 1 (A) and 2 (B) had papillary type 1 RCC, Patient 3 had clear cell RCC (C) and patient 4 had chromophobe RCC (D).
Each patient had evidence of macrocephaly, occipito-frontal head circumference measurement >97% for their sex and height (Figure 4A). Pathognomonic dermatologic manifestations were identified in each patient (Figure 4B,C). All patients had a confirmed germline alteration in PTEN and met clinical criteria for CS (Table 1). Pathologic staging demonstrated lesions were T1 or T2, N0M0 (Table 2). Currently no patient has evidence of recurrence (follow-up 1-8 years).
Figure 4. Clinical Manifestations (Head Circumference and Dermatologic Lesions) that may be seen on Physical Exam of Cowden/PTEN Syndrome Patients.
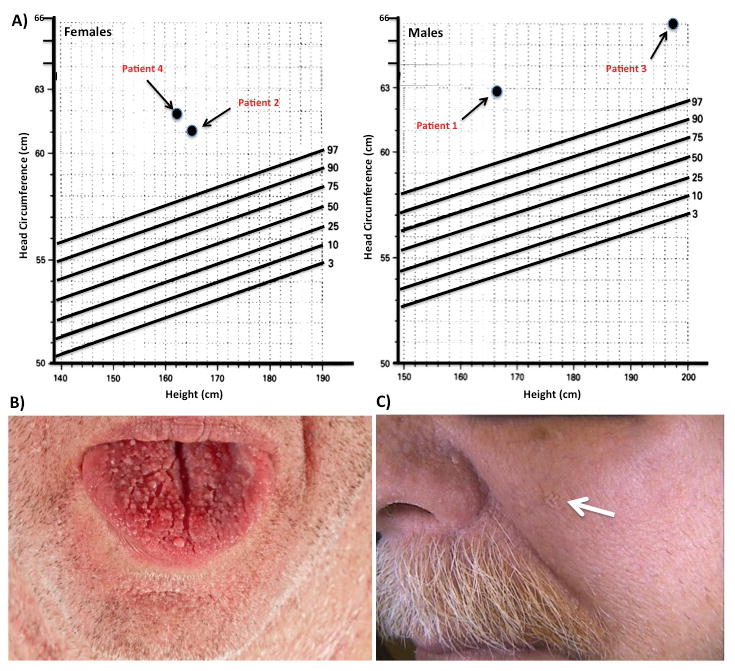
A) Measurement of occipito-frontal head circumferences demonstrated that all four patients had macrocephaly (>97% by height). Macrocephaly is considered major criteria for the diagnosis of Cowden Syndrome. (Adapted from Bushby, KM, et al., Arch. Dis. Child. 1992, 67:1286-1287). A variety of dermatologic manifestations are pathopneumonic for Cowden Syndrome present in Patient #1 B) Oromucosal papillomatous papules creating a cobblestone appearance on the dorsal aspect of the patients tongue C) Cutaneous verroucous papule (white arrow) located at its characteristic distribution, over the central portion of the face.
Table 2.
The diagnosis, genetics and management of the Cowden/PTEN Syndrome Associated RCC (CS-RCC) in our patient cohort.
| Patient | Sex | Age | Diagnosis | Size | Mutation | TNM Stage | Histology | Nephrectomy | Follow-Up |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 53 | Incidental | 1.0cm | c.150delT, p.I50MfsX4 | T1aN0M0 | Papillary Type 1 | Radical | NED 5 yrs |
| 2 | F | 57 | Incidental | 1.3cm | c.70G>C, p.D24H | T1aN0M0 | Papillary Type 1 | Partial | NED 8 yrs |
| 3 | M | 56 | Incidental | 3.0cm | c.388C>T, p.R130X | T1aN0M0 | Clear Cell | Partial | NED 1 yrs |
| 4 | F | 32 | Symptomatic | 9.0cm 4.5cm |
c.388C>T, p.R130X c.388C>T, p.R130X |
T2N0M0 T1bN0M0 |
Chromophobe Chromophobe |
Radical Partial |
NED 7 yrs |
NED – No Evidence of Disease
Review of germline PTEN mutations in demonstrated that missense, nonsense, and splice site mutations were associated with RCC development (Figure 5A). Several exons were involved, including both functional domains (phosphatase tensin-type and the C2 tensin-type). Specific germline mutations could not be associated with distinct histologic types. Notably, one nonsense mutation in exon 5 (p.R130X) was associated with three tumors in three different individuals that presented with three different histologies (one chromophobe, one papillary type I, and one clear cell) (Figure 5A).
Figure 5. PTEN mutation map for CS-RCC and Evidence of PTEN loss of heterozygosity (LOH).

A) A schematic of the PTEN demonstrates the 9 coding exons (fat blue boxes) and the 5′ and 3′ UTRs (thin blue boxes) with the known functions domains shown below linked to their relevant coding exons (P60484 – http://www.uniprot.org/). The mutations from this report (red arrows) were combined with the recent series reported by Mester and colleagues10 (white arrows) and mapped to the PTEN gene with annotation of the reviewed pathology. Pap I – Papillary Type 1, Pap II – Papillary Type 2, Chrom – Chromophobe, Clear – Clear Cell.
B) The loss of heterozygosity (LOH) was assessed in all tumors and two examples are shown here. The left panel demonstrates normal control sequence followed by heterozygous deletion of a T in the blood DNA of Patient 1, creating a frameshift, and LOH of the frameshift in the tumor DNA. The right panel demonstrates normal control sequence followed by heterozygous substitution of a C for a T in the blood DNA of Patient 4 and LOH of this mutation in the DNA of both the assessed tumors.
LOH of PTEN by PCR was found in 4 of the 5 tumors (80%), including one example from each RCC histology (Figure 5B). The wild-type allele was retained in the remaining tumor (patient 2, papillary type I). However, other mechanisms that could account for loss of wild-type PTEN function in this sample such as epigenetic silencing or an additional mutation were not evaluated. Analysis of VHL status in the clear cell tumor demonstrated a somatic mutation (c.404delT in exon 2, causing a p.Leu135fs) in a single allele, but no evidence of LOH.
Discussion
Clinicians managing individuals with kidney cancer should be familiar with the hereditary RCC syndromes, as the role of inherited predisposition to kidney cancer is most likely underestimated.12 The diagnosis of an inherited form of RCC is important in the management of not only the patient, but also for members of the family who may be disease gene carriers. In our hereditary kidney cancer program, affected individuals as well as at-risk family members are routinely screened for the detection of altered germline kidney cancer susceptibility genes, as potentially aggressive renal tumors cancers can be detected at a treatable stage. In addition to RCC screening, other syndromic manifestations can be evaluated and managed. Quality of life may be improved by early detection of disease manifestations, best exemplified by early detection of VHL-associated retinal and endolymphatic sac tumors, respectively.
CS may be a challenging diagnosis for urologic oncologists due to the complexity of a clinical diagnosis that requires a combination of pathognomonic, major, and minor criteria (Table 1).9 Family history of RCC may not provide an indication of a hereditary component for CS, as patients in the present series, as well as those reported by Mester et al. had no additional family members with RCC.10 While a family history may allow recognition of a hereditary syndrome, at times it may not. This scenario may occur more frequently in CS than with other hereditary cancer syndromes as the rate of de novo germline PTEN is estimated to be between 10.7-47.6%.13 Therefore, clinicians should be aware that family history of RCC or other cancers does not exclude individuals from CS. Those individuals with a personal history or clinical manifestations should be considered for genetic counseling. To aid clinicians and genetic counselors with the identification of patients with CS, a useful nomogram has been developed to predict the presence of a PTEN germline alteration (http://www.lerner.ccf.org/gmi/ccscore/).14
In our series we observed a higher CS-RCC penetrance (16.7%) than that reported by Mester and colleagues (4.1%).10 This higher incidence is possibly due to our referral patterns selecting for individuals with more prominent manifestations who may be candidates for a therapeutic trial. CS-RCC penetrance is significantly lower than the kidney cancer penetrance in other hereditary RCC syndromes such as HPRC, where nearly 100% of patients are predicted to develop RCC by their ninth decade.15 Previous reports of CS-RCC demonstrated an association with chromophobe and papillary RCC. In the current study we determined that numerous histologic subtypes, including clear cell can occur in CS-RCC, in contrast with a number of other hereditary RCC syndromes that have histology-specific phenotypes (e.g., VHL, HPRC).
The genes associated with RCC appear to be linked to common metabolic pathways with PTEN positioned at a central regulatory site (Figure 6). Similar to other hereditary cancer syndromes associated with tumor suppressor genes (TSC1, TSC2, FLCN), dysregulation at upstream regulatory points may be associated with a more variable histology16,17 Those syndromes associated with tumor suppressor genes responsible for regulatory proteins at distal positions in the RCC metabolic pathway (VHL, FH, SDHB, SDHC) may have less variable histology.18-20
Figure 6. The Kidney Cancer Gene Pathways.

Kidney cancer is not a single disease; it is made up of a number of different types of cancer with different histologies, having different clinical courses, responding differently to therapy and caused by different genes (e.g., VHL, MET, FLCN, FH, SDHB/C/D, TFE3, TFEB, MITF, TSC1, TSC2 and PTEN).2,29 The PTEN gene, which is frequently found to be mutated in the germline of patients affected with Cowden Syndrome, encodes for a plasma membrane lipid phosphatase that inhibits the PI3K signaling pathway. Loss of PTEN leads to accumulation of phosphatidlyinositol 3,4,5 triphosphate (PIP3), which activates AKT.30 Activation of AKT affects the TSC1/2 pathway, resulting in activation of the mTOR pathway. (Adapted from Linehan.2)
With some kidney cancer hereditary syndromes, a genotype-phenotype correlation may depend on mutation type and location, best exemplified by the VHL mutations; where genotype-phenotype association in VHL patients correlates with propensity to develop pheochromocytomas in patients with missense mutations (Type II VHL). Our preliminary observations do not suggest a correlation of germline mutation with specific histology in CS-RCC. For example, the same PTEN mutation (p.R130X) was associated with chromophobe, papillary, and clear RCC (Figure 5B). Similarly, two other germline PTEN mutations were associated with both type I and type II papillary RCC (p.D24H and p.I50MfsX4). While other genetic modifiers may contribute to an individual's development of a particular histology, in syndromes such as BHD, several histologic types can be observed with a single genotype, even within the same kidney.21 Additional genetic events of key regulatory genes may push a tumor towards a particular phenotype. An event such as the loss of one copy of VHL could account for a clear cell phenotype, as observed with BHD cell line UOK257, derived from a clear cell BHD renal tumor where chromosome 3p is deleted.22 While complete loss of functional VHL is present in the majority of sporadic and hereditary clear cell RCC, this may not be necessary in other hereditary syndromes. Similar to the UOK 257 model, our clear cell CS-RCC did not have detectable LOH despite a VHL mutation. While this finding is not consistent with the traditional Knudson two-hit model for VHL, it raises the question that perhaps cumulative alterations in an overlapping or different metabolic pathway can cooperate to bring about dysregulation of RCC gene pathways, resulting in RCC tumorigenesis. Several preclinical models support cooperative dysregulation between PTEN and VHL, with liver hemangioblastomas and renal cysts resulting from mutation of both genes, but not either alone.23,24 Despite the two-hit model generally considered to have both genetic events happening in consecutive order, recent evidence suggests that haploinsufficiency in tumor suppressor genes can facilitate other genetic events prior to the development of a second hit.25 Along these lines, in CS-RCC, PTEN haploinsufficiency could coordinate with secondary events to promote transformation down separate histologic pathways. This requirement for additional genetic alterations for tumorigenesis could also explain the low penetrance of CS-RCC.
Examining the mechanism associated with the development of CS-RCC as an example of hereditary RCC could improve our understanding of sporadic RCC. Although PTEN expression is commonly decreased in RCC, complete loss occurs in less than 10% of sporadic RCC.26 Mester and colleagues found loss of PTEN by immunohistochemistry in all patients evaluated with CS-RCC.10 The precise mechanism causing the loss of protein expression was not elucidated, but several mechanisms could underlie the second hit such as LOH, somatic mutation or promoter hypermethylation of the wild-type allele. We demonstrate that LOH, present in four of five of our tumor samples, may be a common mechanism associated with RCC tumorigenesis in with this disease.10 The remaining tumor may have a secondary event in the PTEN wild-type allele, however we did not perform full exon sequencing or epigenetic analysis.
Despite a low incidence of PTEN mutations, sporadic clear and non-clear renal tumors frequently have activation of the PI3K/AKT pathway, the downstream targets of PTEN. Targeting this pathway with mTOR agents has shown clinical benefit and 2 agents (everolimus and temsirolimus) are approved for clinical use in RCC. For clear cell RCC, the efficacy of these agents is believed related both to inhibition of the PI3K/AKT pathway as well as its known inhibitory effect on HIF1 translation. In patients with advanced sporadic RCC treated with temsirolimus, pS6 and pAKT are potential biomarkers for response.27 As both pS6 and pAKT are dysregulated by the loss of PTEN, those with CS-RCC who develop advanced disease may also benefit from mTOR inhibition. Even for patients without metastatic disease, targeting the mTOR pathway in this population may serve as cancer chemoprevention, demonstrated in preclinical animal models.28
Detailed characterization on the renal cancer manifestations of a rare disease must be cautioned. The low incidence (1 in 200,000) of CS compounded with the low RCC penetrance limits definitive conclusions on disease biology. Genotype-phenotype relationships could greatly influence the aggressiveness and age of onset. Despite these recognized limitations, our series may aid recognition of clinical features associated with this hereditary cancer syndrome allowing genetic counseling referral and identification of non-urologic disease manifestations in the individuals and affected family members.
Conclusion
CS is a hereditary cancer syndrome associated with dermatologic manifestations and multiple types of cancers including breast, uterine, thyroid, and RCC. Patients often do not have a family history of RCC due to low disease penetrance and a high rate of de novo mutation. Recognition of associated cancers, dermatologic features, as well as macrocephaly should lead to consideration for genetic counseling. Classic histologic types including clear cell, chromophobe, and papillary RCC can be seen in CS-RCC. PTEN LOH is common in these tumors and may lead to activation of the mTOR pathway. Targeting this pathway may have an important therapeutic role for those with advanced CS-RCC and/or PTEN-deficient sporadic RCC.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors acknowledge the outstanding editorial and graphics support by Georgia Shaw.
References
- 1.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22:2089. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanharanta S, Buchta M, McWhinney SR, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 5.Bertolotto C, Lesueur F, Giuliano S, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 6.Lloyd KM, Dennis M. Cowden's disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963;58:136. doi: 10.7326/0003-4819-58-1-136. [DOI] [PubMed] [Google Scholar]
- 7.Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet. 1999;7:267. doi: 10.1038/sj.ejhg.5200289. [DOI] [PubMed] [Google Scholar]
- 8.Lynch ED, Ostermeyer EA, Lee MK, et al. Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am J Hum Genet. 1997;61:1254. doi: 10.1086/301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41:323. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mester JL, Zhou M, Prescott N, et al. Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. Urology. 2012;79:1187. doi: 10.1016/j.urology.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eble J, Sauter G, Epstein J. Pathology and Genetics. Lyon, France: International Agency for Research on Cancer; 2004. [Google Scholar]
- 12.Gudbjartsson T, Jonasdottir TJ, Thoroddsen A, et al. A population-based familial aggregation analysis indicates genetic contribution in a majority of renal cell carcinomas. Int J Cancer. 2002;100:476. doi: 10.1002/ijc.10513. [DOI] [PubMed] [Google Scholar]
- 13.Mester J, Eng C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet Med. 2012 doi: 10.1038/gim.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan MH, Mester J, Peterson C, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88:42. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt L, Junker K, Weirich G, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58:1719. [PubMed] [Google Scholar]
- 16.Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol. 2002;26:1542. doi: 10.1097/00000478-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 18.Gill AJ, Pachter NS, Chou A, et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol. 2011;35:1578. doi: 10.1097/PAS.0b013e318227e7f4. [DOI] [PubMed] [Google Scholar]
- 19.Merino MJ, Torres-Cabala C, Pinto P, et al. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578. doi: 10.1097/PAS.0b013e31804375b8. [DOI] [PubMed] [Google Scholar]
- 20.Ricketts CJ, Shuch B, Vocke CD, et al. Succinate dehydrogenase kidney cancer: an aggressive example of the warburg effect in cancer. J Urol. 2012;188:2063. doi: 10.1016/j.juro.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vocke CD, Yang Y, Pavlovich CP, et al. High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dube-associated renal tumors. J Natl Cancer Inst. 2005;97:931. doi: 10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Padilla-Nash HM, Vira MA, et al. The UOK 257 cell line: a novel model for studies of the human Birt-Hogg-Dube gene pathway. Cancer Genet Cytogenet. 2008;180:100. doi: 10.1016/j.cancergencyto.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Sanford CA, Sun J, et al. VHL and PTEN loss coordinate to promote mouse liver vascular lesions. Angiogenesis. 2010;13:59. doi: 10.1007/s10456-010-9164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frew IJ, Thoma CR, Georgiev S, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008;27:1747. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konishi H, Mohseni M, Tamaki A. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci U S A. 2011;108:17773. doi: 10.1073/pnas.1110969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abou YT, Fahmy MA, Koumakpayi IH, et al. The mammalian target of rapamycin pathway is widely activated without PTEN deletion in renal cell carcinoma metastases. Cancer. 2011;117:290. doi: 10.1002/cncr.25402. [DOI] [PubMed] [Google Scholar]
- 27.Cho D, Signoretti S, Dabora S, et al. Potential histologic and molecular predictors of response to temsirolimus in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2007;5:379. doi: 10.3816/CGC.2007.n.020. [DOI] [PubMed] [Google Scholar]
- 28.Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden's disease by mTOR inhibition with rapamycin. Cancer Res. 2008;68:7066. doi: 10.1158/0008-5472.CAN-08-0922. [DOI] [PubMed] [Google Scholar]
- 29.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature Reviews Urology. 2010;7:277. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]