

Abstract
Background
The calcium sensing receptor (CaSR) regulates serum calcium by suppressing secretion of parathyroid hormone; it also regulates renal tubular calcium excretion. Inactivating mutations of CaSR raise serum calcium and reduce urine calcium excretion. Thyroid C-cells (which make calcitonin) express CaSR and may, therefore, be regulated by it. Since calcium stimulates release of calcitonin, the higher blood calcium caused by inactivation of CaSR should increase serum calcitonin, unless CaSR mutations alter the responsiveness of calcitonin to calcium.
To demonstrate regulatory effects of CaSR on calcitonin release, we studied calcitonin responsiveness to calcium in normal and CaSR heterozygous-ablated (Casr+/-) mice. Casr+/- mice have hypercalcemia and hypocalciuria, and live normal life spans. Each mouse received either 500 μl of normal saline or one of two doses of elemental calcium (500 μmol/kg or 5 mmol/kg) by intraperitoneal injection. Ionized calcium was measured at baseline and 10 minutes, and serum calcitonin was measured on the 10 minute sample.
Results
At baseline, Casr+/- mice had a higher blood calcium, and in response to the two doses of elemental calcium, had greater increments and peak levels of ionized calcium than their wild type littermates. Despite significantly higher ionized calcium levels, the calcitonin levels of Casr+/- mice were consistently lower than wild type at any ionized calcium level, indicating that the dose-response curve of calcitonin to increases in ionized calcium had been significantly blunted or shifted to the right in Casr+/- mice.
Conclusions
These results confirm that the CaSR is a physiological regulator of calcitonin; therefore, in response to increases in ionized calcium, the CaSR inhibits parathyroid hormone secretion and stimulates calcitonin secretion.
Keywords: Animal models/rodent, calcitonin, familial hypocalciuric hypocalcemia, calcium receptor, gene knock-out
Background
Ionized calcium concentration of extracellular fluid is tightly regulated, subject to minute-to-minute control in order to maintain such diverse processes as cell membrane integrity, neuromuscular transmission and blood coagulation [1]. Small decreases in ionized calcium provoke excitability of the nervous system that can manifest as paresthesias and seizures; in contrast, increases in ionized calcium dampen nervous system responses and lead to such manifestations as constipation, obtundation, and coma. Parathyroid hormone (PTH) and calcitonin have long been recognized to respond quickly (but in opposite directions) to increases or decreases in ionized calcium [1-3].
The "homeostat" that controls ionized calcium has been recognized to be the calcium sensing receptor (CaSR), a G protein-coupled, seven transmembrane-spanning receptor that binds calcium ions [4]. CaSR mRNA and protein are expressed by parathyroid chief cells that produce PTH. Binding of calcium activates the CaSR; consequently, an increase in ionized calcium leads to more activation of the receptor and, thereby, inhibition of PTH secretion. Conversely, a fall in ionized calcium leads to less activation of the receptor, and PTH escapes from inhibitory control. In the long-term, the CaSR also regulates the growth of parathyroid cells and the synthesis of PTH.
Since the original cloning of the CaSR by Dr. Edward Brown and colleagues at Harvard [5], it has been recognized that CaSR mRNA is also expressed by the C-cells of the thyroid, the source of calcitonin [6]. Furthermore, cells from a medullary thyroid carcinoma cell line express CaSR mRNA and release calcitonin in response to calcium [7]. The presence of the CaSR on C-cells raises the possibility that the CaSR might directly regulate calcitonin release; however, Dr. Brown has noted that the presence of the CaSR in a cell whose function is modulated by calcium is not sufficient proof that the CaSR mediates that particular action of calcium [4].
The regulation of calcitonin is opposite to that of PTH, in that increases in ionized calcium stimulate calcitonin, and decreases in ionized calcium lead to a fall in calcitonin. This observation led to the general assumption that the mechanisms mediating calcium sensing in parathyroid and C cells are distinctly different, with the latter likely involving some form of voltage-sensitive calcium channels [4,8]. It was, therefore, somewhat unexpected that the parathyroid CaSR would turn out to be expressed by C cells of the thyroid. Because calcium is the primary stimulus for changes in calcitonin, and the C cells express the CaSR, the CaSR is a logical and obvious candidate to fulfill the homeostatic regulatory function that it also does for PTH [4,8,9].
To address the issue of a role for the CaSR in regulating calcitonin in vivo, we utilized the calcium receptor gene ablation model [10]. Heterozygous (Casr+/-) mice have increased ionized calcium, modest hyperparathyroidism, and reduced renal calcium excretion. Homozygous (Casr null) mice have severe hypercalcemia and hyperparathyroidism, and die within several weeks of birth. These two conditions mimic the corresponding human conditions in which CaSR function is impaired or absent, respectively familial hypocalciuric hypercalcemia and neonatal severe hypercalcemia [5].
We addressed the hypothesis that the CaSR stimulates calcitonin release in response to an increase in the blood calcium concentration. Thus, in Casr+/- mice, we expected to see that the calcitonin response to an increase in blood calcium would be blunted compared to the normal response, or shifted to the right. This is in fact what was observed.
Results
It was anticipated that provoking a calcitonin response by calcium injection would demonstrate that ablation of the CaSR had altered the normal relationship between ionized calcium and calcitonin levels. Such an experiment was not practical in fetuses because 3–4 fetal samples would have to be pooled for each calcitonin measurement. Instead, adults were used so that larger blood samples suitable for assay would be obtained. This meant that only wild type and Casr+/- mice could be studied, because Casr null mice are visibly ill within days of birth and die by two to three weeks of age.
Casr+/- mice had higher ionized calcium levels at baseline compared to wild type, consistent with the known phenotype of these mice in both the original C57BL/6 background [10] and in the Black Swiss background [11]. The saline injection did not significantly alter ionized calcium, although the mean value dropped for both wild type and Casr+/- mice, and appeared to be proportionately greater for Casr+/- mice (Table 1). The two different doses of calcium resulted in significant elevations in ionized calcium (Table 1) compared to baseline and compared to the effect of the saline injection. Casr+/- mice had proportionately greater increments in ionized calcium and reached higher peak ionized calcium levels as compared to wild type. Thus, the higher baseline ionized calcium, the greater increments in ionized calcium, and the higher peak values in ionized calcium achieved by Casr+/- mice biased the experiment in favour of obtaining a higher peak calcitonin response from Casr+/- as compared to wild type.
Table 1.
Mean (± SE) ionized calcium levels before (pre) and after (post) injection in wild type and Casr+/- mice. P values apply to the comparison of respective post versus pre values, and to the post values versus the corresponding post-saline value.
| Genotype | Injection | Pre | Post | p value |
| Wild type | Saline (16) | 1.28 ± 0.03 | 1.26 ± 0.02 | - |
| Wild type | 500 μmol/kg (13) | 1.29 ± 0.02 | 1.34 ± 0.04 | < 0.01 |
| Wild type | 5 mmol/kg (8) | 1.29 ± 0.02 | 1.55 ± 0.06 | < 0.01 |
| Casr+/- | Saline (20) | 1.42 ± 0.02 | 1.34 ± 0.04 | - |
| Casr+/- | 500 μmol/kg (25) | 1.42 ± 0.02 | 1.50 ± 0.02 | < 0.01 |
| Casr+/- | 5 mmol/kg (23) | 1.40 ± 0.01 | 1.71 ± 0.03 | < 0.01 |
The mean baseline (post-saline) serum calcitonin levels were slightly lower in Casr+/- mice as compared to wild type, at 12.1 ± 8.0 versus 20.4 ± 8.9 pg/ml, respectively (p = 0.49). In response to calcium injection, the calcitonin level rose in wild type and Casr+/- mice as expected (Figure 1). A logarithmic scatterplot of the data in Figure 1A demonstrates that Casr+/- mice achieved lower calcitonin levels for each level of ionized calcium, and regression analysis confirmed that the dose-response curve for calcitonin was shifted significantly to the right in Casr+/- mice. The mean (± SE) data are plotted in Figure 1B, and confirm shift of calcitonin responsiveness in Casr+/- mice.
Figure 1.
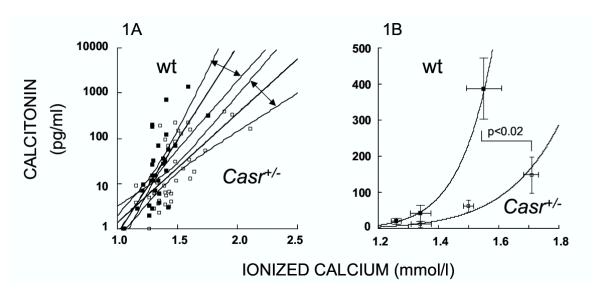
Blunted calcitonin responsiveness in Casr+/- mice. On the left (1A), a logarithmic scatterplot of serum calcitonin versus ionized calcium level in wild type and Casr+/- mice. The straight lines indicate the respective best-fit regression lines; the respective 95% confidence intervals for the fitted lines are also shown, and have been demarcated by double-headed arrows for easier recognition. On the right (1B), a normal plot of mean serum calcitonin (± SE) versus mean ionized calcium (± SE) in wild type and Casr+/- mice. Vertical SE bars correspond to the calcitonin values, and horizontal error bars correspond to ionized calcium values. The peak calcitonin response of wild type and Casr+/- was significantly different for both ionized calcium and calcitonin (p < 0.02). In both graphs, the closed squares indicate wild type values, and the open squares indicate Casr+/- values.
Discussion
These observations in adult wild type and Casr+/- mice demonstrate a definite shift in the physiological response of calcitonin to acute increments in ionized calcium. Since this response was dependent upon the genotype (wild type versus Casr+/-), it is clear that ablation of the CaSR was the determinant that shifted calcitonin responsiveness. The "shift to the right" in calcitonin responsiveness between wild type and Casr+/- mice confirms that the CaSR is a physiological regulator of calcitonin release. These observations complement and confirm the earlier observations of Lavigne et al, who demonstrated that the calcimimetic compound NPS R-467 stimulates calcitonin release in rats [12]. NPS R-467 activates the CaSR and suppresses PTH release; it was reasonably inferred from those experiments that NPS R-467 was stimulating the release of calcitonin through direct actions on the CaSR. The possibility that calcitonin was not stimulated directly by NPS R-467 could not be completely excluded in those experiments, and thus, our experiments in Casr+/- mice provide physiological evidence that the calcitonin response is dependent upon the CaSR.
The effect of inactivating mutations of the CaSR on serum calcitonin in humans has not been extensively or rigorously examined, apart from noting that individuals with hypercalcemia due to familial hypocalciuric hypercalcemia (FHH) had apparently normal baseline calcitonin levels using older calcitonin radioimmunoassays [13-15]. In the first study [13], baseline calcitonin levels in 12 individuals with FHH were no different than in 9 unaffected individuals from the same kindred. In the second study [14], no calcitonin measurements were obtained from normal or unaffected individuals, but instead the results of FHH individuals were simply reported as being within the (at the time) wide normal range of < 69 pg/ml. In the third cited study, the baseline calcitonin levels in 26 individuals with FHH were compared to 20 unrelated healthy controls. The lack of a difference in baseline serum calcitonin levels in all three of these studies is similar to our observation that, at baseline, the calcitonin levels in Casr+/- mice are slightly lower but not significantly different than in related wild type mice. The presence of hypercalcemia at baseline in both Casr+/- mice and in patients with FHH should have caused a higher calcitonin level if the CaSR had normal sensitivity to calcium and normal function. It is only with the calcium challenge that the blunted calcitonin responsiveness became definitively apparent in our studies of Casr+/- mice. A 5 minute calcium challenge with calcitonin response was performed in only one of the studies in humans with FHH but was reported to show no difference in calcitonin response compared to unrelated normal controls [15]. Interestingly, in that study the mean results were actually lower in affected individuals, being 33.6 ± 7.0 pg/ml in 7 normal males versus 18.6 ± 5.4 pg/ml in 6 affected males with FHH, and 9.8 ± 5.4 pg/ml in 7 normal females versus 7.9 ± 2.7 pg/ml in 7 affected females with FHH. Thus, the low sample size number is likely a key reason why that study did not demonstrate these apparent differences to be statistically significant. That study, performed more than 13 years ago, may also have failed to demonstrate a difference due to the earlier-generation calcitonin assay used, and the heterogeneity of the patients under study (unrelated controls) as compared to the current study that used genetically related mice. The possibility of a species difference cannot be excluded; that is, that the CaSR is a significant regulator of calcitonin release in mice but not in humans.
An alternative explanation for our findings is that chronic hypercalcemia in Casr+/- mice downregulated the responsiveness of calcitonin through indirect effects, such as by downregulation of another calcium sensor that in turn might regulate calcitonin. As shown in Table 1, the Casr+/- mice normally maintain a 0.1 mmol/l higher ionized calcium concentration than in wild-type, but acutely achieved a peak increment in ionized calcium that was approximately 0.4 mmol/l higher than the post-saline value. Therefore, because the acute stimulus (increase in ionized calcium) was much greater than the chronic stimulus (mild hypercalcemia), it is likely that the blunted calcitonin response is specific to heterozygous ablation of the CaSR, and not due to chronic hypercalcemia.
Our current findings that ablation of the CaSR shifts the responsiveness of calcitonin to acute changes in the serum calcium predicts that the CaSR will be coupled to different signal transduction pathways depending upon the tissue in which it is expressed. In that way, the same receptor can have an inhibitory response to its activation in parathyroid cells, but a stimulatory response to its activation in parafollicular cells of the thyroid.
Casr+/- mice had greater increments in ionized calcium in response to the same dose of calcium given to wild type mice. The slightly greater (but statistically insignificant) drop in ionized calcium provoked by saline injection in Casr+/- mice may indicate a difference in intravascular volume that was not apparent by the normal hematocrit and body weight that Casr+/- mice have as compared to their siblings [10](and data not shown). Alternatively, the blunted calcitonin response of Casr+/- mice may in turn lead to loss of effect of calcitonin to promote calciuresis and minimize increases in the blood calcium. The role of calcitonin to minimize increases in the blood calcium in response to a calcium or PTH challenge has been demonstrated in mice lacking calcitonin through a genetic ablation [16]; and in rats rendered calcitonin deficient through surgical or immunological techniques [17,18]. The renal tubular action of the CaSR may also partly account for the greater calcemic response of Casr+/- mice. Casr+/- mice have reduced renal calcium excretion due to ablation of the CaSR [10,11], and thereby would be expected to have an impaired ability to excrete calcium in response to a calcium load. Furthermore, PTH-mediated bone resorption is increased in Casr+/- mice, which indicates that efflux of calcium from bone is increased, and bone matrix will be less able to take up calcium in response to a calcium load [10,11].
Although calcitonin has often been considered to be a vestigial hormone with no certain role in mammalian calcium and bone homeostasis, this study demonstrates that calcitonin is subject to physiological regulation by the CaSR. Recent studies in mice indicate that calcitonin has important roles in calcium and bone homeostasis that have not been previously recognized. Mice lacking the gene encoding calcitonin and calcitonin gene-related peptide-α (CGRP-α) develop a phenotype of increased bone mass as adults [16], a phenotype that is not present at the end of fetal development [19]. Mice that are heterozygous for loss of the calcitonin receptor also have a phenotype of increased bone mass as adults [20]. Preliminary studies have indicated that mice lacking the gene encoding calcitonin and CGRP-α lose more than twice as much skeletal calcium content as their wild-type sisters during three weeks of lactation [21], confirming a previously postulated role that calcitonin might protect the maternal skeleton from excessive resorption during lactation. Thus, calcitonin has new and emerging evidence of its importance in regulating calcium homeostasis and bone mass. It may be inferred that the CaSR regulates bone mass through its effect on the parathyroid glands to produce PTH, and through its effect on C-cells of the thyroid to produce calcitonin. In the short-term, the CaSR regulates ionized calcium concentration in blood by altering the release of PTH and calcitonin; in the long-term, it influences the maintenance of bone mass through these same hormones. In the future, pharmacological agents that can manipulate the CaSR to achieve an ideal balance of calcitonin and PTH in the circulation may provide a novel approach to treating disorders such as osteoporosis, secondary (uremic) hyperparathyroidism, hypercalcemia, and hypocalcemia [22,23].
Conclusions
In summary, we found that in adult wild type and Casr+/- mice an acute calcium injection stimulated the release of calcitonin, but the response curve was blunted or shifted to the right in Casr+/- mice. We have confirmed that the CaSR stimulates calcitonin release in response to an increase in the blood calcium concentration. Therefore, the CaSR has a dual physiological response to acute increases in ionized calcium, which includes inhibition of PTH release and stimulation of calcitonin release. In the long-term, the actions of the CaSR to regulate PTH and calcitonin will influence the maintenance of skeletal mineral content.
Methods
Casr mice
Casr-ablated mice were the gift of Jon Seidman (Harvard) and were obtained by targeted disruption of the murine Casr gene in stem cells, as previously described [10]. The mice were back-crossed into outbred Black Swiss strain (Taconic Labs) for at least five generations. Adult wild type and Casr+/- mice were used that were all first degree relatives of each other, and between 2 to 4 months of age. All mice were given a standard chow diet (1% calcium) and water. All studies were performed with the prior approval of the Animal Care Committee of Memorial University of Newfoundland.
Calcium challenge experiments
105 adult mice (37 wild type and 68 Casr+/-) were weighed and distributed into tertiles, to be given an intraperitoneal injection of either 500 μl of normal saline (0.9% sodium chloride), or one of two doses of elemental calcium (500 μmol/kg or 5 mmol/kg in 500 μl of normal saline). These doses of elemental calcium had been determined in preliminary experiments to result in significant increases in ionized calcium and calcitonin of both wild type and Casr+/- mice. Saline injection was used as the control group in order to control for alterations in ionized calcium that would occur due to the injection of fluid in the peritoneum and due to any stress on the animal caused by the experimental procedure. Tail blood was obtained at baseline and 10 minutes after the injection of elemental calcium to assay ionized calcium. Whole blood (near total blood volume) was obtained at 10 minutes by intracardiac needle for the calcitonin assay. The two separate blood collections (tail blood and intracardiac puncture) were performed at the 10 minute time point in order that the 10 minute ionized calcium measurements would be comparable to the baseline ionized calcium measurements. Tail blood and intracardiac blood do not provide comparable ionized calcium levels.
Assays
Ionized calcium was measured with a specific calcium ion electrode (Chiron 634 Ca++/pH Analyzer). Serum calcitonin was measured in duplicate using a specific IRMA for rat calcitonin that cross-reacts with murine calcitonin (Immutopics, San Clemente, CA) [12], and which has previously been validated in our laboratory for use on mouse serum [11,24]. In lieu of mouse calcitonin standards, values were expressed as pg-equivalents rat calcitonin/ml; the stated detection limit of the assay was 1.0 pg/ml.
Statistical analysis
Data was analyzed using SYSTAT 5.2.1 for Macintosh (SYSTAT Inc, Evanston, IL). Two-tailed probabilities are reported, and all data are presented as mean ± SE.
List of abbreviations
CaSR Calcium sensing receptor
PTH Parathyroid hormone
Casr+/- Mice that are heterozygous for deletion of CaSR gene
Casr null Mice that are homozygous for deletion of CaSR gene
CGRP-α calcitonin gene-related peptide-α
FHH Familial hypocalciuric hypercalcemia
SE standard error
Authors' contributions
NF carried out the physiological studies and participated in the writing of each draft of the manuscript. CK conceived of the study, directed it, and did the primary writing of the manuscript. Both authors approved the final draft of the manuscript.
Acknowledgments
Acknowledgements
Supported by operating grants and a 5-year Scholarship (New Investigator Award) from the Canadian Institutes for Health Research (formerly Medical Research Council of Canada), and by funds from the Discipline of Medicine in the Faculty of Medicine at Memorial University of Newfoundland. The work described herein garnered the 2002 Antoni Nalecz Award from the Canadian Society of Endocrinology and Metabolism.
Contributor Information
Neva J Fudge, Email: nevafudge@hotmail.com.
Christopher S Kovacs, Email: ckovacs@mun.ca.
References
- Broadus AE. Mineral balance and homeostasis. In: Favus MJ, editor. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5. Washington, DC: ASBMR Press; 2003. pp. 105–111. [Google Scholar]
- Jüppner H, Kronenberg HM. Parathyroid hormone. In: Favus MJ, editor. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5. Washington, DC: ASBMR Press; 2003. pp. 117–124. [Google Scholar]
- Deftos LJ. Calcitonin. In: Favus MJ, editor. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5. Washington, DC: ASBMR Press; 2003. pp. 137–141. [Google Scholar]
- Brown EM, MacLeod RJ. Extracellular Calcium Sensing and Extracellular Calcium Signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- Garrett JE, Tamir H, Kifor O, Simin RT, Rogers KV, Mithal A, Gagel RF, Brown EM. Calcitonin-secreting cells of the thyroid express an extracellular calcium receptor gene. Endocrinology. 1995;136:5202–5211. doi: 10.1210/en.136.11.5202. [DOI] [PubMed] [Google Scholar]
- Freichel M, Zink-Lorenz A, Holloschi A, Hafner M, Flockerzi V, Raue F. Expression of a calcium-sensing receptor in a human medullary thyroid carcinoma cell line and its contribution to calcitonin secretion. Endocrinology. 1996;137:3842–3848. doi: 10.1210/en.137.9.3842. [DOI] [PubMed] [Google Scholar]
- Brown EM. Is the calcium receptor a molecular target for the actions of strontium on bone? Osteoporos Int. 2003;14:S25–34. doi: 10.1007/s00198-002-1343-6. [DOI] [PubMed] [Google Scholar]
- Brown EM. Calcium-sensing receptor. In: Favus MJ, editor. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5. Washington, DC: ASBMR Press; 2003. pp. 111–117. [Google Scholar]
- Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, Seidman CE. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet. 1995;11:389–394. doi: 10.1038/ng1295-389. [DOI] [PubMed] [Google Scholar]
- Kovacs CS, Ho-Pao CL, Hunzelman JL, Lanske B, Fox J, Seidman JG, Seidman CE, Kronenberg HM. Regulation of murine fetal-placental calcium metabolism by the calcium-sensing receptor. J Clin Invest. 1998;101:2812–2820. doi: 10.1172/JCI2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne JR, Zahradnik RJ, Conklin RL, Lambert LD, Logan MA, Parihar A, Fox J. Stimulation of calcitonin secretion by calcium receptor activators: evaluation using a new, highly sensitive, homologous immunoradiometric assay for rat calcitonin. Endocrine. 1998;9:293–301. doi: 10.1385/ENDO:9:3:293. [DOI] [PubMed] [Google Scholar]
- Menko FH, Bijvoet OL, Fronen JL, Sandler LM, Adami S, O'Riordan JL, Schopman W, Heynen G. Familial benign hypercalcaemia. Study of a large family. Q J Med. 1983;52:120–140. [PubMed] [Google Scholar]
- Law WM, Heath H. Familial benign hypercalcemia (hypocalciuric hypercalcemia). Clinical and pathogenetic studies in 21 families. Ann Intern Med. 1985;102:511–519. doi: 10.7326/0003-4819-102-4-511. [DOI] [PubMed] [Google Scholar]
- Rajala MM, Klee GG, Heath H. Calcium regulation of parathyroid and C cell function in familial benign hypercalcemia. J Bone Miner Res. 1991;6:117–124. doi: 10.1002/jbmr.5650060204. [DOI] [PubMed] [Google Scholar]
- Hoff AO, Catala-Lehnen P, Thomas PM, Priemel M, Rueger JM, Nasonkin I, Bradley A, Hughes MR, Ordonez N, Cote GJ, Amling M, Gagel RF. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. J Clin Invest. 2002;110:1849–1857. doi: 10.1172/JCI200214218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos BA, Yoon M, Cutshaw SV, Kalu DN. Calcium regulatory action of endogenous rat calcitonin demonstrated by passive immunization with calcitonin antibodies. Endocrinology. 1980;107:1320–1326. doi: 10.1210/endo-107-5-1320. [DOI] [PubMed] [Google Scholar]
- Harper C, Toverud SU. Ability of thyrocalcitonin to protect against hypercalcemia in adult rats. Endocrinology. 1973;93:1354–1359. doi: 10.1210/endo-93-6-1354. [DOI] [PubMed] [Google Scholar]
- McDonald KR, Fudge NJ, Woodrow JP, Friel JK, Hoff AO, Gagel RF, Kovacs CS. Ablation of calcitonin/calcitonin gene related peptide-α impairs fetal magnesium but not calcium homeostasis. Am J Physiol Endocrinol Metab. 2004. [DOI] [PubMed]
- Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR, Gebre-Medhin S, Galson DL, Zajac JD, Karsenty G. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol. 2004;164:509–514. doi: 10.1083/jcb.200312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow JP, Noseworthy CS, Fudge NJ, Hoff AO, Gagel RF, Kovacs CS. Calcitonin/calcitonin gene-related peptide protect the maternal skeleton from excessive resorption during lactation [abstract] J Bone Miner Res. 2003;18:S37. [Google Scholar]
- Nemeth EF. The search for calcium receptor antagonists (calcilytics) J Mol Endocrinol. 2002;29:15–21. doi: 10.1677/jme.0.0290015. [DOI] [PubMed] [Google Scholar]
- Wada M, Nagano N, Nemeth EF. The calcium receptor and calcimimetics. Curr Opin Nephrol Hypertens. 1999;8:429–433. doi: 10.1097/00041552-199907000-00006. [DOI] [PubMed] [Google Scholar]
- Kovacs CS, Manley NR, Moseley JM, Martin TJ, Kronenberg HM. Fetal parathyroids are not required to maintain placental calcium transport. J Clin Invest. 2001;107:1007–1015. doi: 10.1172/JCI11321. [DOI] [PMC free article] [PubMed] [Google Scholar]