

Abstract
Asymmetric gold-catalyzed hydrocarboxylations are reported that show broad substrate scope. The hydrophobic effect associated with in situ-formed aqueous nanomicelles leads to good-to-excellent ee’s of product lactones. In-flask product isolation, along with recycling of the catalyst and reaction medium, combine to arrive at an especially environmentally friendly process.
Keywords: Micellar catalysis, gold catalysis, asymmetric catalysis, designer surfactant TPGS-750-M
Micellar catalysis in water can play an important contributing role in green chemistry. Today, many valued reactions take place within the lipophilic core of self-aggregated nanomicelles, composed of “designer” surfactants tailor-made to best accommodate the synthetic chemistry of interest.[1] The hydrophobic interior of these nanoreactors can function as far more than an alternative, albeit green, solvent for a desired reaction; rather, it offers an opportunity to enhance binding between a charged catalyst and its counterion, especially in such a characteristically nonpolar environment. Synergistic binding between ions might be leveraged, e.g., in asymmetric gold-catalyzed reactions, which is particularly challenging due to the bi-coordinate geometry of Au(I). This orientation places a nonracemic ligand distal to its catalytic site,[2] thereby preventing the substrate from attaining the highly biased mode of chelation desired for maximizing enantioselectivity. Nonetheless, a variety of factors have been found leading to asymmetric induction.[2,3] These include the specifics of the ligand-metal binding, such as with Au-arene interactions as well as the nature of the counterion,[4] reaction temperature,[5] and the solvent.[5,6] Ligands such as phosphines,[7] acyclic diaminocarbenes (ADCs),[8] and N-heterocyclic carbenes (NHCs)[9] function well for such Au-catalyzed processes in organic media.
Hydrocarboxylations of olefins and allenes have been found to be highly valuable in the synthesis of target bioactive molecules.[10] Transition metal catalysts, in particular coinage metal-catalyzed enantioselective hydrocarboxylations of allenes forming valuable nonracemic lactones, however, is still an unsolved problem. Indeed, only one simple example is currently known, done in the no longer used solvent benzene and with moderate ee,.[11] By contrast, related reactions such as intramolecular hydroalkoxylations[5] and hydroaminations[12] of allenes are well established. These are typically done in nonpolar media, and show promising levels of asymmetric induction. Nonetheless, as these reactions, carried out in organic solvents, oftentimes require low temperatures[8,13] to maximize ee’s, reaction times can be significantly increased. Moreover, none offers an opportunity to recycle the metal, ligand, or counterion, thus making the overall process both costly and environmentally unattractive.
On the other hand, synergistic binding between catalyst and counterion might be expected due to the hydrophobic effect found within nanomicelles. This could enhance a defined chiral pocket leading to improved enantiocontrol that otherwise is not always observed in traditional organic solvents.[14] Despite these potential advantages to micellar catalysis, no report on asymmetric gold catalysis within such nanoreactors has been appeared to date. Herein we describe asymmetric gold-catalyzed intramolecular cyclizations of β-allenic acids[15] to arrive at enantio-enriched lactones, carried out within self-aggregated, tailor-made nanoparticles in water at room temperature (Scheme 1).
Scheme 1.
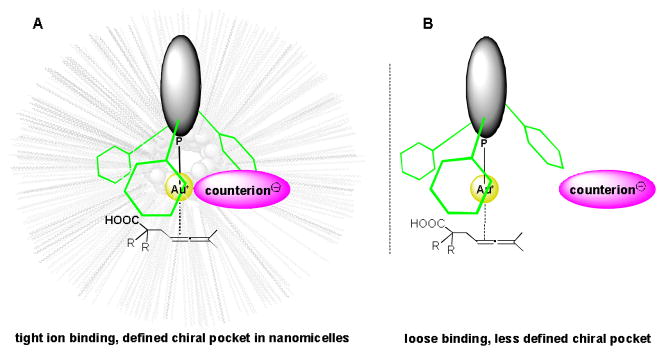
Cationic gold binding (A) within a nanomicellar core in water vs. (B) in many organic solvents.
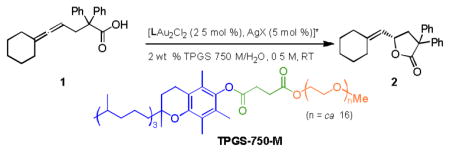
We started our investigations with allenic acid 1 as a model substrate, looking to find a nonracemic catalyst suitable for use in an aqueous micellar medium. An initial reaction in designer surfactant TPGS-750-M/H2O with 5 mol % of catalyst (R)-BINAP-Au2Cl2/AgX afforded 2 in low ee (Table 1, entry 1). Altering the axially chiral core of the ligand to H8-BINAP (4) and synphos (5a), likewise led to no significant improvement in ee (entries 2, 3). Increasing the steric bulk of the aromatic residue on phosphorus contributes toward the observed enantioselectivity, as 3,5-Xyl-synphos (5b) increased the ee to 30% (entry 4). The improvement in ee due to steric effects near the donor atom of the ligand suggested use of monodentate ligands, including phosphoramidite 7 and ADCs 8 and 9. Interestingly, an increased ee to 50% was only seen with the larger ADC 9. The choice of counterion also affected the selectivity of the reaction (entries 7, 8). No significant improvements in terms of ee were observed even with further alterations in the axially chiral core of several bis-phosphine ligands (entries 9–12). A 51% ee was obtained using SEGPHOS ligand 11d when accompanied by p-nitrobenzoate as a counterion (entry 13). The best results were obtained by the fine-tuning of BIPHEP ligands (entries 14–19), with 12f being identified as the best choice so far (entry 19). In the absence of surfactant there was essentially no reaction.
Table 1.
Optimization of Au-catalyzed asymmetric intramolecular hydrocarboxylations of allenic acids
| |||||
|---|---|---|---|---|---|
| Entry | Ligand (L) | X− | Time (h) | % yield 2 | % ee |
| 1 | 3 | BF4 | 78 | 98 | 3 |
| 2 | 4 | SbF6 | 74 | 95 | 11 |
| 3 | 5a | SbF6 | 72 | 95 | 18 |
| 4 | 5b | SbF6 | 78 | 96 | 30 |
| 5 | 7 | SbF6 | 29 | 93 | 1 |
| 6 | 8 | SbF6 | 32 | 92 | 7 |
| 7 | 9 | PMB | 67 | 67 | 40 |
| 8 | 9 | PNB | 70 | 70 | 50 |
| 9 | 10 | SbF6 | 60 | 85 | 29 |
| 10 | 11a | SbF6 | 70 | 90 | 6 |
| 11 | 11b | SbF6 | 75 | 91 | 36 |
| 12 | 11c | SbF6 | 42 | 94 | 7 |
| 13 | 11d | PNB | 72 | 95 | 51 |
| 14 | 12a | SbF6 | 48 | 95 | 12 |
| 15 | 12b | SbF6 | 49 | 91 | 18 |
| 16 | 12c | SbF6 | 40 | 85 | 31 |
| 17 | 12d | PNB | 30 | 97 | 37 |
| 18 | 12e | PNB | 38 | 70 | 30 |
| 19 | 12f | PNB | 48 | 97 | 55 |
LAu2Cl2 = LAuCl (5 mol %) when L = 7, 8, and 9. PNB = p-nitrobenzoate, PMB = p-methoxybenzoate. For ligands, see Scheme 2. For detailed optimization table, see SI.
The impact of counterions on both ee’s and yields in aqueous micelles suggested that further tuning of a catalyst system with focus on the chiral counterion might be productive. Initial match/mismatch studies were performed on both R and S enantiomers of methoxy-BIPHEP ligand 12f, with counterions S-13a, R-13a, S-13b, R-13b, S-14a, and R-14a (Figure 1). A significant mismatch was found between ligand R-12f and counterion R-13a giving only 27% ee of product 2, while the enantiomeric counterion S-13a led to an ee of 51% (Scheme 2). Using a far bulkier BINOL-based counterion S-13b, an insignificant match/mismatch existed, as only a 5% difference in ee was obtained in product 2 (75% vs 70%). An ee of 66% was found in the reaction of 1 using S-12f-Au2 and counterion R-13b-Ag. Spirocyclic phosphate counterion 14 provided slightly better results in terms of ee than did counterion 13a. In order to find the best match for an active catalyst, additional reactions were performed using S-12f-Au2, along with both enantiomers of silver salts of 13a-b and 14. Interestingly, no general trend of match/mismatch between catalyst 12f and different counterions was observed. From all combinations studied using the model reaction, the combinations R-12fAu2–R-13bAg or S-12fAu2–S-13bAg (75% ee) was found to be the best pair between ligand 12f and counterions 13a, 13b, and S- or R-14.
Figure 1.

Ligands and counterions used for optimization studies.
Scheme 2.
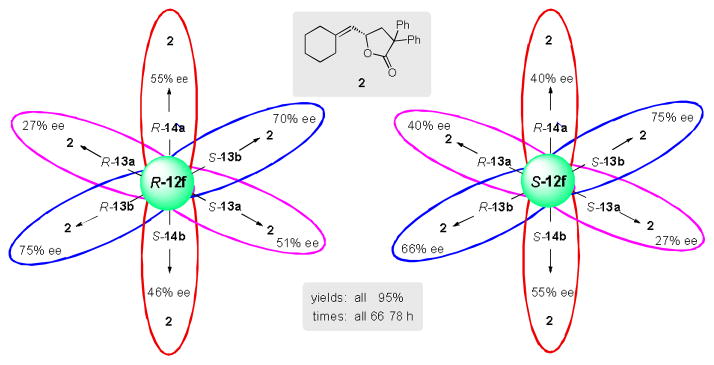
Venn diagram showing match/mismatch combin-ations between ligand 12f and counterions.
Using the catalyst system containing ligand (R)-12f and a counterion (R)-13b, further optimization studies were undertaken. These revealed a dependence of ee on several variables, including (1) Au-nanoparticle-free conditions; (2) the nature of acidic or basic additives; (3) the presence of AgCl; (4) the ratio of L Auto AgX; (5) removal of both chloride ions attached to gold; (6) protection from ambient light; (7) the purity of the catalyst and counterion; (8) the surfactant concentration, and (9) the reaction temperature (see SI for details). Optimal reaction conditions (Scheme 3) were found to be: 3 wt % TPGS-750-M as surfactant in water as the reaction medium, breaking of the crystallinity of solids by the addition of minimal amounts of solvent additives, use of 3 mol % of AgCl-free, pure active gold-catalyst R,R-15, 0.5 M global concentration, extraction of the product with a minimum amount of 10% ether in hexanes to maximize the amount of catalyst that remains in the aqueous medium in the reaction vessel, and ambient temperature.
Scheme 3.

Optimized reaction conditions and X-ray structure of R,R-15 in ball and stick.
Under these optimized conditions, including the “trick” of softening highly crystalline solid substrates with a drop or two of an organic solvent (DMSO, benzene, or toluene) as “additive”, reaction times were decreased significantly; in the case of educt 1, from 72 to 16 hours. The ee could also be increased from 75% to 88% by using isolated AgCl-free catalyst R,R-15. The X-ray structure of R,R-15 shows the defined chiral pocket with weak Au-arene interactions. The binding between cationic gold and the counterion through the phosphate group supports the match between them, which had already been determined experimentally (vide supra).
As illustrated in Scheme 4, a variety of precursor allenic acids were cyclized to afford high yields of product lactones. This new technology is clearly applicable to many substitution patterns associated with both the allenic group, as well as the α-carbon of the carboxylic acid. Unsubstituted as well as dialkyl-substituted allenic termini appear to give ee’s that are among the highest observed (81–92%). Allenes bearing saturated rings at the terminus ranging from 5-to-8 membered in size afforded ee’s that were more variable, and seemingly dependent upon the nature of the substituent(s) located on the α-carbon. Most ee’s are in the 80–90% range, and hence, it’s unclear why spirocyclic lactone 23 was formed in 96% ee. With a single substituent α- to the acid, diastereomers result; the major isomer 32 was isolated and found to have an ee of 90%. The other diastereomer underwent secondary processes leading to unidentified material.
Scheme 4.

Substrate scope.
To confirm that the solvent additive is acting solely to soften the highly crystalline nature of these substrates and catalyst, and neither impacting the role of the micellar environment nor merely acting as the reaction medium, highly crystalline substrate 33 was subjected to the reaction conditions in different reaction media including neat DMF, DMSO, and toluene, as well as in TPGS-750-M with these same solvent additives (Scheme 5). Similar ee’s were observed when reactions were run in aqueous TPGS-750-M with solvent additives, while dissimilar ee’s were obtained in each of the neat organic solvents. Poorer ee’s in polar organic solvents and better ee’s in nonpolar organic solvents and TPGS-750-M is indicative of the surfactant serving as the reaction medium, providing the hydrophobic effect inside the micellar core (for details, see SI).
Scheme 5.
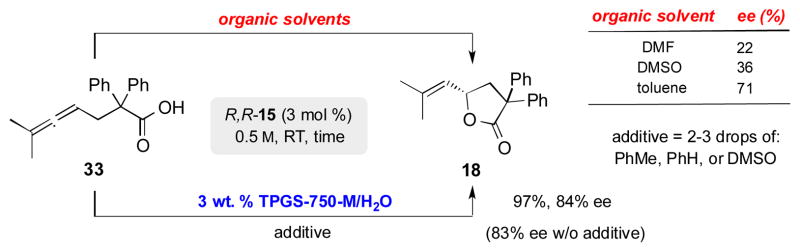
Impact of reaction medium on % ee.
The progress of a representative reaction of allenic acid 33 leading to lactone 19 under the influence of cationic gold catalyst R,R-15 was followed over time. As the reaction progressed, the ee of 19 remained constant, as determined by HPLC analyses of several aliquots. The absolute stereochemistry of the newly created center in the product was determined by single crystal X-ray diffraction analysis, clearly indicating S-stereochemistry (Figure 2).
Figure 2.
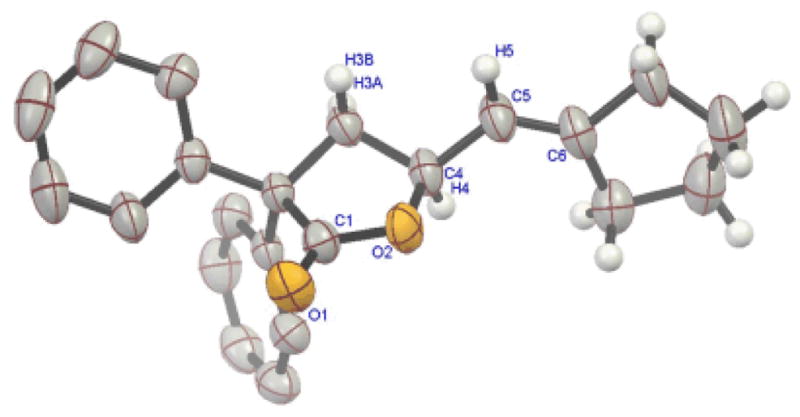
X-ray structure of S-19.
The mechanism by which these Au-catalyzed reactions might proceed has been investigated using DFT calculations, using a PH3 ligand, and with the carboxylic acid precursor to 19 without the phenyl substituents. The calculations showed that direct 5-exo-trig cyclization of A to form a protonated lactone E was substantially uphill, ΔG∘ = 13.2 kcal/mol in toluene (Scheme 6). The 6-endo-dig complex was uphill 10.7 kcal/mole, while the 6-exo-dig and 7-endo-trig products were uphill by 22.6 and 18.8 kcal/mol, respectively, PBE1PBE/6-31+G(d,p)/SDD(Au)/SMD(toluene). On the other hand, cyclization reactions of the deprotonated complex B were downhill energetically by 22.1 kcal/mol for the 5-exo-trig cyclization and 21.9 kcal/mol for the 6-endo-dig cyclization in dichloro-methane (see SI). In toluene, these free energies were considerably larger, 34.6 and 35.2 kcal/mol, respectively, as expected. These observations, together with the fact that addition of catalytic tri-ethylamine or quaternary ammonium hydroxides substantially accelerated the reaction rates in the micelle, suggests that the reaction with base may proceed via the deprotonated complex B to form C. After deprotonation of the carboxylic acid group in the gold-allene complex, the cyclization occurs with little or no barrier in our theoretical modeling. Reaction trajectory modeling is consistent with the observed preference for 5-exo-trig cyclization. This mechanism is in stark contrast to the mechanism proposed for gold-catalyzed cyclization of allenic alcohols, with reversible cyclization and rate-determining protodeauration,[16] but the reactions in the absence of added base may occur via E rather than B. The calculated transition-state energies for cyclization by this pathway also show a preference for the 5-exo-trig pathway. Such a change of mechanism would be consistent with our observation that a low ee is observed in reactions with a chiral counterion in the presence of added triethylamine.
Scheme 6.

Plausible mechanistic scheme with DFT energy diagram at the PBE1PBE/6-31+G(d,p)/SDD(Au)/SMD(toluene) level of theory with relative free energies in toluene at 298K in kcal/mol. The free energy difference between A and B depends upon the base used and is not well known in the micellar media (see Supporting Information).
Studies were also conducted to assess the potential for recycling of the aqueous medium. Given that our portfolio of “designer” surfactants are engineered to remain in the water,[16] the more intriguing question concerned the fate of the gold catalyst with respect to both its recyclability as well as its efficacy. Upon extraction with minimal amounts of 10% ether in hexanes, more than 50% of catalyst remained in the aqueous phase such that only 1 mol % of fresh R,R-15 need be added at each cycle. As illustrated in Scheme 7, the ee’s for product 2 remained essentially constant, as did the yields, throughout six successive recycles. The associated E Factor based on organic solvent usage,[17,18] as a measure of the “greeniness” of this protocol was calculated to be only 4.9, which compares very well with values typically seen from chemistry done in the fine chemicals and pharma arenas.[17]
Scheme 7.

Studies on recycling and E Factor.
In summary, the hydrophobic effect characteristic of nanomicelles has been utilized advantageously to enhance tight ion-pair binding between a nonracemic cationic gold catalyst and its nonracemic counterion. This combination is highly effective in achieving especially challenging asymmetric intramolecular hydrocarboxylation of allenes in high yields and good-to-excellent ee’s. The aqueous micellar reaction medium obviates use of organic solvents, and both the catalyst and surfactant can be recycled. The associated E Factor for the overall process is very low, documenting its environmentally friendly nature.
Supplementary Material
Footnotes
Financial support for our programs in green chemistry, provided by the NIH (GM86485), is warmly acknowledged with thanks. We are pleased to acknowledge support from the Center for Scientific Computing at the CNSI and MRL: an NSF MRSEC (DMR-1121053) and NSF CNS-0960316. We also acknowledge NSF support from the National Center for Supercomputing Applications (NSF TG-CHE100123) utilizing the NCSA Gordon and Blacklight systems. We thank Prof. L. Slaughter (U North Texas) for providing scientific guidance leading to some of the allenic acids utilized in this work.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Dr. Sachin Handa, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106 (USA)
Daniel J. Lippincott, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106 (USA)
Eric D. Slack, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106 (USA)
Donald H. Aue, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106 (USA)
Prof. Bruce H. Lipshutz, Email: lipshutz@chem.ucsb.edu, Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106 (USA)
References
- 1.Lipshutz BH, Ghorai S. Aldrichimica Acta. 2012;45:3. [PMC free article] [PubMed] [Google Scholar]
- 2.Hashmi ASK. Angew Chem, Int Ed. 2005;44:6990. doi: 10.1002/anie.200502735. [DOI] [PubMed] [Google Scholar]
- 3.a) Li Z, Brouwer C, He C. Chem Rev. 2008;108:3239. doi: 10.1021/cr068434l. [DOI] [PubMed] [Google Scholar]; b) Yavari K, Aillard P, Zhang Y, Nuter F, Retailleau P, Voituriez A, Marinetti A. Angew Chem, Int Ed. 2014;53:861. doi: 10.1002/anie.201308377. [DOI] [PubMed] [Google Scholar]; c) Widenhoefer RA. Chem-Eur J. 2008;14:5382. doi: 10.1002/chem.200800219. [DOI] [PubMed] [Google Scholar]; d) Huguet N, Echavarren AM. Asymmetric Synthesis II: More Methods and Applications. 2013:205–211. [Google Scholar]; e) Fürstner A. Acc Chem Res. 2014;45:925. doi: 10.1021/ar4001789. [DOI] [PubMed] [Google Scholar]; f) Gu P, Xu Q, Shi M. Tetrahedron Lett. 2014;55:577. [Google Scholar]
- 4.a) Wang YM, Lackner AD, Toste FD. Acc Chem Res. 2014;47:889. doi: 10.1021/ar400188g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Caracelli I, Zukerman-Schpector J, Tiekink ER. Gold Bull. 2013;46:81. [Google Scholar]; c) Handa S, Slaughter LM. Angew Chem, Int Ed. 2012;51:2912. doi: 10.1002/anie.201107789. [DOI] [PubMed] [Google Scholar]; d) Ohmatsu K, Ito M, Kunieda T, Ooi T. Nat Chem. 2012;4:473. doi: 10.1038/nchem.1311. [DOI] [PubMed] [Google Scholar]; e) Phipps RJ, Hamilton GL, Toste FD. Nat Chem. 2012;4:603. doi: 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]; f) Aikawa K, Kojima M, Mikamia K. Adv Synth Catal. 2010;352:3131. [Google Scholar]; g) Lacour J, Moraleda D. Chem Commun. 2009:7073. doi: 10.1039/b912530b. [DOI] [PubMed] [Google Scholar]; h) Aikawa K, Kojima M, Mikami K. Angew Chem Int Ed. 2009;48:6073. doi: 10.1002/anie.200902084. [DOI] [PubMed] [Google Scholar]; i) Mukherjee S, List B. J Am Chem Soc. 2007;129:11336. doi: 10.1021/ja074678r. [DOI] [PubMed] [Google Scholar]; j) Ooi T, Maruoka K. Angew Chem, Int Ed. 2007;46:4222. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]; k) Lacour J, Hebbe-Viton V. Chem Soc Rev. 2003;32:373. doi: 10.1039/b205251m. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Widenhoefer ZRA. Angew Chem, Int Ed. 2007;46:283. doi: 10.1002/anie.200603260. [DOI] [PubMed] [Google Scholar]
- 6.Liu C, Widenhoefer RA. Org Lett. 2007;9:1935. doi: 10.1021/ol070483c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Ito Y, Sawamura M, Hayashi T. J Am Chem Soc. 1986;108:6405. [Google Scholar]; b) Sun YW, Xu Q, Shi M. Beilstein J Org Chem. 2013;9:2224. doi: 10.3762/bjoc.9.261. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sengupta S, Shi X. Chem Cat Chem. 2010;2:609. [Google Scholar]; d) Zhang ZM, Chen P, Li W, Niu Y, Zhao X, Zhang J. Angew Chem, Int Ed. 2014;53:4350. doi: 10.1002/anie.201401067. [DOI] [PubMed] [Google Scholar]
- 8.a) Raubenheimer HG. Angew Chem, Int Ed. 2012;51:5042. doi: 10.1002/anie.201200739. [DOI] [PubMed] [Google Scholar]; b) Slaughter LM. ACS Catal. 2012;2:1802. [Google Scholar]; c) Barbazanges M, Fensterbank L. Chem Cat Chem. 2012;4:1065. [Google Scholar]; d) Cera G, Bandini M. Isr J Chem. 2013;53:848. [Google Scholar]
- 9.a) Francos J, Grande-Carmona F, Faustino H, Iglesias-Sigüenza J, Díez E, Alonso I, Fernández R, Lassaletta JM, López F, Mascareñas JL. J Am Chem Soc. 2012;134:14322. doi: 10.1021/ja3065446. [DOI] [PubMed] [Google Scholar]; b) Gu P, Xu X, Shi M. Tetrahedron Lett. 2014;55:577. [Google Scholar]
- 10.Andrushko V, Andrushko N. Stereoselective Synthesis of Drugs and Natural Products II. Wiley-VCH; 2013. pp. 1280–1836. [Google Scholar]
- 11.a) Arbour JL, Rzepa HS, Contreras-García J, Adrio LA, Barreiro EM, Hii KK. Chem Eur J. 2012;18:11317. doi: 10.1002/chem.201200547. [DOI] [PubMed] [Google Scholar]; b) Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Bender CF, Widenhoefer RA. J Am Chem Soc. 2007;129:14148. doi: 10.1021/ja0760731. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Bender CF, Widenhoefer RA. Org Lett. 2007;9:2887. doi: 10.1021/ol071108n. [DOI] [PubMed] [Google Scholar]
- 14.Huang S, Voigtritter KR, Unger JB, Lipshutz BH. Synlett. 2010:2041. [Google Scholar]
- 15.This class of allenic acid substrates has recently been developed in related lactonization reactions catalyzed by achiral gold complexes; see Handa S, Subramanium SS, Ruch AA, Tanski JM, Slaughter LM. doi: 10.1039/c4ob02640c. submitted.
- 16.Brown TJ, Weber D, Gagné MR, Widenhoefer RA. J Am Chem Soc. 2012;134:9134. doi: 10.1021/ja303844h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipshutz BH, Isley NA, Fennewald JC, Slack ED. Angew Chem, Int Ed. 2013;52:10952. doi: 10.1002/anie.201302020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Sheldon RA, Arends IWCE, Hanefeld U. Green Chemistry and Catalysis. Wiley-VCH; 2007. pp. 1–47. [Google Scholar]; b) Sheldon RA. Green Chem. 2007;9:1273. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.