

Abstract
Background
The GBGT1 gene encodes the globoside alpha-1,3-N-acetylgalactosaminyltransferase 1. This enzyme catalyzes the last step in the multi-step biosynthesis of the Forssman (Fs) antigen, a pentaglycosyl ceramide of the globo series glycosphingolipids. While differential GBGT1 mRNA expression has been observed in a variety of human tissues being highest in placenta and ovary, the expression of GBGT1 and the genes encoding the glycosyltransferases and glycosidases involved in the biosynthesis of Fs as well as the possible involvement of DNA methylation in transcriptional regulation of GBGT1 expression have not yet been investigated.
Results
RT-qPCR profiling showed high GBGT1 expression in normal ovary surface epithelial (HOSE) cell lines and low GBGT1 expression in all (e.g. A2780, SKOV3) except one (OVCAR3) investigated ovarian cancer cell lines, a finding that was confirmed by Western blot analysis. Hierarchical cluster analysis showed that GBGT1 was even the most variably expressed gene of Fs biosynthesis-relevant glycogenes and among the investigated cell lines, whereas NAGA which encodes the alpha-N-acetylgalactosaminidase hydrolyzing Fs was not differentially expressed. Bisulfite- and COBRA-analysis of the CpG island methylation status in the GBGT1 promoter region demonstrated high or intermediate levels of GBGT1 DNA methylation in all ovarian cancer cell lines (except for OVCAR3) but marginal levels of DNA methylation in the two HOSE cell lines. The extent of DNA methylation inversely correlated with GBGT1 mRNA and protein expression. Bioinformatic analysis of GBGT1 in The Cancer Genome Atlas ovarian cancer dataset demonstrated that this inverse correlation was also found in primary ovarian cancer tissue samples confirming our cell line-based findings. Restoration of GBGT1 mRNA and protein expression in low GBGT1-expressing A2780 cells was achieved by 5-aza-2’-deoxycytidine treatment and these treated cells exhibited increased helix pomatia agglutinin-staining, reflecting the elevated presence of Fs disaccharide on these cells.
Conclusions
GBGT1 expression is epigenetically silenced through promoter hypermethylation in ovarian cancer. Our findings not only suggest an involvement of DNA methylation in the synthesis of Fs antigen but may also explain earlier studies showing differential GBGT1 expression in various human tissue samples and disease stages.
Keywords: Globo series, Glycosphingolipids, Forssman antigen, Epigenetics, DNA methylation, Transcriptional regulation
Background
The GBGT1 gene was first described in canine kidney cells by Haslam et al. in 1996 [1] and three years later cloned in human [2]. GBGT1 mRNA expression has been observed in a broad variety of human tissues including small and large intestines, placenta, and ovary [2]. GBGT1 encodes the Forssman synthetase (globoside alpha-1,3-N-acetylgalactosaminyltransferase 1). Although differential methylation of GBGT1 has been associated with inflammatory bowel disease [3] and GBGT1 nonsense and inactivating missense mutations have been identified that produce a truncated or enzymatically inactive enzyme [4], it is not understood how GBGT1 expression is regulated.
Forssman synthetase catalyzes the last step in the biosynthesis of Forssman (Fs) antigen that involves a series of sequential attachments of monosaccharides catalyzed by the glycosyltransferase UGCG, B4GALT6, A4GALT, B3GALNT1, and finally GBGT1 (Figure 1A). However, it is unknown how and to what extent the expression levels of not only each one of these glycosyltransferases but also of the respective glycosidases such as NAGA (alpha-N-acetylgalactosaminidase; hydrolyses Fs antigen) affect the biosynthesis of Fs antigen in the cells. Likewise, it has not yet been investigated whether the expression levels of these glycosyltransferases and glycosidases differ between cancer and normal cells.
Figure 1.

Expression of glycosyltransferase and glycosidase genes (glycogenes) involved in the biosynthesis of Fs antigen. (A) Schematic presentation of the biosynthesis pathway of Fs pentasaccharide glycosphingolipid from glycosylceramide and of the responsible glycosyltransferases (downward arrows) and glycosidases (upward arrows): 1. glycosylceramide; 2. lactosylceramide; 3. globotriaosylceramide (Gb3, Pk); 4. globoside (P antigen, Gb4); 5. Forssman (Fs, IV3GalNAcα-Gb4Cer). Filled circles (glucose, Glc); open circles (galactose, Gal); squares (N-acetylgalactosamine, GalNAc). B) Heat map with the color key representing normalized (ΔCq) and clustering of glycogene expression (rows) among the investigated cell lines (columns). Color key indicates relative level of expression from high (white) to low (black). Dendrogram shows the result of the hierarchical clustering calculation for the glycogene expression. (C) Normalized expression of GBGT1 and NAGA; Mean ± SD (three independent experiments) in the presented cell lines. A high ΔCq value indicates low level of normalized gene expression. (D) Western blot analysis (autoradiograph and corresponding quantitative analysis) showing GBGT1 protein expression in the presented cell lines (actin is the sample loading control).
Fs antigen is a pentaglycosyl ceramide (GalNAcα1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-1Cer) terminated by GalNAc and belongs to the globo series of glycosphingolipids (GSL). GSL comprise a heterogeneous group of membrane lipids formed by a ceramide (Cer) backbone covalently linked to a glycan moiety: either galactose or glucose to form galactosylceramide (GalCer) or glucosylceramide (GlcCer), respectively. GlcCer is the precursor of at least four different series of GSL. Among those are the globo series GSL which play roles in various biological processes: they are present within lipid raft microdomains [5], they associate with multidrug resistance [6], with angiogenesis [7], and with malignant diseases [8,9], with proliferation [10]. and serve as receptors for human pathogens [11]. Only recently, naturally occurring anti-glycan antibodies against globo series GSL have been identified the levels of which were lower in the plasma of ovarian cancer patients compared to healthy women [9,12,13], an observation that allows discriminating cancer patients from healthy women.
The appearance of Fs antigen is controversial. It was thought to be absent in humans and to be expressed exclusively on animal red blood cells [14]. This is in contrast to studies demonstrating that Fs antigen is present in human cancer [15,16]. Svensson and colleagues found the Fs GSL expressed also on human erythrocytes [17].
In the present study we (i) profiled a panel of ovarian cancer and normal ovary surface epithelial cell lines for the expression of GBGT1, NAGA, and the other glycogenes encoding the glycosyltransferases and glycosidases involved in the biosynthesis of Fs, (ii) investigated whether GBGT1 expression is regulated through DNA methylation and whether the degree of DNA methylation correlates with GBGT1 expression in cell lines and tissue, and (iii) determined whether GBGT1 expression is an outcome predictor in ovarian cancer.
Our results show that GBGT1 is the most variably expressed gene among the Fs-relevant glycogenes and among the investigated cell lines, DNA methylation is involved in the regulation of GBGT1 expression in ovarian cancer cell lines and tissue, and GBGT1 expression does not predict survival.
Results
Differential expression of Forssman antigen biosynthesis-relevant glycogenes in ovarian cell lines
We investigated whether the expression level of GBGT1 differs between ovarian cancer and normal ovarian surface epithelial cells. Because the biosynthesis of Fs antigen requires multiple glycan-processing enzymes, this investigation also includes the glycogenes encoding the corresponding glycosyltransferases and glycosidases. The biosynthesis pathway of Fs pentasaccharide is outlined in Figure 1A. To this aim we profiled the transcriptional activity of these 11 genes in a panel of ovarian cancer cell lines (TOV112D, TOV21G, OVCAR3, SKOV3, A2780, IGROV1) and normal ovarian surface epithelium cell lines (HOSE6-3, HOSE17-1) using RT-qPCR in concordance with MIQE guidelines [18].
The hierarchical cluster analysis of the ΔCq values for each glycogene, portrayed as a heat map, produced two branches (Figure 1B). One branch clustered eight genes (NAGA, B4GALT6, HEXA, GLB1, GBA, GLA, UGCG, HEXB) with generally comparable expression levels among the cell lines, except for the UGCG gene which was expressed at considerably higher levels in both HOSE cell lines compared with the ovarian cancer cell lines. The other branch clustered three genes (B3GALNT1, A4GALT, and GBGT1) with a wide variation in gene expression levels among the cell lines tested. B3GALNT1 was lowest expressed in TOV112D cells (ΔCq = 17.8), moderately in IGROV1 (ΔCq = 12.9), and highly expressed in the remaining cell lines (ΔCq ranging from 5.5-9.9). A4GALT was generally expressed at high levels in all the cell lines (ΔCq ranging from 5.62-9.54), except for A2780 (ΔCq = 11.98). GBGT1 was highly expressed in both HOSE cell lines (HOSE6-3 ΔCq = 8.86; HOSE17-1 ΔCq = 8.00) and in the OVCAR3 ovarian cancer cell line (ΔCq = 8.74) but expressed at low levels in the remaining cell lines (ΔCqs ranging from 14.44-14.99). Cluster analysis also demonstrated that GBGT1 branched first in the respective cluster, indicating that GBGT1 is the most significantly differentially expressed gene in this cluster among the cell lines, being expressed at low levels in ovarian cancer cells (except OVCAR3) and at high levels in normal ovarian surface epithelial cells.
The preferential expression of GBGT1 in ovarian cancer cell lines was fully confirmed in an additional set of independent experiments (Figure 1C). These results showed that GBGT1 expression is significantly different among the tested cell lines (P < 0.001). GBGT1 expression is high (indicated by low ΔCqmean ± ΔCqSD values) in both HOSE cells (ΔCqHOSE6–3 = 9.07 ± 0.52; ΔCqHOSE17–1 = 8.55 ± 0.55) and in OVCAR3 cells (ΔCqOVCAR3 = 7.88 ± 0.70) and low in A2780 cells (ΔCqA2780 = 15.41 ± 0.62), showing that GBGT1 expression is 185-times (ΔΔCq) lower in A2780 cells than in OVCAR3 cells. The comparison of the two normal cell lines (HOSE6-3 and HOSE17-1) with the panel of ovarian cancer cell lines (TOV112D, TOV21G, SKOV3, OVCAR3, IGROV1 and A2780) revealed a significantly differential GBGT1 expression (P < 0.001). In contrast to GBGT1, NAGA (encodes the alpha-N-acetylgalactosaminidase that hydrolyses Fs pentasaccharide) was not differentially expressed among the cell lines. Western blot analysis and densitometry data (Figure 1D) demonstrated that HOSE6-3, HOSE17-1 and OVCAR3 cells display higher levels of GBGT1 protein than A2780, TOV112D, TOV21G, and IGROV1 cells. These data were (apart from SKOV3 showing high GBGT1 protein level despite low mRNA levels) largely consistent with the RT-qPCR data.
Silencing of GBGT1 expression in ovarian cancer cells by DNA methylation
We next investigated whether the observed differential expression levels of GBGT1 among cell lines correlated with the DNA methylation status of its CpG island promoter region. Combined bisulfite and restriction analysis (COBRA) was performed to determine the methylation status within a 217 bp fragment of the CpG island located on chromosome 9: 136038417–136039577. This CpG island contains 105 individual CpG sites spanning 1161 bp, according to the UCSC Genome Bioinformatics data base (http://www.genome.ucsc.edu). The targeted genomic DNA sequence for the COBRA assay is located within the CpG island from position +28 bp to +245 bp downstream from the transcription start site (+1 bp) (Figure 2A). The results showed substantial levels of DNA methylation in the three investigated CpG sites in TOV112D, TOV21G, and A2780 cells (Figure 2B), consistent with the qPCR and protein data showing significantly reduced GBGT1 expression (Figure 1C, D). In contrast, no methylation was found in OVCAR3 and in both HOSE cells (Figure 2B) which exhibited high levels of GBGT1 expression. An intermediate level of methylation was found in SKOV3 and IGROV1 cells (Figure 2B).
Figure 2.
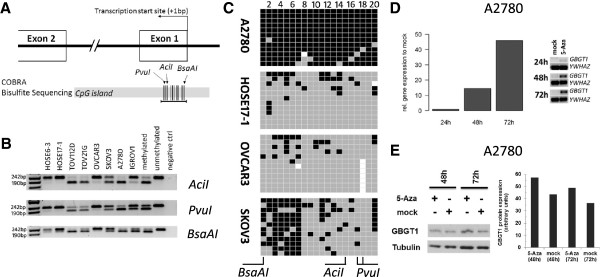
DNA methylation of GBGT1 promoter region. (A) Illustration of the organization of the CpG island encompassing the transcription start site of the GBGT1 gene. The horizontal black line represents the 217 bp amplicon generated using COBRA and bisulfite sequencing. Vertical lines represent the organization of individual CpG dinucleotides within the amplified region. Black triangles indicate the respective recognition sites of the restriction endonucleases (Acil, Pvul and BsaAl). Each of these enzymes digested the amplicon only when the respective CpG dinucleotide(s) within the enzyme recognition site was methylated in the original DNA prior to bisulfite conversion (AciI-active 50 + 176 bp; PvuI-active 37 + 180 bp; BsaAI-active 192 + 25 bp). (B) COBRA with each of the three different endonucleases (Acil, Pvul, BsaAl) revealed considerable variation in the methylation status among the cell lines. The degree of methylation (which relates to the intensity of the lower digested bands as compared to the upper undigested band) was consistent between each restriction enzyme for each cell line. (C) Methylation profiles of individual CpG sites from single DNA strands derived from bisulfite sequencing in A2780, HOSE17-1, OVCAR3, and SKOV3. Columns represent individual CpG sites. Rows represent number of sequenced clones (n = 12). Methylated CpG (black), unmethylated (grey), unknown status (white). (D) Restoration of GBGT1 expression in A2780 induced by treatment with 2.5 μM 5-Aza. RT-qPCR shows a time-dependent increase in GBGT1 transcription. Left, data are presented as the number of PCR products in 5-Aza treated samples relative to the mock-treated control (y-axis) as a function of time (24 h, 48 h, 72 h) after treatment (x-axis). Right, RT-qPCR products of GBGT1 and normalization control YWHAZ. (E) Western blot (autoradiograph and corresponding quantitative analysis) showing 5-Aza -induced increase in GBGT1 protein expression as a function of time after treatment in A2780.
Bisulfite sequencing was performed to quantify the GBGT1 promoter methylation levels in A2780 (high level), HOSE17-1 (low level), OVCAR3 (low level), and SKOV3 (intermediate level), and also to determine the extent of methylation across the CpG island fragment within individual DNA strands. Figure 2C showed that 95.8% of all CpGs are methylated in A2780, whereas only 12.1% were methylated in HOSE17-1 and 9.5% in OVCAR3 cells. The SKOV3 cell line displayed an intermediate 40% methylation of CpGs, preferentially at the first 7 CpG sites. Bisulfite sequencing therefore showed a strong inverse correlation between GBGT1 expression and DNA methylation (r = −0.86; Pearson correlation), indicating that the majority of ovarian cancer cells have silenced GBGT1 expression through DNA methylation.
Increased GBGT1 expression induced by 5-aza-2’-deoxycytidine treatment
We determined whether silenced GBGT1 expression in A2780 cells (95.8% methylation) can be reversed by 5-aza-2’-deoxycytidine (5-Aza) treatment. 5-Aza is a DNA methyltransferase inhibitor that de-methylates the DNA and therefore reactivates or enhances the transcription of genes suppressed by DNA methylation [19]. We found that 5-Aza produced a 15-fold increase in GBGT1 expression after 48 h and a 46-fold increase after 72 h in A2780 cells (Figure 2D), which is also reflected on the GBGT1 protein level (Figure 2E). A 5-Aza-induced effect was also observed for SKOV3 cells (40% methylation) though to a lesser extent (data not shown).
Elevated HPA-staining in A2780 cells after 5-aza-2’-deoxycytidine treatment
In order to determine whether the 5-Aza-induced increase in GBGT1 expression results in elevated presence of Fs antigen on A2780 cells we used HPA (Helix pomatia agglutinin)-staining as a “surrogate marker” for Fs antigen. HPA is a lectin which preferentially binds the Fsdi disaccharide [20], the terminal structure of the Fs antigen pentasaccharide. Flow cytometry analysis data showed increased HPA-staining in 5-Aza treated A2780 cells (48 h p = 0.25; 72 h p = 0.08; student t test; Figure 3) which reflects the elevated presence of Fs on these cells.
Figure 3.
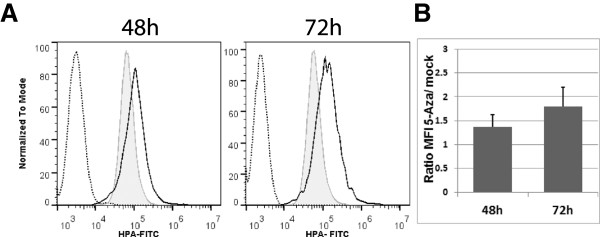
Elevated HPA-staining in A2780 cells treated with 5-Aza. (A) Flow cytometry data (representative of three independent experiments) showing a rightwards peak shift (black solid line) after 48 h and 72 h of 5-Aza-treatment relative to the mock control (grey line). The unstained control is shown by the dashed line. (B) Plot illustrates the increase in HPA-staining manifested as the increase in the mean fluorescence intensity (MFI) relative to the mock controls as a function of time after 5-Aza -treatment. Data presented as the mean ± SD of three independent experiments.
Differential DNA methylation of GBGT1 in ovarian tumor and normal adjacent tissue (analysis of The Cancer Genome Atlas (TCGA) data)
We wondered whether the observed differential GBGT1 methylation pattern, i.e. largely unmethylated GBGT1 in the normal HOSE cells and hypermethylated in ovarian cancer cells, was also found in primary and recurrent ovarian carcinoma and in normal adjacent tissue. To this aim we analyzed the methylation levels of GBGT1 using the publicly accessible data from The Cancer Genome Atlas (TCGA) ovarian cancer sample set [21]. DNA methylation at two CpG sites within the GBGT1 CpG island promoter, cg18089000 (located 403 bp upstream of the GBGT1 transcription start site,) and cg01169778 (located 613 bp downstream of it), were represented on the Illumina Infinium HumanMethylation27 BeadChip array (27 k array). Methylation between these two sites was significantly correlated within primary tumor data (n = 528, Spearman rho = 0.32, P = 1.352e-15). Within the TCGA ovarian tissue dataset, DNA methylation data for normal adjacent ovarian tissue were available for a small number (n = 10) of individuals, of which only four had matched tumor. The levels of DNA methylation in ovarian tumors from the TCGA dataset varied widely, which was consistent with our findings in the ovarian cancer cell lines. GBGT1 methylation levels at both cg01169778 and cg18089000 in adjacent normal ovarian tissues were consistently high (Figure 4A,B). When all normal samples (n = 10) were compared with all primary tumors samples (n = 528), no significant difference in methylation at either cg01169778 (P = 0.5694) or cg18089000 (P = 0.1136) was found (Figure 4A,B). However, across individuals for which paired tumor and normal-adjacent tissue methylation data was available, cg01169778 was higher in tumors compared with normal adjacent tissue in all four individuals (mean difference = 19.1% between tumors and normal adjacent) and this difference was borderline statistically significant (P = 0.056) (Figure 4C). Methylation of cg18089000 was not significantly different between these four matched normal and tumor pairs (P = 0.43).
Figure 4.

GBGT1 methylation and RNA expression levels in The Cancer Genome Atlas (TCGA) ovarian cancer data. Methylation levels within the GBGT1 CpG island, detected at the (A) cg01169778 and (B) cg18089000 probes on the Illumina Infinium HumanMethylation27 BeadChip array in normal-adjacent ovarian tissue (n = 10), primary ovarian tumor (n = 528) and recurrent tumor (n = 33), do not show a difference among these three groups. (C) Box-plot showing a borderline significant difference in methylation between matched normal adjacent tissue and primary tumor pairs (n = 4 cases). (D)GBGT1 RNA expression, determined by the Illumina HiSeq RNA-Seq platform, stratified by GBGT1 methylation levels at the cg18089000 probe divided into quartiles, (n = Q1(lowest):67, Q2:64, Q3:64, Q4:70). (E)GBGT1 gene expression, determined by the Huex-1_0-st-V2 Affymetrix exon array, stratified by quartiles of methylation levels at the GBGT1 cg18089000 probe (n = Q1:141, Q2:145, Q3:144, Q4:141). **P-value (linear regression) <0.01, ***P < 0.001.
Association between DNA methylation and expression
We now investigated the dependence of GBGT1 expression on DNA methylation in tissue samples to translate our cell line-based findings.
Integrated analysis of GBGT1 DNA methylation and gene expression in the TCGA ovarian cancer data set showed that DNA methylation at cg18089000 was inversely correlated with GBGT1 expression in primary tumors, measured using either the Huex-1_0-st-V2 Affymetrix exon array (n = 583, rho = −0.27, P = 1.869e-11), or the Illumina HiSeq RNA-Seq platform (n = 265, rho = −0.42, P = 1.312e-12) (Additional file 1: Table S1). GBGT1 cg18089000 methylation was not significantly correlated with GBGT1 exon expression in normal adjacent tissue (n = 10, Spearman rho = 0.55, P = 0.10) in this small sample size analysis. In linear models adjusted for age at pathological diagnosis and clinical stage, cg18089000 methylation was significantly associated with GBGT1 expression, measured by the exon array (n = 571, P = 0.0005), and RNA-seq data (n = 265, P = 2.05e-05). A dose-effect relationship between cg18089000 methylation and gene expression is apparent when GBGT1 expression is stratified by DNA methylation quartiles (Figure 4D,E). In contrast, cg01169778 methylation did not significantly correlate with GBGT1 expression or associate in linear models in any sample type using either gene expression measurement (Additional file 1: Table S1).
Discussion
The present study shows that (i) GBGT1 is the most differentially expressed glycogene involved in the biosynthesis of Fs antigen and (ii) that GBGT1 expression is silenced through DNA hypermethylation in ovarian cancer cells and tissue but re-activated by 5-Aza treatment. In contrast GBGT1 (iii) does not correlate with clinical outcome (survival) in ovarian cancer patients. We therefore conclude that GBGT1 expression is epigenetically regulated in ovarian cancer but that it is not a prognostic predictor for survival.
The major finding from the present study is DNA methylation is one mechanism by which GBGT1 expression is regulated in both ovarian cancer cells and tissue. This is a novel finding and expands on a previous study reporting that GBGT1 is differentially methylated and associated with inflammatory bowel disease [3]. DNA methylation is one out of at least four epigenetic mechanisms by which eukaryotic organisms regulate the transcription of a significant number of genes, and aberrant hypermethylation of CpG island promoters associated with transcriptional silencing is a frequent occurrence in various types of cancer [22]. Epigenetic silencing is, however, only one mechanism by which GBGT1 expression is regulated. GBGT1 nonsense and inactivating missense mutations have been identified that produce a truncated or enzymatically inactive protein [4].
Interestingly, a correlation between GBGT1 repression and DNA hypermethylation was notably not only found in ovarian cancer cell lines. A bioinformatical analysis of the Ovarian TCGA dataset showed a clear-cut inverse correlation between the degree of methylation at the probe closest to the transcription start site and GBGT1 mRNA expression levels within the TCGA primary ovarian cancers, although the degree of GBGT1 methylation varied considerably among samples (ranging from nearly unmethylated to hypermethylated) and levels of GBGT1 methylation were also detected in adjacent normal ovarian tissue. Notably, however, in the four TCGA cases with matched tumor and normal adjacent tissues higher methylation levels were found in tumor samples compared to normal adjacent tissues, being in concordance with our cell line-based observation. The present study is therefore not only the first one addressing GBGT1 expression and its regulation in ovarian cancer but also suggest that GBGT1 is differentially expressed in cancer and healthy conditions i.e. lower in ovarian cancer patients and higher in normal cells. This observation is consistent with a previous study showing GBGT1 mRNA expression in various tissues [2].
From a clinical point of view it is interesting whether GBGT1 methylation and/or expression levels associated with clinical outcome parameters such as survival and therefore serve as a prognostic factor. However, our ovarian cancer TCGA dataset analysis did not reveal an association between methylation or expression levels and survival (neither in univariate models nor when age- and stage-adjusted), indicating that GBGT1 methylation or expression levels are not outcome parameters in ovarian cancer.
GBGT1 is not the only glycosyltransferase-encoding gene regulated by DNA methylation. B4GALNT2 and ST3GAL6 have been found to be epigenetically silenced in colon and gastric cancer cells and shown to be re-expressed after 5-Aza treatment [23]. Both are involved in the biosynthesis of the blood group carbohydrate Sda (GalNAcβ1-4(NeuAcα2-3)Galβ1-4GlcNAc) which is abundantly present on normal gastrointestinal tract mucosa cells but decreased on gastric cancers [24]. FUT7 has been shown to be differentially methylated in patients with ulcerative colitis and Crohn's disease [3]. Various genes implicated in the glycosylation of secreted N-linked glycoproteins may also be epigenetically regulate because 5-Aza treated ovarian cancer cells displayed altered glycosylation of these proteins [25].
Another important finding is that A2780 cells incubated with 5-Aza exhibited an elevated HPA staining. HPA is a lectin from Helix pomatia agglutinin that preferentially binds Fsdi (GalNAcα1-3GalNAc-R), the terminal disaccharide (the product of GBGT1) of Fs antigen [20,26,27], and therefore HPA staining may serve as a “surrogate marker” for Fs antigen expression. In this case, this finding together with the 5-Aza-induced increase in GBGT1 expression suggests that the elevated HPA staining may be a result of the increased GBGT1 expression and Fs synthesis in these cells. On the other hand, we cannot exclude that 5-Aza also affects the expression of glycogenes other than GBGT1 that, in turn, produces an “elevated binding” of HPA to glycan structures other than Fsdi. The latter may explain the relatively high level of HPA-staining in “mock”-treated A2780 cells in the respective experiment.
HPA expression has previously been proposed as a determinant for poor prognosis in breast, gastric, and colon cancer [26,28,29]. Given that elevated HPA staining is a result of increased GBGT1 expression and that HPA staining is a determinant for poor prognosis also in ovarian cancer, we would expect increased GBGT1 expression in cancer cells. The observation that GBGT1 expression is lower by trend in ovarian cancer does neither support nor rules out the idea that the HPA-staining may be a prognostic determinant in ovarian cancer. Earlier studies have suggested Fs antibody titers as disease determinants: high Fs autoantibody titers were found in Graves' disease and Hashimoto's thyroiditis [30] and low were found in patients with cancer of the gastrointestinal tract and breast [31].
Conclusions
Our results identify GBGT1 as the most differentially expressed Fs-biosynthesis-relevant glycogene among the cell lines investigated and provide evidence that DNA methylation at its promoter region is one mechanism by which GBGT1 expression is regulated on the transcriptional level in ovarian cancer. This mechanism may explain the observed differential GBGT1 expression in earlier studies.
Methods
Cell lines
The serous ovarian cancer cell lines OVCAR3, SKOV3, A2780 and IGROV1 were cultured in RPMI 1640 medium, the endometrioid ovarian cancer cell line TOV112D and the clear cell ovarian cancer cell line TOV21G in DMEM, and the (normal) human ovarian surface epithelial cells HOSE6-3 and HOSE17-1 in 1:1 Medium 199:MCDB 105. All media (Sigma-Aldrich Pty. Ltd, Castle Hill, Australia) contained 10% FCS (Sigma-Aldrich Pty. Ltd) and penicillin/streptomycin (Sigma-Aldrich Pty. Ltd). Cells were cultured at 37°C in 5% CO2. All cultures were free of mycoplasma (routinely checked as by qualitative PCR using VenorGeM® Mycoplasma Detection Kit, Biocene Pty Ltd, Rozelle, Australia).
RNA and DNA extraction
To examine the expression of the glycogenes and the reference genes, cell cultures were grown in 6-well plates (NUNC, Thermo Fisher Scientific, Roskilde, Denmark), washed twice with DPBS (Invitrogen Pty Ltd, Thornten, NSW, Australia), and lysed by the addition of lysis buffer directly onto the cells (NucleoSpin RNAII Kit, Macherey&Nagel, Scientifix, Clayton, VIC, Australia). Total RNA was extracted according to the manufacturer’s protocol (NucleoSpin RNAII Kit, Macherey&Nagel). After the elution of total RNA in 50 μl RNase-free water, RNA concentration and the ratios for A260/230nm and A260/280nm were measured (NanoDrop ND-1000, Thermo Fisher Scientific). The integrity of the RNA samples, i.e. the RNA integrity number (RIN) and 28S/18S rRNA ratio, was determined by electropherograms (Agilent Bioanalyzer RNA 6000 Nano).
Genomic DNA was extracted from cell cultures grown in 6-well plates. Cells were lysed with 250 μl lysis buffer (20 mM Tris–HCl, 4 mM Na2EDTA, 100 mM NaCl), followed by the addition of 25 μl of 10% (w/v) sodium dodecyl sulfate (SDS). The lysate was transferred into an Eppendorf tube and vigorously vortexed. Then 2.5 μl Proteinase K (Finnzymes, Genesearch Pty Ltd., Arundal, Queensland, Australia) was added and protein digestion was performed for at least 2 h at 55°C. Proteins were then precipitated by adding 200 μl of 5.3 M NaCl, followed by centrifugation at 13’000xg for 30 min at 4°C. The supernatant was transferred to a different tube and an equal volume of ice-cold isopropanol was added in order to precipitate the DNA by inverting the tubes several times. After centrifugation for 15 min (13’000xg) the DNA was washed with 70% ethanol and re-centrifuged for 10 min. Genomic DNA was dissolved in 10 mM Tris HCl pH 8.5 and the DNA concentration was measured with the NanoDrop ND-1000 (Thermo Fisher Scientific).
All RNA extracts were of high quality and purity: the RNA integrity number (RIN) was 10, the values for A260/280 and A260/230 ranged from 2.08-2.16 and 1.80-2.21, respectively, and the 28 s/18 s ratio ranged from 1.9-2.3.
Reverse Transcription (RT) and quantitative Polymerase Chain Reaction (qPCR)
An amount of 500 ng to 1 μg total RNA was reverse-transcribed using the iScript Reverse Transcription Supermix for RT-qPCR according to the manufacturer’s instructions (Bio-Rad Laboratories (Pacific) Pty Ltd, Gladesville, Australia). The complementary DNA (cDNA) was stored at −20°C until further use.
QPCR was performed on the selected glycogenes and reference genes (HSPCB, SDHA, YWHAZ) in order to investigate the relative expression of the glycogenes in the selected cell lines and the patient samples. The variation of reference genes for normalization (mean coefficient of variation of 4.75%) reflecting biological and inter-assay variability was in full concordance with our previous observation on selected reference genes in Jacob et al. 2013 [32]. A listing of the accession number, the name of the gene, the sequence of forward and reverse primers, and the amplicon size is presented in Table 1. The reference gene primers were selected according to their specificity to human cDNA and their stable expression pattern. The ‘glyco gene’ primers were designed using QuantPrime [33] (purchased from Sigma-Aldrich Pty. Ltd). QPCR was performed with Stratagene Mx3005® (Integrated Sciences Pty. Ltd, Chatswood, Australia) in 96-well microtiter plates using the 2x SensiFAST™ SYBR Lo-ROX kit (Bioline Pty Ltd, Alexandria, Australia) with low ROX as the reference dye. Optimum reaction conditions were obtained with 400 nM specific sense primer, 400 nM specific antisense primer, RNase/DNase-free water, and cDNA template up to final volume of 20 μl. QPCR setups were based on 10 ng of the previously isolated and reverse-transcribed total RNA. Amplifications were performed starting with a 30 sec enzyme activation at 95°C, followed by 40 cycles of denaturation at 95°C for 5 sec, and an annealing/extension step at 60°C for 30 sec. A melt curve was produced from 65-95°C (continuous measurement). All samples including the negative controls were amplified in triplicates. Mean, standard deviation (SD) and coefficient of variation were calculated. For inter-assay comparability a 0.1 threshold was set for all runs for the determination of the Cq values. Quantification cycle (Cq) values of >36 were excluded from further mathematical calculations, because Cq > 36 do not represent quantitative information of the cDNA/RNA amount and therefore presents the end of the qPCR. All RT-qPCR experiments were performed in compliance to MIQE guidelines [18].
Table 1.
qPCR primers
| Gene | Accession number | Gene Name | Forward Primer 5’-3’ | Reverse Primer 5’-3’ | Amplicon size in bp |
|---|---|---|---|---|---|
|
A4GALT |
NM_017436 |
α1,4-galactosyl transferase |
CGCTGGAGCTAGAGATGGATTTGC |
AGCCGACCTTCTTTGCCAACAC |
78 |
|
GLA |
NM_000169 |
α galactosidase A precursor |
TCTAATGACCTCCGACACATCAGC |
ACACTTCAAAGTTGTCTCCCTGTC |
130 |
|
HEXA |
NM_000520.4 |
β hexosaminidase α chain precursor |
TTTGTCACACTTCCGCTGTGAG |
ACTCCTGCTCACAGAAGCCTAC |
80 |
|
GBA |
NM_001171812 |
glucosidase, β, acid |
ACAGCCACAGCATCATCACGAAC |
TGGGACTGTCGACAAAGTTACGC |
117 |
|
UGCG |
NM_003358 |
UDP-glucose ceramide glucosyltransferase |
TGTGTTGGATCAAGCAGGAGGAC |
AACCTCCAACCTCGGTCAGCTATC |
96 |
|
B4GALT6 |
NM_004775 |
UDP-Gal:βGlcNAcβ1,4- galactosyltransferase, polypeptide 6 |
AGGAGGTCCCTATGGCACTAAC |
TCTCTACAGACAGGCCCATTAGTC |
89 |
|
GLB1 |
NM_001079811 |
galactosidase, β1, transcript variant 1 |
TGGCCAGCCATTTCGCTACATC |
TGAAAGTTCCAGGGCACATACGTC |
135 |
|
HEXB |
NM_000521 |
β-hexosaminidase β chain precursor |
GCAAGTGCTGTTGGTGAGAGAC |
GTTGTGCAGCTATTCCACGTTCG |
118 |
|
GBGT1 |
NM_021996.4 |
Globoside α-1,3-N-acetylgalactosaminyltransferase 1 |
GCACAAGCTTCAGTGTCCTGTG |
TGGCTTCTCCCTCTTGTAGTGC |
122 |
|
B3GALNT1 |
NM_033169 |
β1,3-N-acetylgalactosaminyl transferase 1 |
TGCTCTATCACGTGGTGCTCTC |
ACGCGAGCCGAAGGTTCTTTAC |
62 |
|
NAGA |
NM_000262.2 |
N-acetylgalactosaminidase, α |
AGCTTCCAGAGCCCAACACATAC |
ACATGTCCCAGCAAGAGCACTG |
113 |
|
HSPCB |
NM_007355 |
Heat shock protein 90 kDa α |
TCTGGGTATCGGAAAGCAAGCC |
GTGCACTTCCTCAGGCATCTTG |
80 |
|
YWHAZ |
NM_001135702 |
Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypetide |
ACTTTTGGTACATTGTGGCTTCAA |
CCGCCAGGACAAACCAGTAT |
94 |
| SDHA | NM_004168 | Succinate dehydrogenase complex, subunit A | TGGGAACAAGAGGGCATCTG | CCACCACTGCATCAAATTCATG | 86 |
The qPCR assay performance for each glycogene and three reference genes (HSPCB, SDHA and YWHAZ) was determined in four consecutive 10-fold dilution steps. The value for PCR efficiency ranged from 97.0% (GBGT1) to 107.3% (HEXA), and the coefficient of determination (R2) was always higher than 0.995, indicating that the qPCR assay performance was reliable (Table 2). An amount of 10 ng RNA was used for each of the gene transcripts in the profiling experiments.The ΔCq values for each glycogene, obtained by normalization against the logarithmic mean of the three reference genes profiled in parallel, and for each cell line were subjected to clustering analysis and presented as a heat map (Figure 1).
Table 2.
qPCR parameters describing the standard curve for each primer pair on eleven glycogenes and three reference genes
| Gene | Slope | Intercept | Efficiency in % | R 2 | Dilution range |
|---|---|---|---|---|---|
|
UGCG |
−3.1875 |
21.58 |
105.9 |
0.999 |
10 ng-100 ng |
|
GBA |
−3.2175 |
24.48 |
104.6 |
0.998 |
10 pg-100 ng |
|
B4GALT6* |
−3.1905 |
29.01 |
105.8 |
0.998 |
5 pg-50 ng |
|
GLB1 |
−3.1640 |
24.29 |
107.0 |
0.991 |
1 pg-10 ng |
|
A4GALT |
−3.3425 |
28.63 |
99.1 |
0.998 |
10 pg-100 ng |
|
GLA |
−3.2025 |
25.75 |
105.2 |
0.998 |
10 pg-100 ng |
|
B3GALNT1 |
−3.2425 |
27.92 |
103.4 |
0.999 |
10 pg-100 ng |
|
HEXA |
−3.1575 |
23.92 |
107.3 |
0.999 |
10 pg-100 ng |
|
HEXB |
−3.2300 |
22.51 |
104.0 |
0.999 |
10 pg-100 ng |
|
GBGT1 |
−3.395 |
32.69 |
97.0 |
0.995 |
100 pg-100 ng |
|
NAGA |
−3.152 |
28.36 |
107.0 |
0.999 |
100 pg-100 ng |
|
HSPCB* |
−3.250 |
20.09 |
103.1 |
0.998 |
1 pg-100 ng |
|
YWHAZ* |
−3.294 |
20.35 |
101.2 |
0.998 |
1 pg-100 ng |
| SDHA* | −3.194 | 24.64 | 105.6 | 0.994 | 1 pg-100 ng |
*Standard curve was established previously [32].
Bisulfite conversion and COBRA (Combined Bisulfite Restriction Analysis)
Bisulfite conversion was performed using the EZ DNA Methylation-Gold™ Kit according to the manufacturer’s protocol (ZYMO RESEARCH, Integrated Sciences, Pty. Ltd, Chatswood, Australia). An amount of 500 ng genomic DNA was used for bisulfite conversion.
Bisulfite converted genomic DNA was subjected to polymerase chain reaction (PCR) using Platinum® Taq DNA Polymerase (Invitrogen Pty. Ltd., Blackburn, Australia). The primers, designed using MethPrimer [34] (purchased from Sigma-Aldrich Pty. Ltd., Castle-Hill, Australia). PCR was set up as follows: 1x Platinum Taq reaction buffer, 1 mM MgCl2, 100 nM dNTP mix, 400 nM forward (GBGT1_COBRAfor6; 5’-TGT TTG TTT TGT TTT AGG TTT TAT TTG-3’) and reverse primer (GBGT1_COBRArev6; 5’-AAA ACC CTC CTC CTT AAC CCC TTA C-3’) and 0.05 units of Taq polymerase made up with Ultra Pure™ distilled water (Invitrogen Pty. Ltd., Blackburn, Australia) to a final volume of 20 μl. PCR was performed by initial denaturation at 94.0°C for 5 min, followed by 40 cycles of 94°C for 30 sec, 58°C for 30 sec, 72°C for 45 sec, and finally by amplicon elongation at 72°C for 10 min. The PCR product (size 217 bp) was visualized in a 1.7% agarose gel. Restriction fragment length analysis was performed on PCR products incubated with the restriction endonucleases AciI (methylated 50 bp + 167 bp, unmethylated 217 bp), PvuI-HF (methylated 37 bp + 180 bp, unmethylated 217 bp) and BsaAI (methylated 192 bp + 25 bp, unmethylated 217 bp) (New England BioLabs Inc., Genesearch Pty Ltd., Arundal, Queensland, Australia). Genomic DNA isolated from human blood serum served as an unmethylated COBRA control. Genomic DNA from the same sample treated with CpG methyltransferase M.SssI (New England BioLabs Inc., Genesearch Pty Ltd.) served as the positive methylation control.
Bisulfite Sequencing
COBRA-based PCR for GBGT1 was performed as described above. For subsequent ligation, the PCR products were extracted from the 1.7% agarose gel using the QIAquick® Gel Extraction Kit (Qiagen Pty Ltd, Doncaster, VIC, Australia) and ligated into pGEM®-T Easy Vector System I according to the manufacturer’s protocol (Promega, Alexandria, NSW, Australia). Afterwards, the entire ligation was heat shock transformed into E.coli XL10 GOLD strain. Bacterial cells were held together with the ligation on ice for 30 min, followed by incubation at 42°C for 35 sec. Immediately after heat shock, bacterial cells were placed on ice for 2 min and pre-warmed medium (Sigma-Aldrich Pty. Ltd., Castle-Hill, Australia) was added, followed by incubation at 37°C for 1 h. Transformed bacteria were plated on LB agar plates containing 50 μg/ml carbenicillin, IPTG, and X-Gal, and were incubated overnight at 37°C. White colonies were picked and colony PCR was performed using the T7 universal primer (5’-TAA TAC GAC TCA CTA TAG GG-3’) and SP6 universal primer (5’-ATT TAG GTG ACA CTA TAG-3’) (Sigma-Aldrich Pty) to amplify the plasmid inserts. The conditions were as follows: 1x Platinum Taq reaction buffer, 3 mM MgCl2, 100nM dNTP mix, 400nM forward and reverse primer, and 0.05 units of Taq polymerase made up with Ultra Pure™ distilled water up to a final volume of 20 μl. PCR samples were separated on a 1.7% agarose gel. Free dNTPs were removed by enzymatic PCR clean-up and the remaining primers were incubated with the PCR products for 30 min at 37°C with 2.5U Antarctic phosphatase and 10U Exonuclease I (New England BioLabs Inc., Genesearch Pty Ltd., Arundal, QLD, Australia) dissolved in 1× Antarctic Phosphatase Reaction buffer. The reaction was heat-inactivated at 80°C for 20 min. The PCR clean-up reaction was applied to the sequencing reaction, containing 0.5 μl BigDye®, Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems Australia Pty Ltd, Mulgrave, VIC, Australia), 2 μl 5× sequencing reaction buffer, 0.35 μl SP6 universal primer, and 2.5 μl distilled water. The sequencing reaction was performed using the following conditions: 94°C for 20 sec, 50°C for 20 sec, and 60°C for 4 min. All steps were repeated for 25 cycles. DNA was precipitated by 100% ethanol and 3 M sodium acetate pH5.2, washed with 70% ethanol, and air-dried. Samples were sequenced (ABI 3730 Capillary Sequencer, The Ramaciotti Centre for Gene Functional Analysis, University of New South Wales, Sydney, NSW, Australia). Bisulfite sequencing data were visualized and analyzed using the web based software BISMA, Bisulfite Sequencing Data Presentation and Compilation [35].
5-aza-2’-deoxycytidine treatment
Cells carrying a hypermethylated GBGT1 gene were treated with 5-Aza (Sigma-Aldrich Pty. Ltd; stock solution prepared in 50% (v/v) acetic acid) in order to restore or increase GBGT1 expression. An amount of 105 cells were seeded in 6-well plates (NUNC, Thermo Fisher Scientific Pty. Ltd., Scoresby, Australia) and treated with 5-Aza (2.5 μM final concentration) on the next day. The culture medium was removed every 24 h and replaced by fresh culture medium containing 2.5 μM 5-Aza. Samples were harvested after 24 h, 48 h and 72 h of treatment. Genomic DNA and total RNA were extracted as described above. Mock controls contained 50% (v/v) acetic acid at concentrations identical to 5-Aza.
Preparation of cell lysates and Western blot analysis
Whole cell lysates were obtained from subconfluent cultures. The cultures were treated with 5-Aza or a corresponding concentration of acetic acid (mock control) 24 hours after seeding. Cells were then harvested 48 h or 72 h after treatment. Cells were lysed for Western blot analysis according to standard laboratory protocols. The protein concentration of cell lysates was determined by the BCA Protein Assay Kit (Pierce, Perbio Science, Lausanne, Switzerland). Twenty μg protein was loaded and separated using SDS-PAGE, followed by blotting onto a polyvinylidene difluoride membrane (Amersham Biosciences, Otelfingen, Switzerland). Proteins were detected with specific primary antibodies (goat anti-GBGT1, Lab Force, Nunningen, Switzerland; anti-mouse β-actin, Sigma-Aldrich, Buchs, Switzerland; anti-rabbit tubulin, Cell Signaling, BioConcept, Allschwil, Switzerland) and the appropriate horseradish peroxidase-conjugated secondary antibodies (anti-goat IgG-HRP, Lab Force, Basel, Switzerland; anti-rabbit IgG-HRP and anti-mouse IgG-HRP, Cell Signaling, BioConcept, Basel, Switzerland). Complexes were visualized by enhanced chemiluminescence (Amersham Biosciences, Otelfingen, Switzerland) and autoradiography. Densitometry (quantitative analysis of the intensity of the complexes on the autoradiograph, normalized against actin orv tubulin) was performed using the XXXXX software.
Flow cytometry analysis
The presence of Fsdi on the cell membrane was analyzed by flow cytometry (BD Accuri C6 flow cytometer, BD Bioscience, Basel, Switzerland). Biotin-conjugated helix pomatia agglutinin (HPA; Sigma-Aldrich Pty. Ltd., Castle-Hill, Australia) followed by streptavidin conjugated to FITC (BD Bioscience, Basel, Switzerland) was used for fluorescence detection. Single cells were gated to quantify HPA-positive cells (cell debris excluded). Data acquisition was performed using BD Accuri C6 software (BD Bioscience, Basel, Switzerland). Data analysis was performed using the FlowJo v10 software (Tree Star Inc., Ashland, USA).
TCGA clinical sample data analyses
The Cancer Genome Atlas ovarian cancer molecular and clinical data was downloaded from the Broad Institute TCGA Data and Analysis website (https://confluence.broadinstitute.org/display/GDAC/Home) which provides data subjected to standardized preprocessing and normalization, as described in detail on their website.
DNA methylation was measured using the Illumina Infinium HumanMethylation27 BeadChip array (n = 582 primary ovarian tumor sample, 10 normal adjacent tissue samples and 18 recurrent tumor samples). GBGT1 expression was measured using the Huex-1_0-st-V2 exon array (n = 583 primary tumors overlapping with DNA methylation data) and Illumina HiSeq RNA-Seq (n = 265 primary tumors overlapping with DNA methylation data) platforms. Technical analysis parameters were as described on the TCGA website (http://cancergenome.nih.gov/).
Statistical analysis
Basic R packages and R package ‘qPCR’ [36] from the open source statistical programming language R (http://CRAN.R-project.org/, version 2.13.2) were used for data analysis of qPCR (including the calculations of PCR efficiency, slope, intercept and r squared (r2)). The one-way ANOVA test with a significance level of P < 0.01 was applied to compare gene expression among the cell lines. Student’s t test was applied to compare gene expression levels between normal and cancer cell lines. Pearson correlation (R package “stats”) was performed for the relationship between RT-qPCR data and bisulfite sequencing data.
For TCGA data analyses, the Shapiro-Wilks test was used to test for normal distribution within molecular data. DNA methylation deviated significantly from a normal distribution at GBGT1 cg01169778 (P < 2.2e-16) and cg18089000 (P < 2.2e-16), and RNA-Seq data for GBGT1 was not normally distributed (Shapiro Wilks P = 8.538e-16), therefore data for these variables were log-transformed for all parametric tests, whereas exon array data, which was not abnormally distributed (P = 0.09), was not log transformed. Spearman correlations were used to test for correlations between DNA methylation and gene expression, and linear regression was used to test for associations between methylation and expression, adjusting for other variables, with methylation treated either as a continuous variable or as a categorical variable based on methylation quartiles. The Student’s t-test was used to test for DNA methylation differences between tumor and adjacent normal tissue, with matching for matched pairs. Cox proportional hazard models were used to test for associations between DNA methylation (or gene expression) and survival time, measured as days until death from pathological diagnosis, with methylation treated either as a continuous variable or as a categorical variable based on methylation quartiles. Cox models were adjusted for age at pathological diagnosis and clinical stage, as these factors were significantly associated with survival time (age: P = 0.0007, HR = 1.02, stage: P = 0.0008, HR = 1.3). Kaplan-Meier plots were used to visually compare survival time differences between DNA methylation categories.
Competing interests
The authors have declared that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: FJ MH AF KB SN VHS; performed the experiments: FJ, AF, SN; analyzed the data: FJ MH SN VHS; contributed reagents/ materials/ analysis tools: FJ MH NH RW VHS; drafted and wrote the manuscript: FJ AF MH VHS. All authors read and approved the final manuscript.
Supplementary Material
Association between DNA methylation and gene expression in the TCGA ovarian cancer dataset.
Contributor Information
Francis Jacob, Email: francis.jacob@unibas.ch.
Megan P Hitchins, Email: mhitchin@stanford.edu.
André Fedier, Email: andre.fedier@usb.ch.
Kevin Brennan, Email: kbren@stanford.edu.
Sheri Nixdorf, Email: s.nixdorf@unsw.edu.au.
Neville F Hacker, Email: N.Hacker@unsw.edu.au.
Robyn Ward, Email: robyn@unsw.edu.au.
Viola A Heinzelmann-Schwarz, Email: viola.heinzelmann@usb.ch.
Acknowledgements
We thank members of the Adult Cancer Program, Lowy Cancer Research Centre for suggestions regarding performing and optimizing experiments. We also acknowledge Rea Guertler, Stephanie Naim and Laurence Luu (University students) for their support.
Funding
This work was supported by the Swiss National Foundation (PBZHP3-133289 and −138752 to F.J.); Cancer Institute New South Wales (09/CRF/2-02 to VHS); Royal Australian and New Zealand College of Obstetricians and Gynaecologists (to VHS); William Maxwell Trust (to VHS) and the Royal Hospital for Women Foundation (to VHS).
References
- Haslam DB, Baenziger JU. Expression cloning of Forssman glycolipid synthetase: a novel member of the histo-blood group ABO gene family. Proc Natl Acad Sci U S A. 1996;93(20):10697–10702. doi: 10.1073/pnas.93.20.10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Storch T, Yu M, Elliott SP, Haslam DB. Characterization of the human Forssman synthetase gene. An evolving association between glycolipid synthesis and host-microbial interactions. J Biol Chem. 1999;274(41):29390–29398. doi: 10.1074/jbc.274.41.29390. [DOI] [PubMed] [Google Scholar]
- Cooke J, Zhang H, Greger L, Silva AL, Massey D, Dawson C, Metz A, Ibrahim A, Parkes M. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18(11):2128–2137. doi: 10.1002/ibd.22942. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Cid E, Yamamoto F. Molecular genetic basis of the human Forssman glycolipid antigen negativity. Sci Rep-Uk. 2012;2:975. doi: 10.1038/srep00975. doi:10.1038/srep00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzel IU, Pohlentz G, Storck W, Radamm L, Hoffmann P, Bielaszewska M, Bauwens A, Cichon C, Schmidt MA, Mormann M, Karch H, Müthing J. Association of Shiga toxin glycosphingolipid receptors with membrane microdomains of toxin-sensitive lymphoid and myeloid cells. J Lipid Res. 2013;54(3):692–710. doi: 10.1194/jlr.M031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnam-Motlagh P, Tyler A, Grankvist K, Johansson A. Verotoxin-1 treatment or manipulation of its receptor globotriaosylceramide (gb3) for reversal of multidrug resistance to cancer chemotherapy. Toxins. 2010;2(10):2467–2477. doi: 10.3390/toxins2102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desselle A, Chaumette T, Gaugler MH, Cochonneau D, Fleurence J, Dubois N, Hulin P, Aubry J, Birkle S, Paris F. Anti-gb3 monoclonal antibody inhibits angiogenesis and tumor development. PLoS One. 2012;7(11):e45423. doi: 10.1371/journal.pone.0045423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler U, Souady J, Hulsewig M, Drmic-Hofman I, Haier J, Friedrich AW, Karch H, Senninger N, Dreisewerd K, Berkenkamp S, Schmidt MA, Peter-Katalinic J, Müthing J. Shiga toxin receptor Gb3Cer/CD77: tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS One. 2009;4(8):e6813. doi: 10.1371/journal.pone.0006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Goldstein DR, Bovin NV, Pochechueva T, Spengler M, Caduff R, Fink D, Vuskovic MI, Huflejt ME, Heinzelmann-Schwarz V. Serum antiglycan antibody detection of nonmucinous ovarian cancers by using a printed glycan array. Int J Cancer. 2012;130(1):138–146. doi: 10.1002/ijc.26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Kwak CY, Shayman JA, Kim JH. Globoside promotes activation of ERK by interaction with the epidermal growth factor receptor. Biochim Biophys Acta. 2012;1820(7):1141–1148. doi: 10.1016/j.bbagen.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- Pochechueva T, Jacob F, Goldstein DR, Huflejt ME, Chinarev A, Caduff R, Fink D, Hacker N, Bovin NV, Heinzelmann-Schwarz V. Comparison of printed glycan array, suspension array and ELISA in the detection of human anti-glycan antibodies. Glycoconj J. 2011;28(8–9):507–517. doi: 10.1007/s10719-011-9349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Anugraham M, Pochechueva T, Tse BW, Alam S, Guertler R, Bovin NV, Fedier A, Hacker NF, Huflejt ME, Packer N, Heinzelmann-Schwarz VA. The glycosphingolipid P1 is an ovarian cancer-associated carbohydrate antigen involved in migration. Br J Cancer. 2014. doi:10.1038/bjc.2014.455. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Tillack TW, Allietta M, Moran RE, Young WW Jr. Localization of globoside and Forssman glycolipids on erythrocyte membranes. Biochim Biophys Acta. 1983;733(1):15–24. doi: 10.1016/0005-2736(83)90086-X. [DOI] [PubMed] [Google Scholar]
- Hakomori S, Wang SM, Young WW Jr. Isoantigenic expression of Forssman glycolipid in human gastric and colonic mucosa: its possible identity with "A-like antigen" in human cancer. Proc Natl Acad Sci U S A. 1977;74(7):3023–3027. doi: 10.1073/pnas.74.7.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanami J. The appearance of Forssman hapten in human tumor. J Biochem. 1972;72(3):783–785. doi: 10.1093/oxfordjournals.jbchem.a129960. [DOI] [PubMed] [Google Scholar]
- Svensson L, Hult AK, Stamps R, Angstrom J, Teneberg S, Storry JR, Jorgensen R, Rydberg L, Henry SM, Olsson ML. Forssman expression on human erythrocytes: biochemical and genetic evidence of a new histo-blood group system. Blood. 2013;121(8):1459–1468. doi: 10.1182/blood-2012-10-455055. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Christman JK. 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- Lescar J, Sanchez JF, Audfray A, Coll JL, Breton C, Mitchell EP, Imberty A. Structural basis for recognition of breast and colon cancer epitopes Tn antigen and Forssman disaccharide by Helix pomatia lectin. Glycobiology. 2007;17(10):1077–1083. doi: 10.1093/glycob/cwm077. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura YI, Toyota M, Kawashima R, Hagiwara T, Suzuki H, Imai K, Shinomura Y, Tokino T, Kannagi R, Dohi T. DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology. 2008;135(1):142–151. doi: 10.1053/j.gastro.2008.03.031. e143. [DOI] [PubMed] [Google Scholar]
- Dohi T, Ohta S, Hanai N, Yamaguchi K, Oshima M. Sialylpentaosylceramide detected with anti-GM2 monoclonal antibody. Structural characterization and complementary expression with GM2 in gastric cancer and normal gastric mucosa. J Biol Chem. 1990;265(14):7880–7885. [PubMed] [Google Scholar]
- Saldova R, Dempsey E, Perez-Garay M, Marino K, Watson JA, Blanco-Fernandez A, Struwe WB, Harvey DJ, Madden SF, Peracaula R, McCann A, Rudd PM. 5-AZA-2'-deoxycytidine induced demethylation influences N-glycosylation of secreted glycoproteins in ovarian cancer. Epigenetics. 2011;6(11):1362–1372. doi: 10.4161/epi.6.11.17977. [DOI] [PubMed] [Google Scholar]
- Brooks SA. The involvement of Helix pomatia lectin (HPA) binding N-acetylgalactosamine glycans in cancer progression. Histol Histopathol. 2000;15(1):143–158. doi: 10.14670/HH-15.143. [DOI] [PubMed] [Google Scholar]
- Mitchell BS, Schumacher U. The use of the lectin Helix pomatia agglutinin (HPA) as a prognostic indicator and as a tool in cancer research. Histol Histopathol. 1999;14(1):217–226. doi: 10.14670/HH-14.217. [DOI] [PubMed] [Google Scholar]
- Hakomori S, Nudelman E, Levery SB, Kannagi R. Novel fucolipids accumulating in human adenocarcinoma. I. Glycolipids with di- or trifucosylated type 2 chain. J Biol Chem. 1984;259(7):4672–4680. [PubMed] [Google Scholar]
- Mitchell BS, Brooks SA, Leathem AJ, Schumacher U. Do HPA and PHA-L have the same binding pattern in metastasizing human breast and colon cancers? Cancer Lett. 1998;123(1):113–119. doi: 10.1016/S0304-3835(97)00414-X. [DOI] [PubMed] [Google Scholar]
- Ariga T, Yoshida T, Mimori T, Yu RK. Autoantibodies against Forssman glycolipids in Graves' disease and Hashimoto's thyroiditis. Clin Exp Immunol. 1991;86(3):483–488. doi: 10.1111/j.1365-2249.1991.tb02957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artamonova SI, Ozherel'ev AS, Ryzhkov AD. [Antibodies to Forssman antigen in patients with cancer of the gastrointestinal tract and breast] Ter Arkh. 1986;58(9):99–102. [PubMed] [Google Scholar]
- Jacob F, Guertler R, Naim S, Nixdorf S, Fedier A, Hacker NF, Heinzelmann-Schwarz V. Careful Selection of Reference Genes Is Required for Reliable Performance of RT-qPCR in Human Normal and Cancer Cell Lines. PLoS One. 2013;8(3):e59180. doi: 10.1371/journal.pone.0059180. doi:59110.51371/journal.pone.0059180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson S, Kwasniewski M, Riano-Pachon DM, Mueller-Roeber B. QuantPrime–a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics. 2008;9:465. doi: 10.1186/1471-2105-9-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA–fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC bioinformatics. 2010;11:230. doi: 10.1186/1471-2105-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Spiess AN. qpcR: an R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics. 2008;24(13):1549–1551. doi: 10.1093/bioinformatics/btn227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Association between DNA methylation and gene expression in the TCGA ovarian cancer dataset.