

Abstract
Macrophages contribute to tissue homeostasis and influence inflammatory responses by modulating their phenotype in response to the local environment. Understanding the molecular mechanisms governing this plasticity would open new avenues for the treatment for inflammatory disorders. We show that deletion of calcineurin (CN) or its inhibition with LxVP peptide in macrophages induces an anti-inflammatory population that confers resistance to arthritis and contact hypersensitivity. Transfer of CN-targeted macrophages or direct injection of LxVP-encoding lentivirus has anti-inflammatory effects in these models. Specific CN targeting in macrophages induces p38 MAPK activity by downregulating MKP-1 expression. However, pharmacological CN inhibition with cyclosporin A (CsA) or FK506 did not reproduce these effects and failed to induce p38 activity. The CN-inhibitory peptide VIVIT also failed to reproduce the effects of LxVP. p38 inhibition prevented the anti-inflammatory phenotype of CN-targeted macrophages, and mice with defective p38-activation were resistant to the anti-inflammatory effect of LxVP. Our results identify a key role for CN and p38 in the modulation of macrophage phenotype and suggest an alternative treatment for inflammation based on redirecting macrophages toward an anti-inflammatory status.
Keywords: anti-inflammatory therapy, calcineurin, inflammation, macrophage, p38 MAPK
See also: JL Schultze (May 2014).
Introduction
The phosphatase calcineurin (CN) couples calcium-mobilizing signals to cell responses and is the target of the immunosuppressive (IS) drugs cyclosporin A (CsA) and FK506 (Liu et al, 1991). Each of these drugs forms a complex with a specific immunophilin (IP) (cyclophilin A and FK506 binding protein, respectively), and it is these IS/IP complexes that bind and inhibit CN (Schreiber & Crabtree, 1992). These drugs have been widely used in molecular studies of CN function and are used therapeutically to prevent transplant rejection and treat inflammatory diseases such as atopic dermatitis (Lee et al, 2004), severe asthma (Niven & Argyros, 2003), and rheumatoid arthritis (Tugwell et al, 1995). However, IS/IP complexes have CN-independent effects on other signaling pathways (Matsuda et al, 2000) and many side effects, including hepatotoxicity, nephrotoxicity, and high blood pressure (Kiani et al, 2000; Martinez-Martinez & Redondo, 2004).
Many CN-dependent processes described in mammals involve the regulation of the nuclear factor of activated T cells (NFAT) family of transcription factors (Crabtree & Olson, 2002; Hogan et al, 2003; Aramburu et al, 2004). Structural and functional analyses of NFAT proteins have identified PxIxIT and LxVP motifs as docking sites involved in the interaction with CN (Li et al 2011; Liu et al, 2001; Martinez-Martinez et al, 2006; Park et al, 2000). We recently showed that a peptide based on LxVP interferes with the CN-NFAT interaction by binding to the same docking site on CN as the IS/IP complexes (Rodriguez et al, 2009). In addition to inhibiting the binding of CN to LxVP-containing substrates, LxVP inhibits the phosphatase activity of CN (Martinez-Martinez et al, 2006; Rodriguez et al, 2009). In contrast, the PxIxIT peptide does not affect CN activity and only inhibits its binding to PxIxIT-containing substrates (Aramburu et al, 1998).
A central role for the CN-NFAT pathway in adaptive immune responses has been documented through extensive studies in T cells (Macian, 2005); however, much less is known about its role in cells of the innate immune system such as macrophages. Macrophages not only constitute the first line of defense to an inflammatory insult, but also regulate the specific immune response by conditioning the cytokine milieu (Mantovani et al, 2004). Macrophages are reciprocally influenced by the surrounding environment and adopt pro-inflammatory or anti-inflammatory properties in response to it. Anti-inflammatory macrophages contribute to the suppressive microenvironment during tumorigenesis (Mantovani et al, 2002), maintain adipose tissue homeostasis (Lumeng et al, 2007), and promote resolution of inflammation in atherosclerosis (Mantovani et al, 2009) and myocardial infarction (Frangogiannis, 2012).
Here, we reveal a novel role for CN in macrophage activation and show that specific targeting of CN in macrophages confers resistance to inflammation by preventing MKP-1-mediated suppression of p38 MAPK activity. We moreover report a macrophage-based anti-inflammatory treatment that specifically targets CN through mechanisms unrelated to those of conventional IS drugs.
Results
Constitutive deletion of CN, but not inhibition with CsA or FK506, drives macrophages toward an anti-inflammatory phenotype
To study the influence of CN activity on macrophage phenotype, we generated Cnb1Δ/flox LysM-Cre mice, in which CN is constitutively deleted specifically in the myeloid lineage. CN expression was efficiently suppressed in peritoneal macrophages (Fig1A), and CN-deficient cells showed increased expression of several anti-inflammatory markers, such as IL-10, Arg1, and Mrc1 (Fig1B–D), and decreased levels of the pro-inflammatory marker iNOS (Fig1E). To test the impact of CN-negative macrophages in vivo, we examined two models of acute inflammation: oxazolone-induced contact hypersensitivity in the ear and zymosan-induced inflammation in the paws. In both models, Cnb1Δ/flox LysM-Cre mice were resistant to inflammation (Fig1F and G). These results suggest that suppression of CN activity induces anti-inflammatory properties in macrophages. However, this phenotype was not reproduced by treatment of wild-type peritoneal macrophages with the classical CN inhibitors CsA or FK506; indeed, these inhibitors reduced the levels of the Arg1, Mrc1, and IL-10 anti-inflammatory markers (Fig1H). These results suggest either that the phenotype of macrophages constitutively deficient for CN is not a direct result of CN-deletion or that CsA and FK506 have off-target effects that mask the effect of CN inhibition.
Figure 1. Constitutive CN deficiency, but not IS treatment, confers anti-inflammatory properties to macrophages.

A Western blot confirming deletion of CnB and destabilization of CnA in macrophages from Cnb1Δ/flox LysMCre+ mice (CN KO) compared with Cnb1flox/flox LysMcre− mice (Control).
B ELISA analysis of IL-10 protein in supernatants of CN-deleted and control macrophages (mean ± s.d.; n = 3).
C–E mRNA levels of Arg1 (C) and Mrc1 (D) and iNOS protein (E) in peritoneal macrophages from CN KO and control mice.
F, G In vivo imaging analysis of inflammation in CN KO and control mice after (F) sensitization with oxazolone in the right ear and (G) zymosan inoculation in the right hindpaw (n = 9 animals per genotype and condition).
H mRNA levels of IL-10, Arg1, Mrc1, and iNOS in peritoneal macrophages treated with CsA or FK506 (mean ± s.e.m.; n > 3).
Data information: *P < 0.05, **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
Previous reports showed that CsA and FK506 inhibit iNOS expression in macrophages and other cell types (Hortelano et al, 1999; Hamalainen et al, 2002). We confirmed that high doses of CsA (3–10 μg/ml) inhibit iNOS gene expression in macrophages (Supplementary Fig S1A). However, this effect is not associated with the inhibition of the CN activity, since lower CsA doses (200 ng/ml) that efficiently inhibit CN-NFAT signaling (Supplementary Fig S1B) not only failed to inhibit iNOS expression but significantly increased it (Supplementary Fig S1A).
Inducible CN deletion results in a population of anti-inflammatory macrophages with therapeutic effects in collagen-induced arthritis
To bypass potential indirect effects of constitutive CN deficiency on macrophages, we deleted CN ex vivo by transducing CnB1Δ/flox macrophages with CRE-encoding lentivirus (Fig2A). These CN-deleted macrophages, but not control CRE-transduced cells, upregulated IL-10, Arg1, and Mrc1, and downregulated iNOS (Fig2B–E and Supplementary Fig S2A–D), mirroring the phenotype of constitutively CN-deficient macrophages. To further examine the anti-inflammatory properties of induced CN-deleted macrophages, we transferred them to a mouse model of collagen-induced arthritis (CIA), in which disease is triggered in the susceptible DBA1J mouse strain by two intradermal injections of collagen (Supplementary Fig S3). Local transfer of CN-deleted macrophages into the footpads of arthritic mice before the second collagen injection significantly reduced disease severity compared with mice inoculated with control macrophages (Fig2F). Consistent with this, paws inoculated with CN-deleted macrophages expressed higher levels of the anti-inflammatory markers Mrc1 and IL-10 (Fig2G).
Figure 2. Inducible CN deletion generates anti-inflammatory macrophages that protect against CIA.

A Western blot confirming CnB deletion and CnA destabilization in macrophages from Cnb1Δ/flox mice transduced ex vivo with CRE-encoding lentivirus. Tubulin expression is shown as a loading control.
B ELISA analysis of IL-10 protein in supernatants of CN-deleted and control macrophages (mean ± s.d.; n = 3).
C, D mRNA expression of Arg1 (C) and Mrc1 (D) in control and CN-deleted macrophages.
E Representative flow cytometry analysis of iNOS protein in CN-deleted and control macrophages.
F CIA score in arthritic mice inoculated in the footpad with CN-deleted or control macrophages. Data are means ± s.e.m. from a representative experiment; n = 10 mice per group.
G Immunofluorescence staining (red) of Mrc1 (top) and IL-10 (bottom) in sections from paws inoculated with CN-deleted or control macrophages. Nuclei are stained blue.
Data information: *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Specific CN targeting by LxVP induces anti-inflammatory properties in macrophages
The failure of CsA and FK506 treatment to reproduce the consistent results obtained with both CN-deletion strategies indicated that IS drugs may have off-target effects. To clarify this issue, we tested the effect of an alternative strategy for CN inhibition with the peptide LxVP, which inhibits CN independently of IPs and therefore could avoid many of the side effects of IS drugs. Macrophages were transduced with lentivirus encoding LxVP or the mutant version AxAA (mutLxVP), both fused to GFP. Expression of LxVP in peritoneal macrophages strongly inhibited the phosphatase activity of CN (Fig3A). Moreover, LxVP-transduced macrophages isolated from transgenic mice expressing an NFAT-luciferase reporter showed decreased NFAT activity under basal conditions and in response to the NFAT-inducer zymosan (Fig3B; Goodridge et al, 2007).
Figure 3. LxVP expression induces an anti-inflammatory phenotype in macrophages.
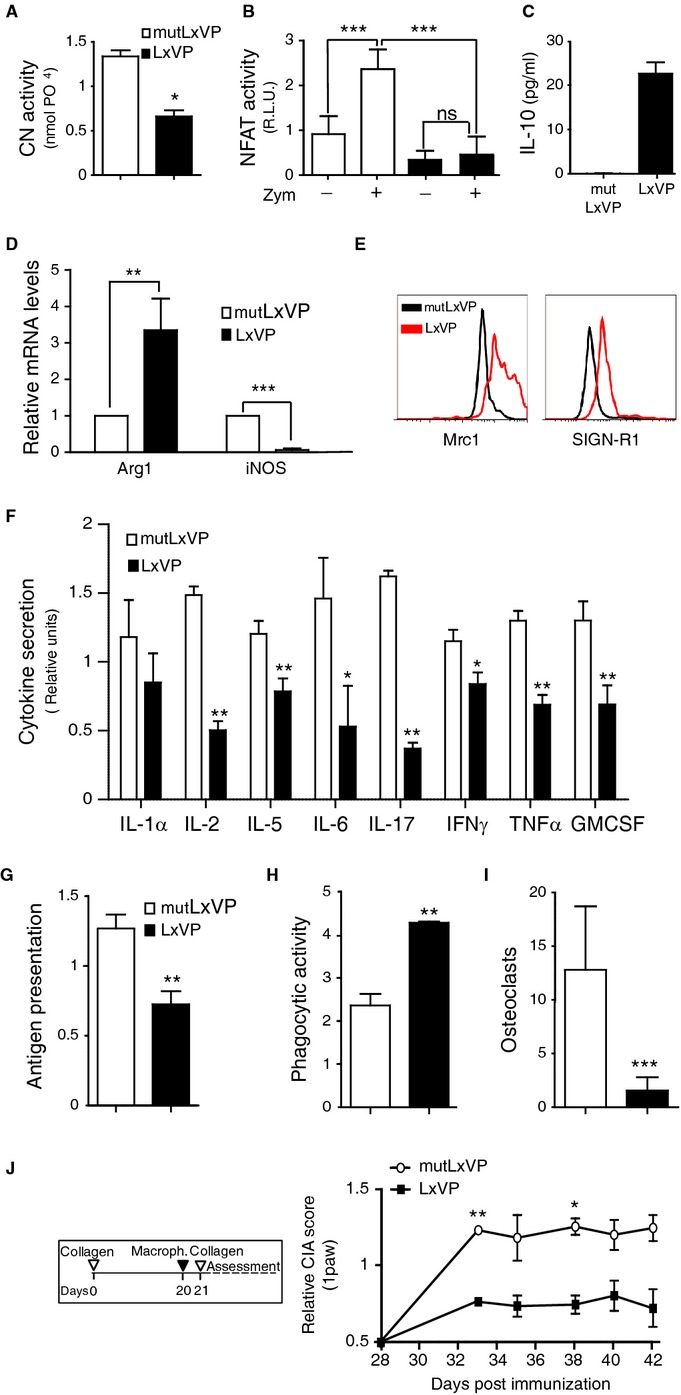
- CN activity in total protein extracts of peritoneal macrophages isolated 5 days after i.p. injection with LxVP or mutLxVP lentivirus (mean ± s.d.; n = 3).
- NFAT transcriptional activity (relative luciferase units) in untreated and zymosan-stimulated (Zym) macrophages from NFAT-luc transgenic mice i.p. injected with LxVP or mutLxVP lentivirus (mean ± s.d.; n = 5).
- IL-10 protein levels in culture supernatants of macrophages isolated from mice 5 days after i.p. injection with LxVP or mutLxVP lentivirus.
- Real-time PCR analysis of Arg1 and iNOS mRNA in isolated LxVP- and mutLxVP-transduced macrophages (mean ± s.d.; n = 3).
- Flow cytometry analysis of Mrc1 and SIGNR1 expression in isolated LxVP- and mutLxVP-transduced macrophages.
- Multiplex analysis of pro-inflammatory cytokines in culture supernatants of macrophages from mice i.p. injected with LxVP or mutLxVP lentivirus.
- Presentation of antigen (ovalbumin) to B3Z T-cell hybridoma cells by macrophages expressing LxVP or mutLxVP.
- Phagocytosis of opsonized red blood cells by LxVP- or mutLxVP-expressing macrophages.
- Osteoclast differentiation, recorded as the number of multinucleated TRAP+ cells.
- CIA score in arthritic mice inoculated in the footpads with LxVP- or mutLxVP-transduced macrophages. Macrophages were injected on day 20 to ensure their exposure to the booster collagen treatment. Data are means ± s.e.m. of three independent experiments; n = 30 mice per group.
Data information: *P < 0.05; **P < 0.01; ***P < 0.001.
Like the CN-deleted macrophages, LxVP-transduced macrophages had a typical anti-inflammatory expression profile, including upregulated expression of IL-10 and Arg1, downregulated iNOS, and increased cell-surface expression of Mrc1 and SIGNR1 (Fig3C–E). Moreover, LxVP-transduced macrophages had downregulated expression of pro-inflammatory cytokines including IL-17, TNF-α, IFN-γ, and IL-6 (Fig3F). LxVP also conferred anti-inflammatory functions, such as reduced antigen-presentation capacity, increased phagocytic activity, and impaired differentiation to osteoclasts (Fig3G–I and Supplementary Fig S4A and B), three hallmarks of the resolution of inflammation. Consistent with the ability of the VIVIT peptide (a high-affinity PxIxIT-derived peptide) to interfere with CN-NFAT interaction, lentiviral-mediated VIVIT expression in macrophages inhibited zymosan-induced NFAT-dependent transcription (Supplementary Fig S5A). However, VIVIT failed to induce the anti-inflammatory phenotype displayed by the LxVP-transduced macrophages (Supplementary Fig S5B–D).
To evaluate the anti-inflammatory potential of LxVP-transduced macrophages in vivo, we performed cell therapy assays in the CIA model, in which transduced macrophages were injected into the footpads of arthritic mice. Paws injected with LxVP-transduced macrophages had significantly lower arthritic scores than those injected with control-transduced macrophages (Fig3J). These results, together with the cell therapy experiments with CN-deleted macrophages, indicate that macrophages with suppressed CN-activity are competent to mediate anti-inflammatory effects in vivo.
The anti-inflammatory actions of CN-targeted macrophages are mediated by releasing p38 MAPK from MKP-1-mediated repression
Since CN modulates p38 activity in other cell types (Lim et al, 2001; Braz et al, 2003), we analyzed whether the anti-inflammatory macrophage phenotype triggered by CN gene deletion and LxVP administration was mediated by p38 activity, and whether p38 activation was implicated in the differences from the effect of IS drugs. LxVP-transduced and CN-deficient macrophages, but not control or IS-drug-treated macrophages, showed sustained activation of p38 (Fig4A–D). Treatment of LxVP-transduced or CN-deficient macrophages with the p38-chemical inhibitor SB203580 (SB) reduced the expression of the anti-inflammatory markers Mrc1, Arg1, and IL-10 to basal levels (Fig4E and F and Supplementary Fig S6A and B). Since IS drugs have been reported to activate p38 activation in macrophages (Kang et al, 2007), we again determined whether this was dependent on the IS doses used. As with iNOS expression, p38 was only activated at very high concentrations of CsA (50 μg/ml) and FK506 (10 μg/ml), but not at pharmacological doses of the drugs able to inhibit CN activity (Supplementary Fig S7A and B).
Figure 4. p38 MAPK activity mediates the induction of anti-inflammatory macrophages upon specific CN targeting.

A Representative Western blot showing expression of phosphorylated P-p38 and P-ERK in peritoneal macrophages treated with CsA or FK506 (FK) or transduced with LxVP or control lentivirus in vitro. p38 and tubulin were used as loading controls.
B Quantification of P-p38 expression in the experimental conditions mentioned in (A) (mean ± s.e.m.; n = 3).
C P-p38 (top) and CNB (center) protein expression in peritoneal macrophages from Cnb1 flox/flox LysMCre− (control) and Cnb1 Δ/flox LysMCre+ (CN KO) mice treated with CsA or FK506. Tubulin was used as a loading control (Tub).
D Quantification of P-p38 expression in the experimental conditions in (C) (mean ± s.e.m.; n = 3).
E, F Effect of p38 inhibition by SB203580 (SB) treatment on Mrc1, Arg1, and IL-10 mRNA levels in (E) LxVP-transduced macrophages and (F) CN KO macrophages.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
p38 activation in macrophages is regulated by the phosphatase MKP-1 (Perdiguero et al, 2011; Comalada et al, 2012; Liu et al, 2013). Consistent with the p38-activation induced by CN targeting, both gene deletion and LxVP-mediated CN inhibition reduced the expression of MKP-1 (Fig5A and B). In MKP-1-deficient macrophages, p38 activation was enhanced to a similar extent as in LxVP-treated wild-type macrophages (Fig5C and D), and LxVP treatment did not further activate p38 in MKP-1-deficient macrophages (Fig5D). These results implicate MKP-1 in the CN-mediated regulation of p38 activation and indicate that p38 contributes to the anti-inflammatory phenotype of CN-targeted macrophages.
Figure 5. MKP-1 links CN targeting with p38 MAPK activation.
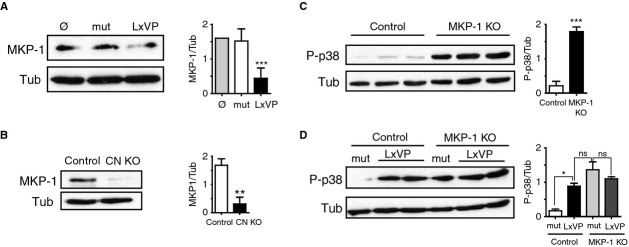
- MKP-1 protein expression in control and mutLxVP- or LxVP-transduced macrophages (n = 5).
- MKP-1 protein expression in control and CN KO macrophages (n = 3).
- P-p38 expression in wild-type and MKP-1-deficient macrophages (n = 3).
- P-p38 expression in wild-type and MKP-1-deficient macrophages transduced with mutLxVP or LxVP lentivirus (n = 2).
Data information: Quantifications (relative units) are shown next to the corresponding Western blots in all panels. *P < 0.05, **P < 0.01, ***P < 0.001.
Source data are available online for this figure.
Systemically delivered LxVP targets macrophages, which migrate to sites of inflammation
The above results establish the potential of LxVP as an anti-inflammatory treatment based on cell therapy. To test the therapeutic potential of LxVP in a gene therapy approach, we assessed the anti-inflammatory effects of LxVP lentivirus in the CIA model after systemic administration by intraperitoneal (i.p.) injection. To determine lentivirus tropism after i.p. injection, we examined the peritoneal exudate 5 days after inoculation of naive mice. At this time, all detected GFP+ cells are transduced by the lentivirus, and we can exclude false positives due to phagocytosis of GFP molecules or lentiviral particles. Although peritoneal exudates were enriched in cell populations such as T and B cells, GFP was undetectable in CD4+, CD8+, or B220+ cells, and all GFP+ cells were triple positive for F4/80, CD11b, and CD11c and also expressed Mertk (Fig6A) clearly identifying these cells as macrophages.
Figure 6. Systemically delivered LxVP lentivirus transduces macrophages, which then migrate to inflammatory foci.
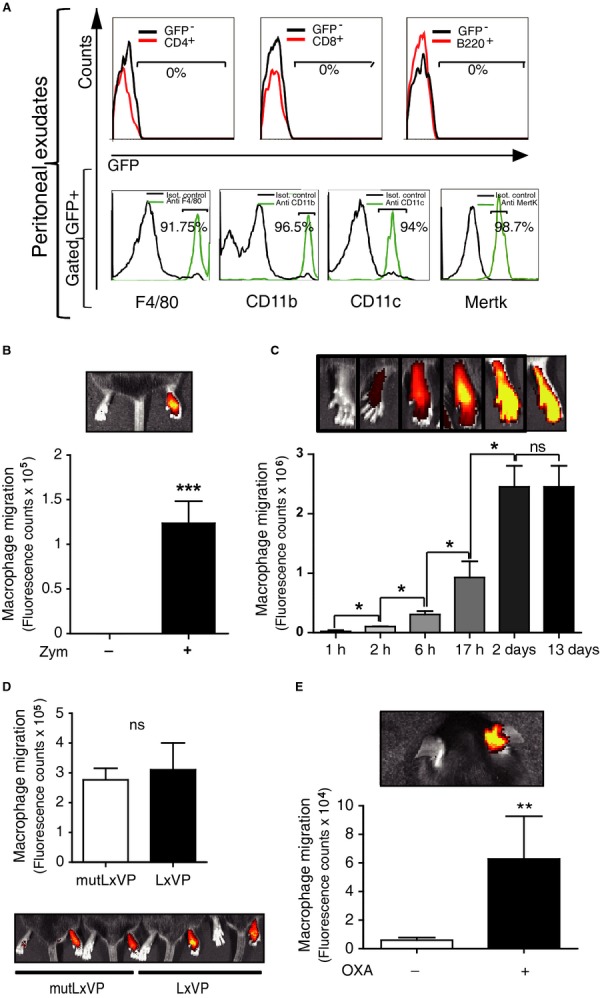
- Top, Flow cytometry analysis of GFP expression in gated CD4+, CD8+, or B220+ cells from peritoneal exudate obtained 5 days after i.p. injection with GFP-encoding lentivirus. Bottom, F4/80, CD11b, CD11c, and MertK expression in gated GFP+ cells from peritoneal exudate. Data are from a representative experiment (n ≥ 3).
- In vivo tracking of i.p.-injected DIR-labeled non-transduced macrophages in mice with zymosan-induced acute inflammation in the right hindpaw. In vivo images (top) and quantification (bottom). Zym, zymosan. (means ± s.d.; n = 3).
- Time profile of DIR signal in inflamed paws after i.p. injection of DIR-labeled non-transduced macrophages (means ± s.d.; n ≥ 3).
- In vivo imaging analysis of the capacity of DIR-labeled LxVP- or mutLxVP-transduced macrophages to migrate to inflamed paws.
- In vivo tracking of i.p.-injected DIR-labeled non-transduced macrophages in mice with oxazolone-induced contact hypersensitivity in the right ear. OXA, oxazolone (means ± s.d.; n ≥ 3).
Data information: *P < 0.05; **P < 0.01; ***P < 0.001.
Since macrophages transduced upon i.p. administration of LxVP lentivirus were located in the peritoneal cavity, and therefore far from the site of action in the inflamed paw, we tested whether these macrophages migrated toward the inflammation focus to have their anti-inflammatory effects. CIA affects all four limbs, so we used a model of zymosan-induced acute paw inflammation to generate a single inflammation focus in one paw, leaving the contralateral paw as a non-inflammation control. We inoculated mice with zymosan in the right hind footpad and subsequently injected (i.p.) donor macrophages labeled ex vivo with the fluorescent tracer DIR. Macrophages migrated selectively from the peritoneal cavity to the inflamed paw, with no signal detected in control paws (Fig6B). Migration increased progressively from 2 h to 2 days after macrophage transfer, and migrated cells were detectable for at least 13 days (Fig6C). LxVP- and control-transduced macrophages had the same migratory capacity, as revealed by in vivo analysis of DIR signal in inflamed paws at 24 h post-injection (Fig6D) and by GFP staining in tissue sections (Supplementary Fig S8). Homing of DIR-labeled macrophages to the inflammation site was also seen in the model of oxazolone-induced contact hypersensitivity in mouse ears, suggesting that peritoneal macrophages can migrate to different inflamed locations (Fig6E).
Gene therapy with LxVP lentivirus resolves remote inflammation in a p38-dependent manner
Systemic i.p. treatment with LxVP lentivirus prevented the progression of CIA, maintaining the low arthritic scores recorded at the time of treatment (Fig7A). Histological analysis of joint sections confirmed the near absence of inflammation in LxVP-treated mice, whereas joints of control-transduced animals showed abundant inflammatory cell infiltrates and severe bone and cartilage damage (Fig7B). Injection of control lentivirus did not exacerbate disease symptoms (Supplementary Fig S9). A clear anti-inflammatory effect was also observed after systemic treatment with LxVP lentivirus in the oxazolone-induced contact hypersensitivity model in mouse ears (Fig7C).
Figure 7. Systemically delivered LxVP lentivirus protects against CIA and contact hypersensitivity in a p38-dependent manner.
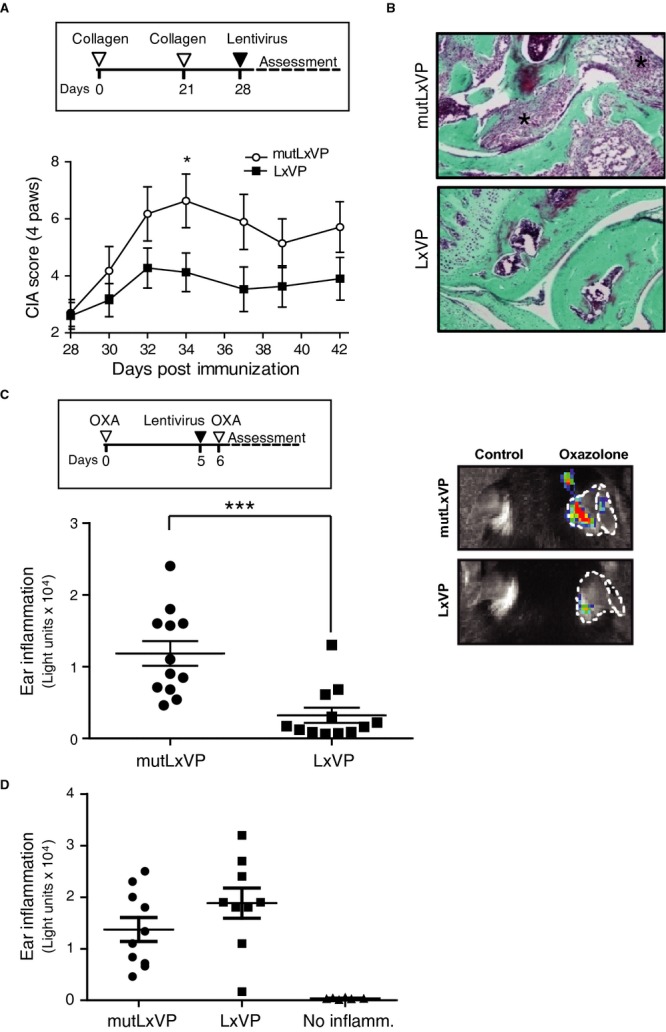
- The scheme shows the CIA protocol, indicating the time of lentivirus injection. The chart shows the time profile of arthritic score in mice inoculated i.p. with LxVP or mutLxVP lentivirus at disease onset (day 28). Data are means ± s.e.m. of three independent experiments; n = 20 mice per group.
- Masson's trichrome staining in paw joints of animals inoculated i.p. at disease onset with mutLxVP or LxVP. Green: bone and collagen; blue: cell nuclei; red: muscle fibers and keratin. Asterisks mark inflammatory cell infiltrates.
- Ear inflammation (luminol signal) in wild-type mice i.p. injected with LxVP or mutLxVP lentivirus (mean ± s.d.; n = 2, 12 mice per group).
- Ear inflammation (luminol signal) in MKK3−/−6+/− mice i.p. injected with LxVP or mutLxVP lentivirus (mean ± s.d.; n = 2, 10 ears per group).
Data information: *P < 0.05; ***P < 0.001.
To further investigate the role of p38 activity in the anti-inflammatory effects observed after in vivo injection of LxVP lentivirus, we i.p. injected LxVP or control lentivirus into MKK3−/−6+/− mice, in which p38 activation is defective (Nagaleekar et al, 2011; Noubade et al 2011). The therapeutic effect of LxVP observed on oxazolone-induced contact hypersensitivity in wild-type mice was abolished in MKK3−/−6+/− mice (Fig7D). The accumulated data thus indicate that LxVP, through p38 activation, induces a population of macrophages in vivo that exert anti-inflammatory effects after migration to remote inflammation sites.
Gene therapy with LxVP lentivirus resolves local inflammation
To determine whether LxVP lentivirus also has anti-inflammatory properties when administered locally, we first tested LxVP tropism in this setting. Footpads were inoculated with LxVP lentivirus 24 h before induction of local inflammation by injection of zymosan. After 5 days, LxVP-transduced cells were detected in the popliteal draining lymph nodes (LN) (Fig8A) and, despite the abundance of lymphocytes in this organ, all sorted GFP+ cells were F4/80+/CD11b+/CD11c+ macrophages (Fig8B). This result was confirmed by immunofluorescence analysis of footpad tissue sections for F4/80 and Mac3, which shows that all GFP+ cells were also F4/80+or Mac3+ (Fig8C and Supplementary Fig S10A).
Figure 8. Locally delivered LxVP targets macrophages and prophylactically protects against CIA.

- Footpad-injected lentivirus targets popliteal lymph nodes (LN). The chart shows luciferase activity in different LN-cell homogenates from mice inoculated in the footpad with luciferase-encoding lentivirus.
- Flow cytometry analysis of F4/80, CD11b, and CD11c expression in sorted GFP+ cells obtained from popliteal LN after inoculation of footpads with GFP-encoding lentivirus.
- Confocal immunofluorescence showing colocalization of virus-encoded GFP (green) and F4/80 (red) in sections of inflamed paws locally injected with lentiviral vectors.
- The scheme shows the CIA protocol, indicating the time of lentivirus injection. Charts show time profiles of arthritic score (left) and incidence (right) in mice transduced in the right hind footpad on day −5 with LxVP or mutLxVP lentivirus. Data are means ± s.e.m. from a representative experiment of two; n = 10 mice (10 paws) per group.
- Masson's trichrome staining in joints of paws locally transduced on day −5 with mutLxVP or LxVP. Green: bone and collagen; blue: cell nuclei; red: muscle fibers and keratin. Asterisk marks inflammatory cell infiltrate.
- Confocal immunofluorescence showing GFP (green) and Mrc1 or IL-10 (red) in sections of arthritic paws locally injected with LxVP or mutLxVP lentivirus.
- Non-transduced cells express Mrc1 in LxVP-inoculated paws. Confocal immunofluorescence showing GFP (green) and Mrc1 (red) in arthritic paws locally injected with LxVP or mutLxVP lentivirus.
Data information: * P < 0.05; **P < 0.01; ***P < 0.001.
To test the effect of locally administered LxVP in the CIA model, we inoculated hindpaws with the lentivirus 5 days before the first collagen injection. LxVP had a clear prophylactic effect, with arthritic scores significantly lower in LxVP-treated mice than in animals inoculated with mutLxVP lentivirus (Fig8D left). Moreover, LxVP delayed the onset of disease and reduced disease incidence from 85 to 40% (Fig8D right). Histological analysis of joint sections confirmed the near absence of inflammation in LxVP-treated mice, whereas joints of control-transduced animals showed abundant inflammatory cell infiltrates and severe bone and cartilage damage (Fig8E). Local injection of the control lentivirus did not exacerbate disease symptoms (Supplementary Fig S10B). Examination of footpad sections at endpoint revealed upregulated expression of Mrc1 and IL-10 in GFP+ cells of LxVP-inoculated inflamed paws (Fig8F), confirming that the anti-inflammatory phenotype of LxVP-transduced macrophages is present in vivo under proinflammatory conditions. Interestingly, Mrc1 expression was increased not only in transduced macrophages (GFP+) but also in neighboring GFP− macrophages (Fig8G).
Discussion
This study identifies CN and p38 as critical modulators of macrophage activation that determine the course of inflammation. CN deletion or LxVP-mediated inhibition, but not inhibition with CsA or FK506, induces a population of anti-inflammatory macrophages with beneficial effects in a variety of inflammatory settings. Moreover, this divergence is due to differing effects on p38 MAPK, suggesting that the commonly used IS drugs are not the best tools for the study of CN function and underlining the need for specific CN inhibitors. Cell therapy with CN-gene-deleted or LxVP-inhibited macrophages has potential in the development of anti-inflammatory strategies. Moreover, direct administration of LxVP-encoding lentiviruses significantly reduced the severity of inflammation in independent inflammatory models, suggesting the utility of this CN-inhibitory peptide in gene therapy approaches.
CN gene deletion or inhibition by LxVP induced a population of macrophages with anti-inflammatory properties. Although the phenotype observed recapitulates some of the properties of standard M2 macrophages, it does not match any of the categories already defined. For instance, CN targeting did not affect TGFβ or Ym1 expression (Supplementary Fig S11A and B), and therefore, this phenotype must be considered distinct from the standard established anti-inflammatory phenotypes. Future work on a global genomic and proteomic profiles will be needed to define the relationship of LxVP (or CN-targeted) macrophages with other well-known activation programs such as those of M1 and M2. Surprisingly, the classical CN inhibitors CsA and FK506 did not reproduce the phenotype obtained upon CN gene deletion or LxVP inhibition, and this was also the case with the VIVIT peptide. VIVIT is a high-affinity improved version of the natural peptide PxIxIT that binds to CN and inhibits the binding to NFAT and other CN-substrates containing PxIxIT sites (Aramburu et al, 1998). However, LxVP but not the PxIxIT peptide, inhibits the phosphatase activity of CN (Martinez-Martinez et al, 2006; Rodriguez et al, 2009). Thus, the LxVP peptide is expected to inhibit signaling mediated by all substrates regulated by CN activity, and not only CN signaling by substrates containing LxVP or LxVP-like sites. This difference between the PxIxIT/VIVIT and the LxVP motifs might be related to the selective ability of LxVP to induce an anti-inflammatory phenotype. In this regard, although the VIVIT peptide inhibits the expression of some pro-inflammatory cytokines and iNOS, it also inhibits the expression of IL-10 induced by LPS in macrophages (Elloumi et al, 2012).
A previous study reported that CN deletion in macrophages abrogates pro-inflammatory macrophage activation upon TLR stimulation (Jennings et al, 2009), but as these effects were also exerted by common IS drugs, they are unlikely to be mediated through the mechanisms we report here. In addition, we show not just inhibition of pro-inflammatory functions, but also the induction of anti-inflammatory properties; although CsA and FK506 inhibit the pro-inflammatory action of macrophages, they do not induce anti-inflammatory properties. Another report claims that FK506 induces alternative macrophage polarization (Bai et al, 2010); however, only one of the analyzed anti-inflammatory markers, Mrc1, was upregulated, whereas Arg1 and IL-10 were unchanged. Moreover, that report did not show downregulation of the pro-inflammatory marker iNOS, and the data do not appear to be consistent with alternative polarization. Our results clearly show that pharmacologically effective doses of IS inhibit the expression of anti-inflammatory markers and do not reduce iNOS expression.
The differences between genetic or LxVP-mediated CN inhibition and CsA/FK506 treatment might be explained by the reported off-target effects of IS drugs on other signaling pathways. Indeed, both CsA and FK506 are reported to inhibit p38 and JNK MAPK in an IP-dependent and CN-independent manner in Jurkat T cells (Matsuda et al, 2000). Our results in macrophages are in line with those reported in T cells: We show that CsA selectively inhibits the activation of p38 induced by the combined treatment of PMA and calcium ionophore, but not that induced by LPS or hyper-osmolar treatment (Supplementary Fig S12). Although these effects were not observed in other non-lymphoid cells (Matsuda et al, 2000), our results indicate that this selective inhibition is not restricted to the T-cell activation process as was initially proposed.
Activation of macrophage p38 was reported in response to a high dose of FK506 (10 μg/ml) (Kang et al, 2007). However, this dose is about 500 times higher than required to inhibit CN, and the observed p38-activation was transient, peaking at 30 min and declining thereafter. In our hands, exposure of macrophages to non-toxic, pharmacological doses of CsA (200 ng/ml), or FK506 (10 ng/ml) for periods ranging from 30 min to 5 days did not induce any sign of p38 activation, but did produce efficient inhibition of CN signaling. This might indicate that the anti-inflammatory phenotype that we observe requires sustained activation of p38 and, in fact, the kinetics and duration of MAPK activation in macrophages is reported to determine their functional outcome (Comalada et al, 2012). Although in our models, p38 inhibition reduced the expression of several anti-inflammatory markers to basal levels, this does not exclude the contribution of a basal activity of other MAPKs or the involvement of other mediators in this phenotypic change mediated by CN inhibition. However, the non-responsiveness of MKK3−/−6+/− mice to LxVP in the contact hypersensitivity model indicates that deficiency in p38 activation is sufficient to abrogate the anti-inflammatory action of LxVP-mediated CN inhibition.
Previous reports showed that CN negatively regulates p38 MAPK activity in cardiomyocytes via upregulation of MKP-1 gene expression (Lim et al, 2001) and that MKP-1 regulates p38 in macrophages (Perdiguero et al, 2011; Comalada et al, 2012; Liu et al, 2013). Our results strongly suggest that MKP-1 mediates the repression of p38 by CN: CN deletion or LxVP-mediated inhibition reduced MKP-1 levels; LxVP activated p38 to a similar extent as displayed by MKP-1-deficient macrophages, and LxVP did not further increase p38 activation in MKP-1-deficient macrophages. However, additional mechanisms could also be involved in the activation of p38 that we have observed. In this regard, we cannot exclude that some secreted mediators might trigger autocrine actions that contribute to p38 activation. However, p38 activation by pro-inflammatory cytokines such as TNF-α or IL-1 can be discounted since CN targeting inhibits their secretion and the opposite effect on p38 would be expected.
While p38 has been implicated in classical macrophage activation (Kang et al, 2007), its role in macrophage polarization is not well characterized. Some reports suggest that p38 activity could be related to the induction of particular anti-inflammatory mediators by macrophages (Lawrence & Natoli, 2011; Perdiguero et al, 2011; Csoka et al, 2012), but these studies do not describe a complete phenotype with upregulated anti-inflammatory markers and downregulated pro-inflammatory markers as we show here. Activation of p38 has also been reported in tumor-associated macrophages (Tjiu et al, 2009). Our results support the involvement of p38 in the anti-inflammatory phenotype induced by CN targeting. To our knowledge, a link between CN and p38 in macrophage activation has not been established before. However, such a link has been identified in the heart, where expression of active CN induces hypertrophy through the inhibition of p38 in cardiomyocytes (Lim et al, 2001).
CN has been also reported to negatively regulate NF-κB and IRF in macrophages (Kang et al, 2007). IRF4 activation has been associated with IL-4 and IL-10 transcription via NFAT in T cells (Hu et al, 2002; Rengarajan et al, 2002; Lee et al, 2009) and with alternative macrophage polarization (Honma et al, 2005; Negishi et al, 2005; Satoh et al, 2010). However, our analysis did not show an effect of LxVP-mediated CN inhibition on IRF4 mRNA expression in macrophages (Supplementary Fig S13A). Reports showing that CN inhibitors activate NF-κB are based on the use of IS doses far higher than those required to inhibit CN signaling. We reproduced the reported effects using these high IS doses, but at lower, pharmacologically active doses or by LxVP-mediated inhibition found no activation of the NF-κB pathway (Supplementary Fig S13B and C). High doses of IS not only result in CN-independent activation of NF-κB and p38 but also induce CN-independent inhibition of iNOS gene expression (Supplementary Fig S1). These findings indicate that it might be wise to revisit assertions about the involvement of CN signaling in physiological and pathophysiological settings when these are based on the use of high doses of IS.
The therapeutic effects of CN-deficient and LxVP-inhibited macrophages when locally transferred to arthritic paws suggest the clinical utility of macrophages for cell therapy-based treatment for inflammatory diseases. Interestingly, our findings show that macrophages migrate specifically from the peritoneal cavity to inflammation sites. Several studies have described macrophage recruitment to the peritoneal cavity in response to an i.p. inflammatory insult (Hopper, 1986), from the peritoneal cavity to draining LN (Rosen & Gordon, 1990; Bellingan et al, 1996), and to inflammation sites after intravenous injection (Audran et al, 1995); however, the migration of macrophages from the peritoneal cavity to peripheral inflamed locations has not been reported previously. Our results indicate that macrophages migrate rapidly toward inflammatory foci, highlighting their attractiveness as targets for anti-inflammatory cell therapy. We have identified a number of chemokines, including MCP1, RANTES, MIP1α, MIP1β, and CCL7 (Supplementary Fig S14A–E), whose expression is increased in inflammatory foci. These chemokines are likely to drive this migration, but future studies will be required to characterize this process and identify the potential contribution of these mediators to the migration process. The preferential migration of macrophages to inflammation sites opens up the possibility of autologous cell therapy, in which macrophages from patients could be switched toward an anti-inflammatory phenotype ex vivo and then reintroduced systemically, resulting in selective migration to inflammatory foci.
For gene therapy experiments, we used lentiviral vectors to engineer LxVP expression in cells. These vectors integrate in the genome and support long-lasting transgene expression and low in vivo immunogenicity (Sakuma et al, 2012). When the lentiviruses were injected i.p. or intradermally into the footpads, the entire GFP+ transduced population found in peritoneal cavity, tissue sections, or LN corresponded to a F4/80+/CD11b+/CD11c+ macrophage population. This population has been previously associated with allergy-induced lung inflammation (Moon et al, 2007), adipose tissue from obese individuals (Lumeng et al, 2007; Nguyen et al, 2007) and tumors (Umemura et al, 2008), but has also been found in non-pathological settings (Denning et al, 2011). Although we identified this macrophage population as the sole target of lentiviral transduction in vivo, we cannot exclude low frequency transduction of other cell types. However, despite this possibility, the cell therapy experiments with macrophages show that these cells are sufficient by themselves to mediate anti-inflammatory effects.
Although our results show a clear selective tropism of lentiviruses for macrophages, it is important to emphasize that the administration route could determine viral tropism. In fact, lentiviruses systemically injected into the jugular vein are able to efficiently transduce the vascular wall (Esteban et al, 2011) and also successfully transduce primary cells in vitro, such as endothelial cells, T-cell blasts and VSMC (Esteban et al, 2011; Urso et al, 2011). The macrophage tropism observed after i.p. inoculation might also reflect the metabolic activity of macrophages, which is much higher than that of other cell types enriched in peritoneal exudate such as T cells. In this regard, we have been unable to obtain lentiviral transgene expression in resting transduced T cells, but have successfully transduced primary T blasts (Urso et al, 2011), suggesting that T cells might be transduced by lentivirus but unable to express the transgene. Another important factor might be the half-life of cells in the peritoneal cavity; granulocytes, for example, might be transduced by lentiviruses, but viral integration and transgene expression likely require longer than their lifespan (Pillay et al, 2010).
The experiments with LxVP lentivirus in CIA highlight the potential of this peptide for the treatment for inflammatory disorders. After i.p. lentiviral delivery, transduced cells can also be found in spleen and liver, which might indicate off-target effects; however, the morphology and location of the transduced cells are typical of macrophages (Supplementary Fig S15). Another possible undesired effect of LxVP might be impaired tissue healing due to hyperactivation of macrophage p38 (Perdiguero et al, 2011), and it will therefore be important to assess whether such effects counter the possible benefits of LxVP in the treatment for inflammatory diseases. However, unlike IS, LxVP induces its effects on CN without binding to immunophilins (IP; Martinez-Martinez et al, 2006) and is thus likely to lack the severe side effects associated with IS/IP complexes. Nevertheless, further progress will be needed to establish the safety and advantages of LxVP treatment.
Our data suggest that the role of CN in macrophage activation, revealed here by LxVP-mediated inhibition or CN gene deletion, was not identified before probably because it was masked by the off-target effects of IS drugs. Given this finding, specific tools for CN targeting should now be used to revise earlier data on the role of CN, particularly in innate immune cells. We propose that CN inhibition or deletion in macrophages might have beneficial effects in pathological settings in which anti-inflammatory macrophages participate in the resolution of inflammation, such as obesity or myocardial infarction. Inhibition of CN without the involvement of IP would avoid many CN-independent side effects, offering new possibilities for the treatment for inflammatory diseases. Future studies will be required to translate these findings into efficient treatments for human disease.
Materials and Methods
Animals
Male DBA1J mice (7–10 weeks old) and C57BL6 mice (6–8 weeks old) were purchased from Charles River. NFAT-luc transgenic mice (in FBV genetic background) were kindly provided by Prof. Jeffery D. Molkentin, calcineurin B1 conditional knockout mice (in C57/BL6 genetic background) by Prof. Gerald R. Crabtree, and MKP-1 knockout mice (in C57/BL6 genetic background) by Dr. Rosario Perona. MKK3−/−6+/− mice (in C57/BL6 genetic background), generated as previously described (Nagaleekar et al, 2011), were kindly provided by Prof. Roger Davis. Animals were housed in a dedicated pathogen-free facility and were fed and watered ad libitum. Animal studies were approved by the local ethics committee. All animal procedures conformed to EU Directive 2010/63EU and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 1201/2005.
Lentivirus production and cell infection
Lentiviruses expressing GFP-peptide fusion proteins and luciferase were previously described (Rodriguez et al, 2009; Garaulet et al, 2013). Lentivirus encoding Cre recombinase was generated by cloning a Cre PCR product in the pHRSIN lentiviral vector. Lentivirus was produced by transient calcium phosphate transfection of HEK-293 cells, using a three-plasmid HIV-derived and VSV-pseudotyped lentiviral system (Addgene plasmid 12259). HEK293T cells were cultured in Dulbecco's modified Eagle medium (Sigma) supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 U/ml). Cells were plated at 30% confluence and transfected the next day. At 48 h and 72 h after transfection, supernatants were collected, concentrated by ultracentrifugation (88,000 g for 2 h at 4°C), and stored at −80°C. For infection of peritoneal macrophages, mice were i.p. inoculated with 300 μl concentrated lentiviral supernatant (7 × 108–2 × 109 TU/ml).
Isolation and culture of peritoneal macrophages
Peritoneal macrophages elicited by i.p. injection of thioglycolate (Difco) or lentivirus were collected after 3 or 5 days, respectively, by peritoneal lavage with PBS (2 × 10 ml) and immediately cultured in AlphaMEM (Lonza) supplemented with 10% FBS, L-glutamine (2 mM) and antibiotics. After overnight culture, non-adherent cells were removed to enrich for macrophages. When indicated, macrophages were treated with CsA (Sigma), FK506 (LC laboratories), SB203580 (10 μM) (Sigma), LPS (10 ng/ml) (Sigma) (40 min), or NaCl (0.7 M) (40 min).
Collagen-induced arthritis and lentiviral treatments
Male DBA1J mice were inoculated intradermally on day 0 and 21 with 200 μg of type II collagen from chicken (Sigma) emulsified 1:1 (v/v) in complete Freund's adjuvant (Difco) containing 10 mg/ml Mycobacterium tuberculosis (Difco). Limbs were graded by blinded examiners for arthritic scores as follows: grade 0 = no swelling, grade 1 = slight swelling and erythema, grade 2 = moderate swelling and edema, grade 3 = extreme swelling and pronounced edema, and grade 4 = joint rigidity. Joint sections were analyzed by Masson's trichrome staining. For prophylactic treatments, lentiviral vectors were injected 5 days before the first collagen immunization (day −5). For therapeutic treatments, vectors were injected on day 28. Lentiviral particles (7 × 108–2 × 109 TU/ml) were injected into the footpad (30 μl) or the peritoneal cavity (300 μl).
Flow cytometry and sorting
Peritoneal cells were washed and suspended in PBS containing 1% BSA and 0.5% EDTA. Before primary antibody labeling, cells were incubated with Fc receptor-blocking antibody (BD Pharmingen). Cells were incubated for 30 min at 4°C with 1:100 dilutions of fluorophore-coupled anti-F4/80, anti-CD11b, anti-CD11c, anti-B220, anti-CD4, or anti-CD8 primary antibodies (BD Pharmingen). For staining with antibodies to Mrc1 and SIGNR1 (Serotec) or Mertk, cells were fixed and permeabilized with methanol. After incubation with secondary antibodies (15 min/4°C), cells were analyzed using BD FACS Canto II. Data were analyzed with FlowJo 1.6. GFP-positive cells in lymph nodes (LN) were separated by FACS (BD FACS Aria II cell sorter).
Real-time qRT-PCR and conventional PCR
Total RNA was extracted from macrophages using TriPure reagent (Roche). For RNA extraction from mice paws, those were processed using the MagNA Lyser equipment (Roche). Complementary DNA was synthesized using M-MLV retro transcriptase (Invitrogen). Gene expression was analyzed by SYBR Green gene expression assay (Applied Biosystems). Arg1:F:CTCCAAGCCAAAGTCCTTAGAG;R:AGGAGCTGTCATTAGGGACATC; iNOS:F:CAGCTGGGCTGTACAAACCTT;R:CATTGGAAGTGAAGCGTTTCG; IL-10:F:TGCTATGCTGCCTGCTCTTA;R:TCATTTCCGATAAGGCTTGG; Mrc1:F:ATGCCAAGTGGGAAAATCTG;R:TGTAGCAGTGGCCTGCATAG; Ym1:F:ACTTTGATGGCCTCAACCTG;R:AATGATTCCTGCTCCTGTGG; TGFβ:F:TTGCTTCAGCTCCACAGAGA;R:TGGTTGTAGAGGGCAAGGAC; MCP1:F:CAGCCAGATGCAGTTAACGC;R:GCCTACTCATTGGGATCATCTTG; RANTES:F:AGTGCTCCAATCTTGCAGTCG;R:CACTTCTTCTCTGGGTTGGCA; CCL7:F:CACATTCCTACAGACAGCTC;R:AGCTACAGAAGGATCACCAG; MIP1α:F:ACTGCCTGCTGCTTCTCCTACA;R:AGGAAAATGACACCTGGCTGG; MIP1β:F:AAACCTAACCCCGAGCAACA;R:CCATTGGTGCTGAGAACCCT.
CRE gene was detected in genomic DNA from lenti-CRE transduced macrophages by conventional PCR using the following primers. CRE: F: AGGTGTAGAGAAGGCACTTAGC; R: CTAATCGCCATCTTCCAGCAGG.
Cell lysates and immunoblotting
Cells were lysed in buffer (20 mM Tris–HCl pH 7.5, 5 mM MgCl2, 50 mM NaF, 10 mM EDTA, 500 mM NaCl, 1% Triton X-100 and protease inhibitors) (15 min/4°C). Protein was quantified by the Bradford assay (Bio-Rad) and separated by 15% SDS–PAGE. After transference, membranes were probed with anti-CNB (Upstate), anti-CNA (Chemicon), anti-phospho p38 (Cell signaling), anti-phospho ERK (Millipore), anti-p38 (Santa Cruz), anti-IκBα (kindly provided by Dr. N. Rice, NIH), anti MKP-1 (C-19, Santa Cruz), or anti-tubulin (Sigma) and then with horseradish peroxidase-conjugated corresponding secondary antibodies. Peroxidase activity was detected with the enhanced chemiluminiscence system (GE Healthcare) and quantified with the software Quantity One.
Cytokine measurements
Cytokines were measured in supernatants of 48 h macrophage cultures by IL-10 ELISA (Diaclone) or with the mouse 10 plex kit Flowcytomix (Bender Medsystems).
Immunohistochemistry and immunofluorescence
For immunohistochemistry, paw, lung, spleen, and liver fragments were fixed in 10% formalin at 4°C for 24 h. After paraffin embedding, 5-μm sections were obtained. Endogenous peroxidase was blocked with 3% H2O2, and putative endogenous avidin-binding sites were blocked with the Avidin/Biotin Blocking kit (Vector laboratories). Slide-mounted sections were incubated (o/n, 4°C) with anti-GFP (Invitrogen) 1:250 in PBS, 1% horse serum and 5% BSA. Slides were incubated with biotinylated secondary antibody (RT/1 h) and then with preformed avidin–biotin peroxidase complexes (ABC Kit, Vector Laboratories), and stain was developed with the peroxidase substrate diaminobenzidine (DAB Kit, Vector Laboratories).
For immunofluorescence, paw fragments were snap-frozen in O.C.T. embedding medium (Tissue-Tek). Anti-GFP was used as before; anti-Mrc1 (Serotec) and anti-F4/80 (AbD Serotec) were diluted 1:50 and anti-IL-10 was diluted 1:100.
CN activity assay
Calcineurin enzyme activity in total macrophage extracts (5 μg) was determined with the Calcineurin Cellular Activity Assay Kit (Biomol).
Reporter gene assays
Peritoneal macrophages from NFAT-luc transgenic mice were stimulated with zymosan (100 μg/ml) (Sigma) or PMA (20 ng/ml) (Sigma) + calcium ionophore (30 μM) (A23187, Calbiochem) for 5 h and then lysed according to the Luciferase Assay System (Promega). LN from animals infected with luciferase-encoding lentivirus were homogenized and processed in a similar way. Luciferase activity was measured with a luminometer (Berthold Detection Systems) and normalized to protein content.
Macrophage migration
Thioglycolate-elicited peritoneal macrophages were collected by peritoneal lavage and labeled ex vivo (5 min RT) with the fluorescent lipophilic tracer DIR (Invitrogen). Labeled peritoneal macrophages were reinjected into the peritoneal cavity of recipient mice with zymosan-induced acute inflammation in the right hindpaws (180 μg zymosan/footpad) or oxazolone-induced contact hypersensitivity in the right ears (see below). DIR signal was tracked in vivo with the IVIS system using ICG/ICG BCK filters (Xenogen). Images were analyzed with Living image 3.1. When indicated, mice were inoculated with 300 μl lentivirus-enriched supernatant (7 × 108–2 × 109 TU/ml).
Antigen presentation
Macrophages were cultured in 6-well plates in the presence of 2 mg/ml ovalbumin antigen for 2 h. After several washes with PBS, macrophages were cultured for 5 h in regular culture medium, after which 2 × 106 β-galactosidase-expressing B3Z T cells were added and incubation continued overnight. Antigen presentation was quantified by monitoring the hydrolysis of chlorophenol red-β-D-galactopyranoside (CPRG) (Calbiochem) at O.D. = 595–655 nm.
Phagocytosis
In vivo transduced macrophages (1 × 106 total cells) were plated on crystal overnight and were serum-starved for 2 h. Red blood cells (20/macrophage) were untreated or opsonized with rabbit IgG (MP Biomedicals) and then added to the macrophage culture for 15 min. Cells were fixed, permeabilized, and stained with phalloidin TxRed (Invitrogen) and Alexa 647 chicken anti-rabbit IgG (Invitrogen). Phagocytosed red blood cells were counted under a confocal microscope.
Osteoclast differentiation
In vivo infected peritoneal macrophages were cultured in AlphaMEM containing 100 ng/ml RANKL and 20 ng/ml MCSF (both from Peprotech) for 7 days. Cells were stained for activity of tartrate-resistant acid phosphatase (TRAP) (Sigma) on day 7, and multinucleated TRAP+ cells were counted. Cell perimeter and area were analyzed with Metamorph software.
Cell therapy
Transduced macrophages were obtained from donor mice as described above. In experiments with CN KO cells, macrophages from Cnb1Δ/flox mice were infected ex vivo with CRE-recombinase-encoding lentivirus to induce CN deletion. Macrophages (4 × 104) were injected into the footpads of receptor mice 1 day before the second collagen immunization.
Zymosan-induced acute inflammation
Acute inflammation was induced in mouse paws by injection of 180 μg zymosan (30 μl) in the footpads. Stock zymosan solution was prepared at 30 mg/ml in endotoxin-free saline by boiling twice and subsequent sonication. Working solution was prepared by diluting in saline.
Oxazolone-Induced contact hypersensitivity
Mice were sensitized on the abdomen with 2% 4-ethoxymethylene-2-phenyl-2-oxazolin-5-one (oxazolone) (Sigma) in absolute ethanol. For migration assays, DIR-labeled macrophages were i.p. injected 6 days later and after 5 h hypersensitivity was elicited in the right ear by application of 1% oxazolone. In experiments to assess the therapeutic effect of lentivirus, mice were inoculated i.p. on day five with lentiviruses. Hypersensitivity was elicited the next day. Ear inflammation was quantified on day nine with the IVIS imaging system after i.p. injection of luminol (200 mg/kg) (Sigma). Luminescence images were analyzed with Living image 3.1.
Statistical analysis
The significance of statistical differences was assessed by one-way ANOVA or unpaired one-or two sample Student's t-test, using GraphPad Prism software 5.01. Differences were considered statistically significant at P < 0.05.
Acknowledgments
We thank Dr. G. R. Crabtree for providing Cnb1Δ/flox mice; Dr. J. D. Molkentin for providing NFAT-luc transgenic mice; Dr. Rosario Perona for providing MKP-1-deficient mice; Dr. M. K. Collins for providing the three-plasmid HIV-derived lentiviral system and Dr. Didier Trono for the pMD2.G vector; Dr. M. Campanero, Dr. I. Álvarez; Dr. A. Castrillo, Dr. A. García-Arroyo, Dr. A. Hidalgo and Dr. F. Sánchez-Madrid for critical reading of the manuscript and support; Dr. S. Bartlett for English editing; and Raquel Sánchez and Beatriz Ornés for technical assistance. J.M.R. is supported by the Spanish Ministry of Economy and Competitiveness (Ministerio de Economía y Competitividad; SAF2009-10708 and SAF 2012 34296), the Fundación Genoma España (MEICA) Fundación La Marató TV3 (080731), and the Spanish Ministry of Health (Ministerio de Sanidad y Consumo) Red de Investigación Cardiovascular (RIC; grant RD12/0042/0022). SM is supported by the Fundación La Marató TV3 (122532). The Centro Nacional de Investigaciones Cardiovasculares (CNIC) is supported by the Spanish Ministry of Economy and Competitiveness and the Pro-CNIC Foundation. A.E. held an FPI fellowship (BES-2007-15342).
Conflict of interest
The authors declare that they have no conflict of interest.
Author contributions
AE performed, analyzed, and interpreted most experiments and generated the figures. SM-M, AA, KU, HMI, MD, FM, GS, DS, and PGA provided ideas and experimental support. JMR supervised the experiments, interpreted, and analyzed results and together with AE wrote the manuscript.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol Cell. 1998;1:627–637. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Heitman J, Crabtree GR. Calcineurin: a central controller of signalling in eukaryotes. EMBO Rep. 2004;5:343–348. doi: 10.1038/sj.embor.7400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audran R, Collet B, Moisan A, Toujas L. Fate of mouse macrophages radiolabelled with PKH-95 and injected intravenously. Nucl Med Biol. 1995;22:817–821. doi: 10.1016/0969-8051(95)00013-n. [DOI] [PubMed] [Google Scholar]
- Bai L, Gabriels K, Wijnands E, Rousch M, Daemen MJ, Tervaert JW, Biessen EA, Heeneman S. Low- but not high-dose FK506 treatment confers atheroprotection due to alternative macrophage activation and unaffected cholesterol levels. Thromb Haemost. 2010;104:143–150. doi: 10.1160/TH09-07-0502. [DOI] [PubMed] [Google Scholar]
- Bellingan GJ, Caldwell H, Howie SE, Dransfield I, Haslett C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J Immunol. 1996;157:2577–2585. [PubMed] [Google Scholar]
- Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comalada M, Lloberas J, Celada A. MKP-1: a critical phosphatase in the biology of macrophages controlling the switch between proliferation and activation. Eur J Immunol. 2012;42:1938–1948. doi: 10.1002/eji.201242441. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, Kepka-Lenhart D, Morris SM, Jr, Gause WC, Leibovich SJ, Hasko G. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012;26:376–386. doi: 10.1096/fj.11-190934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, Karp CL, Pulendran B. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol. 2011;187:733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elloumi HZ, Maharshak N, Rao KN, Kobayashi T, Ryu HS, Muhlbauer M, Li F, Jobin C, Plevy SE. A cell permeable peptide inhibitor of NFAT inhibits macrophage cytokine expression and ameliorates experimental colitis. PLoS ONE. 2012;7:e34172. doi: 10.1371/journal.pone.0034172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban V, Mendez-Barbero N, Jimenez-Borreguero LJ, Roque M, Novensa L, Garcia-Redondo AB, Salaices M, Vila L, Arbones ML, Campanero MR, Redondo JM. Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med. 2011;208:2125–2139. doi: 10.1084/jem.20110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaulet G, Alfranca A, Torrente M, Escolano A, Lopez-Fontal R, Hortelano S, Redondo JM, Rodriguez A. IL10 released by a new inflammation-regulated lentiviral system efficiently attenuates zymosan-induced arthritis. Mol Ther. 2013;21:119–130. doi: 10.1038/mt.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge HS, Simmons RM, Underhill DM. Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J Immunol. 2007;178:3107–3115. doi: 10.4049/jimmunol.178.5.3107. [DOI] [PubMed] [Google Scholar]
- Hamalainen M, Lahti A, Moilanen E. Calcineurin inhibitors, cyclosporin A and tacrolimus inhibit expression of inducible nitric oxide synthase in colon epithelial and macrophage cell lines. Eur J Pharmacol. 2002;448:239–244. doi: 10.1016/s0014-2999(02)01947-7. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, Kumatori A, Suzuki S, Matsuyama T, Yui K. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci USA. 2005;102:16001–16006. doi: 10.1073/pnas.0504226102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper KE. Kinetics of macrophage recruitment and turnover in peritoneal inflammatory exudates induced by Salmonella or thioglycollate broth. J Leukoc Biol. 1986;39:435–446. doi: 10.1002/jlb.39.4.435. [DOI] [PubMed] [Google Scholar]
- Hortelano S, Lopez-Collazo E, Bosca L. Protective effect of cyclosporin A and FK506 from nitric oxide-dependent apoptosis in activated macrophages. Br J Pharmacol. 1999;126:1139–1146. doi: 10.1038/sj.bjp.0702422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CM, Jang SY, Fanzo JC, Pernis AB. Modulation of T cell cytokine production by interferon regulatory factor-4. J Biol Chem. 2002;277:49238–49246. doi: 10.1074/jbc.M205895200. [DOI] [PubMed] [Google Scholar]
- Jennings C, Kusler B, Jones PP. Calcineurin inactivation leads to decreased responsiveness to LPS in macrophages and dendritic cells and protects against LPS-induced toxicity in vivo. Innate Immun. 2009;15:109–120. doi: 10.1177/1753425908100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Kusler B, Otsuka M, Hughes M, Suzuki N, Suzuki S, Yeh WC, Akira S, Han J, Jones PP. Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol. 2007;179:4598–4607. doi: 10.4049/jimmunol.179.7.4598. [DOI] [PubMed] [Google Scholar]
- Kiani A, Rao A, Aramburu J. Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity. 2000;12:359–372. doi: 10.1016/s1074-7613(00)80188-0. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Lee CG, Kang KH, So JS, Kwon HK, Son JS, Song MK, Sahoo A, Yi HJ, Hwang KC, Matsuyama T, Yui K, Im SH. A distal cis-regulatory element, CNS-9, controls NFAT1 and IRF4-mediated IL-10 gene activation in T helper cells. Mol Immunol. 2009;46:613–621. doi: 10.1016/j.molimm.2008.07.037. [DOI] [PubMed] [Google Scholar]
- Lee SS, Tan AW, Giam YC. Cyclosporin in the treatment of severe atopic dermatitis: a retrospective study. Ann Acad Med Singapore. 2004;33:311–313. [PubMed] [Google Scholar]
- Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21:91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HW, New L, Han J, Molkentin JD. Calcineurin enhances MAPK phosphatase-1 expression and p38 MAPK inactivation in cardiac myocytes. J Biol Chem. 2001;276:15913–15919. doi: 10.1074/jbc.M100452200. [DOI] [PubMed] [Google Scholar]
- Liu J, Arai K, Arai N. Inhibition of NFATx activation by an oligopeptide: disrupting the interaction of NFATx with calcineurin. J Immunol. 2001;167:2677–2687. doi: 10.4049/jimmunol.167.5.2677. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Liu WH, Chen YJ, Cheng TL, Lin SR, Chang LS. Cross talk between p38MAPK and ERK is mediated through MAPK-mediated protein phosphatase 2A catalytic subunit alpha and MAPK phosphatase-1 expression in human leukemia U937 cells. Cell Signal. 2013;25:1845–1851. doi: 10.1016/j.cellsig.2013.05.021. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- Martinez-Martinez S, Redondo JM. Inhibitors of the calcineurin/NFAT pathway. Curr Med Chem. 2004;11:997–1007. doi: 10.2174/0929867043455576. [DOI] [PubMed] [Google Scholar]
- Martinez-Martinez S, Rodriguez A, Lopez-Maderuelo MD, Ortega-Perez I, Vazquez J, Redondo JM. Blockade of NFAT activation by the second calcineurin binding site. J Biol Chem. 2006;281:6227–6235. doi: 10.1074/jbc.M513885200. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Shibasaki F, Takehana K, Mori H, Nishida E, Koyasu S. Two distinct action mechanisms of immunophilin-ligand complexes for the blockade of T-cell activation. EMBO Rep. 2000;1:428–434. doi: 10.1093/embo-reports/kvd090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KA, Kim SY, Kim TB, Yun ES, Park CS, Cho YS, Moon HB, Lee KY. Allergen-induced CD11b+ CD11c(int) CCR3 + macrophages in the lung promote eosinophilic airway inflammation in a mouse asthma model. Int Immunol. 2007;19:1371–1381. doi: 10.1093/intimm/dxm108. [DOI] [PubMed] [Google Scholar]
- Nagaleekar VK, Sabio G, Aktan I, Chant A, Howe IW, Thornton TM, Benoit PJ, Davis RJ, Rincon M, Boyson JE. Translational control of NKT cell cytokine production by p38 MAPK. J Immunol. 2011;186:4140–4146. doi: 10.4049/jimmunol.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T, Honda K. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. 2005;102:15989–15994. doi: 10.1073/pnas.0508327102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- Niven AS, Argyros G. Alternate treatments in asthma. Chest. 2003;123:1254–1265. doi: 10.1378/chest.123.4.1254. [DOI] [PubMed] [Google Scholar]
- Noubade R, Krementsov DN, Del Rio R, Thornton T, Nagaleekar V, Saligrama N, Spitzack A, Spach K, Sabio G, Davis RJ, Rincon M, Teuscher C. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118:3290–3300. doi: 10.1182/blood-2011-02-336552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Uesugi M, Verdine GL. A second calcineurin binding site on the NFAT regulatory domain. Proc Natl Acad Sci USA. 2000;97:7130–7135. doi: 10.1073/pnas.97.13.7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardi M, Caelles C, Serrano AL, Munoz-Canoves P. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J Cell Biol. 2011;195:307–322. doi: 10.1083/jcb.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, Tesselaar K, Koenderman L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116:625–627. doi: 10.1182/blood-2010-01-259028. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Roy J, Martinez-Martinez S, Lopez-Maderuelo MD, Nino-Moreno P, Orti L, Pantoja-Uceda D, Pineda-Lucena A, Cyert MS, Redondo JM. A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol Cell. 2009;33:616–626. doi: 10.1016/j.molcel.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Gordon S. Adoptive transfer of fluorescence-labeled cells shows that resident peritoneal macrophages are able to migrate into specialized lymphoid organs and inflammatory sites in the mouse. Eur J Immunol. 1990;20:1251–1258. doi: 10.1002/eji.1830200609. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J. 2012;443:603–618. doi: 10.1042/BJ20120146. [DOI] [PubMed] [Google Scholar]
- Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Tjiu JW, Chen JS, Shun CT, Lin SJ, Liao YH, Chu CY, Tsai TF, Chiu HC, Dai YS, Inoue H, Yang PC, Kuo ML, Jee SH. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J Invest Dermatol. 2009;129:1016–1025. doi: 10.1038/jid.2008.310. [DOI] [PubMed] [Google Scholar]
- Tugwell P, Pincus T, Yocum D, Stein M, Gluck O, Kraag G, McKendry R, Tesser J, Baker P, Wells G. Combination therapy with cyclosporine and methotrexate in severe rheumatoid arthritis. The Methotrexate-Cyclosporine Combination Study Group. N Engl J Med. 1995;333:137–141. doi: 10.1056/NEJM199507203330301. [DOI] [PubMed] [Google Scholar]
- Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T, Takami T. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008;83:1136–1144. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- Urso K, Alfranca A, Martinez-Martinez S, Escolano A, Ortega I, Rodriguez A, Redondo JM. NFATc3 regulates the transcription of genes involved in T-cell activation and angiogenesis. Blood. 2011;118:795–803. doi: 10.1182/blood-2010-12-322701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.