

Abstract
eIF5B is a eukaryal translational GTPase that catalyzes ribosomal subunit joining to form elongation-competent ribosomes. Despite its central role in protein synthesis, the mechanistic details that govern the function of eIF5B or its archaeal and bacterial (IF2) orthologs remained unclear. Here, we present six high-resolution crystal structures of eIF5B in its apo, GDP- and GTP-bound form that, together with an analysis of the thermodynamics of nucleotide binding, provide a detailed picture of the entire nucleotide cycle performed by eIF5B. Our data show that GTP binding induces significant conformational changes in the two conserved switch regions of the G domain, resulting in the reorganization of the GTPase center. These rearrangements are accompanied by the rotation of domain II relative to the G domain and release of domain III from its stable contacts with switch 2, causing an increased intrinsic flexibility in the free GTP-bound eIF5B. Based on these data, we propose a novel domain release mechanism for eIF5B/IF2 activation that explains how eIF5B and IF2 fulfill their catalytic role during ribosomal subunit joining.
Keywords: crystal structure, molecular machines, GTPase, ribosome, subunit joining, translation initiation
Introduction
Translation is the fundamental cellular process in which the ribosome synthesizes proteins according to genetically encoded information. Among the individual steps, translation initiation is the most complex and most divergent in the three domains of life, which is highlighted by the different number of initiation factors (IFs) employed by eukaryal (∼12 eIFs) or bacterial cells (three IFs) to accomplish the same goals during ribosome assembly (Marintchev & Wagner, 2004). The major differences between eukaryal and bacterial translation initiation concern the formation of the 48S/30S pre-initiation complex (pre-IC) where the small 40S/30S ribosomal subunit is assembled at the AUG start codon of an mRNA with the charged initiator-tRNA (Met-tRNAiMet/fMet-tRNAfMet) in its P site (Marintchev & Wagner, 2004). This is followed by the formation of the elongation-competent 80S/70S ribosome, which is achieved by the joining of the large 60S/50S ribosomal subunit, catalyzed by the orthologous GTPases eIF5B, aIF5B and IF2 in eukarya, archaea and bacteria, respectively (Gualerzi et al, 2001; Pestova et al, 2000; Roll-Mecak et al, 2001).
Together with the elongation factors EF-Tu and EF-G, the initiation factors eIF5B, aIF5B and IF2 form the set of canonical translational GTPases (trGTPases) that are ubiquitous in extant cellular life. This suggests that the function of eIF5B and its orthologs was fixed at an early stage of cellular evolution before the onset of speciation, reflecting the importance of subunit joining as the final control step in the initiation pathway. Up to now, however, the structural dynamics between the active and inactive factor that govern the process of subunit joining, and the degree of their conservation across the three domains of life remain unclear.
Earlier crystal structures of aIF5B and IF2 revealed similar architectures for both proteins with an N-terminal GTP-binding (G) domain and a β-barrel domain II as structural core, followed by domains III and IV (Eiler et al, 2013; Roll-Mecak et al, 2000; Simonetti et al, 2013). IF2 and eIF5B contain an additional N-domain, which displays little conservation in sequence and length and was shown to be dispensable for the function of yeast eIF5B (Shin et al, 2002).
Like all trGTPases, eIF5B and its orthologs belong to the family of guanine nucleotide-binding (G) proteins (Bourne et al, 1991). Consequently, their mechanism has to be viewed as a specific variation of the classical concept of G proteins as molecular switches that alternate between an inactive GDP- and a structurally distinct active GTP-bound state. The transition between the two states is defined by conformational changes in two dynamic elements of the G domain, termed switch 1 and switch 2, which specifically interact with the γ-phosphate of the GTP molecule (Vetter & Wittinghofer, 2001). Only in the GTP-bound state the G protein interacts tightly and productively with effector molecules. Accordingly, the functional cycle of the G protein ends when it is “switched off” by GTP hydrolysis and the following structural transition to the inactive GDP-bound state (Bourne et al, 1991; Vetter & Wittinghofer, 2001).
For eIF5B, it was demonstrated that it interacts with the ribosomal subunits and catalyzes 80S ribosome formation in a GTP-dependent manner (Acker et al, 2009; Pestova et al, 2000). It was further shown that subunit joining occurs catalytically in the presence of GTP but only stoichiometrically with the non-hydrolyzable GTP analog GDPNP. This indicates that GTP hydrolysis is required for the release of the factor from the 80S ribosome, in line with the observation that GDPNP, but not GTP slows down the dissociation of eIF5B from 80S ribosomes and inhibits the peptidyl-transfer reaction (Pestova et al, 2000; Shin et al, 2002). Similarly, it was found that IF2 promotes subunit joining much more efficiently in the presence of GTP and GDPNP than with GDP and that GTP hydrolysis is required for the dissociation of the factor from the ribosome and subsequent peptide-bond formation (Antoun et al, 2003; Benne et al, 1973; Dubnoff et al, 1972). Based on this evidence, it was suggested that eIF5B and IF2 employ a similar mechanism to promote subunit joining (Antoun et al, 2003) which is compatible with the classical concept of G protein function.
However, this assumption stands in sharp contrast to other structural and biochemical data. It was found that GDP- and GTP-bound IF2 catalyze subunit joining nearly equally well and that GTP hydrolysis is not required for the release of IF2 from the ribosome (Fabbretti et al, 2012; Tomsic et al, 2000). Based on these observations, it was proposed that IF2 functions differently from eIF5B and as a non-classical GTPase with apparently no role for GTP hydrolysis (Eiler et al, 2013; Rodnina et al, 2000; Tomsic et al, 2000), raising the question why the catalytic machinery required for GTP binding and hydrolysis is universally conserved in IF2.
So far available structure-based models for eIF5B/IF2 function do not provide explanations for these contradictory results. The “articulated lever model” for eIF5B/IF2 function, which is based on crystal structures of aIF5B, assumes that a GTP-induced ∼2 Å shift in switch 2 is amplified by an en bloc rearrangement of domains II to IV into a ∼5 Å movement of domain IV (Roll-Mecak et al, 2000). According to this model, neither switch 1 nor switch 2 undergoes the conformational changes or forms the direct contacts with the γ-phosphate that are typical for the classical molecular switch. However, low-resolution cryo-EM reconstructions of bacterial and eukaryal 70S/80S ICs revealed conformations of IF2 and eIF5B that are incompatible with the articulated lever model (Allen et al, 2005; Fernandez et al, 2013). In order to reconcile the contradictory experimental data with the classical concept of molecular switching, it was suggested that eIF5B and IF2 follow a mechanism of “conditional switching,” in which GTP binding alone is insufficient to activate eIF5B/IF2 but requires the ribosome as a cofactor that shifts the equilibrium between an inactive and an active GTP-bound form toward the latter (Hauryliuk et al, 2008; Pavlov et al, 2011). More recently, it was proposed that IF2 does not have an “effecter domain” like other trGTPases and therefore behaves different from eIF5B and not as a classical GTPase (Eiler et al, 2013), which, however, leaves open the question how the nucleotide status of the G domain is communicated into domains III and IV.
For aIF5B as well as IF2, crystal structures have previously been solved in the GDPNP- and GTP-bound state, respectively (Roll-Mecak et al, 2000; Simonetti et al, 2013). However, in both cases, the G domain remained in the apo/GDP conformation despite the presence of the γ-phosphate. Thus, the knowledge of eIF5B and IF2 function is limited by the fact that up to now no high-resolution structural information is available for their GTP-bound forms that is in agreement with the classical concept of molecular switching. Though of paramount importance to the understanding of eIF5B/IF2 function, it is therefore not known what distinguishes the active from the inactive state of the G domain and how these differences modulate the affinity of the overall eIF5B/IF2 to ribosomal effector complexes or influence the mechanism of ribosome-induced GTP hydrolysis.
Here, we present thermodynamic data and high-resolution structures of eIF5B that provide a detailed picture of its entire nucleotide cycle. We determined six crystal structures of eIF5B from Saccharomyces cerevisiae and Chaetomium thermophilum containing the G domain and domains II-IV or substructures thereof in the apo, GDP- and GTP-bound states. These structures unambiguously demonstrate that the G domain of free eIF5B follows the classical switch mechanism involving large structural rearrangements of the two switch regions. The GTP-induced changes result in the formation of a catalytic GTPase center similar to that in EF-Tu, suggesting a possible scenario for ribosome-dependent GTPase activation in eIF5B. Most importantly, the various structures in combination with an analysis of the thermodynamics of nucleotide binding suggest a mechanism for eIF5B activation in which the local switch within the G domain is propagated into the rest of the factor through the release of domain III, resulting in an increase of intrinsic flexibility that is necessary for efficient subunit joining. Based on these observations, we propose a domain release mechanism for eIF5B activation, which represents a novel variation from the classical paradigm of G proteins and suggests a unified picture of subunit joining by a/eIF5B and IF2.
Results
Overall structure and domain arrangement in apo eIF5B
Six different crystal structures of eIF5B from Chaetomium thermophilum (Ct) and Saccharomyces cerevisiae (Sc) were solved either in the apo form or cocrystallized with GDP or GTP. All structures were solved by means of molecular replacement. A summary of structures, crystallographic details and data statistics is presented in Table1 and Supplementary Fig S1.
Table 1.
Crystallization, X-ray data collection and refinement statistics
| Organism | Chaetomium thermophilum | Saccharomyces cerevisiae | ||||
|---|---|---|---|---|---|---|
| Construct purified | elF5B(517C) | elF5B(517C) | elF5B(517–970) | elF5B(517–970) | elF5B(399–852) | elF5B(399–852) |
| Construct in structure | elF5B(517C) apo | elF5B(870C) | elF5B(520–970)·GDP | elF5B(517–860)·GTP | elF5B(401–852)·apo | elF5B(401–852)·GDP |
| Crystallization | ||||||
| Condition | 100 mM MES (pH 6.8) 12% PEG 20000 10 mM Na-lactate | 100 mM MES (pH 6.8) 12% PEG 20000 10 mM Na-lactate | 15% PEG 8000 0.5 M Li2SO4 | 100 mM Hepes (pH 7) 13% PEG 4000 100 mM NaOAc | 20% ethylene glycol 5% PEG 3350 20 mM MgCI2 | 8% PEG 8000 0.37 M Li2SO4 |
| Temperature (°C) | 4 | 4 | 20 | 20 | 4 | 10 |
| Data collection | ||||||
| Space group | P3221 | P3121 | P212121 | P21 | P41 | P212121 |
| Unit cell |
a = b = 111.5 Å c = 115.2 Å |
a = b = 98.2 Å c = 97.4 Å |
a = 66.9 Å b = 72.9 Å c = 199.2 Å |
a = 55.4 Å b = 114.8 Å c = 65.9 Å |
a = b = 118.0 Å c = 77.5 Å |
a = 73.6 Å b = 119.5 Å c = 120.7 Å; |
| α = β = 90° γ = 120° |
α = β = 90° γ = 120° |
α = β = γ = 90° | α = 90° β = 102.3° γ = 90° |
α = β = γ = 90° | α = β = γ = 90° | |
| Molecules/asym. unit | 1 | 1 | 2 | 2 | 2 | 2 |
| Resolution (Å) | 2.75 (2.85–2.75) | 3.2 (3.3–3.2) | 2.12 (2.21–2.12) | 1.87 (1.97–1.87) | 1.83 (1.93–1.83) | 3.02 (3.12–3.02) |
| Observed reflections | 168 769 (15,898) | 59 836 (5506) | 288 203 (38,832) | 251 805 (36,887) | 426 259 (62,736) | 108 724 (10 591) |
| Unique reflections | 22 063 (2217) | 9288 (800) | 56 555 (7213) | 66 297 (9605) | 93 360 (13677) | 21 521 (1972) |
| Completeness (%) | 99.9 (100) | 99.8 (100) | 99.7 (99.9) | 99.8 (99.8) | 99.8 (99.9) | 99.2 (99.5) |
| <l>σ | 32.66 (3.29) | 32.12 (4.31) | 17.79 (2.97) | 20.53 (2.38) | 21.05 (3.11) | 23.3 (3.52) |
| Rsym (%) | 3.7 (58.9) | 3.4 (57.4) | 5.6 (63.1) | 4.4 (62.0) | 3.9 (52.5) | 5.3 (60.6) |
| Refinement | ||||||
| Rwork (%) | 19.3 | 19.0 | 21.9 | 16.7 | 16.8 | 24.9 |
| Rfree (%) | 23.8 | 22.0 | 25.2 | 20.6 | 19.4 | 28.7 |
| Rmsd from standard stereochemistry | ||||||
| Bond length (Å) | 0.005 | 0.003 | 0.005 | 0.019 | 0.008 | 0.004 |
| Bond angles (°) | 0.82 | 0.66 | 0.95 | 1.75 | 1.17 | 0.99 |
| Ramachandran plot statistics | ||||||
| Most favored (%) | 98.0 | 98.0 | 98.8 | 98.3 | 98.5 | 97.0 |
| Allowed regions (%) | 2.0 | 2.0 | 1.2 | 1.7 | 1.5 | 3.0 |
| Disallowed regions (%) | 0 | 0 | 0 | 0 | 0 | 0 |
Values in parentheses refer to the highest resolution shell.
Rwork and Rfree factors are calculated using the formula R = ∑ hkl||F(obs)hkl| − |F(calc)hkl|| / ∑ hkl|F(obs)hkl|, where F(obs)hkl and F(calc)hkl are observed and measured structure factors, respectively.
Rwork and Rfree differ in the set of reflections they are calculated from: Rfree is calculated for the test set, whereas Rwork is calculated for the working set.
The topology of the individual domains as well as the overall domain arrangement of apo eIF5B is similar to that of the archaeal ortholog aIF5B (Roll-Mecak et al, 2000) (Fig1). Domains I-III form the core structure that is assembled as triangle around switch 2 of domain I (G domain). Domains I and II are tightly associated through hydrophobic contact areas composed of β2 (part of switch 1), β3 and β4 in the G domain as well as β9, β10 and β16 in domain II (Fig1). The orientation of domain II relative to the G domain is nearly identical in Ct-eIF5B and Sc-eIF5B and similar to that found in IF2, EF-G or GDPNP-bound EF-Tu (Laurberg et al, 2000; Nissen et al, 1995; Simonetti et al, 2013).
Figure 1. Front view of the overall structure of Chaetomium thermophilum eIF5B(517C) in the apo form.
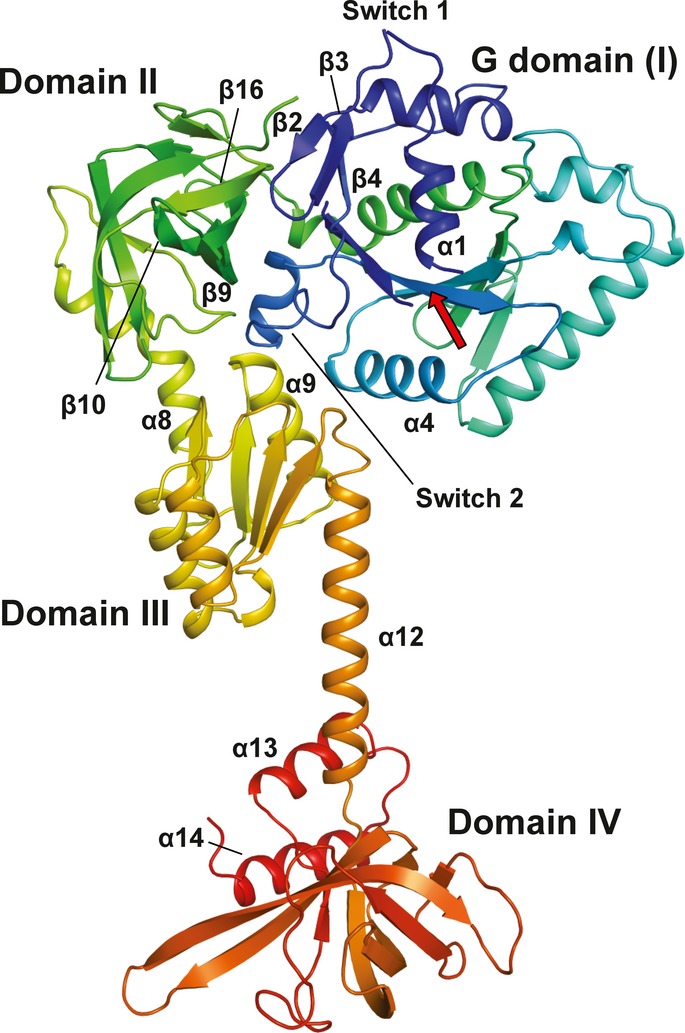
The Cα trace is shown in rainbow coloring from the N- (blue) to the C-terminus (red). The functional core of eIF5B is composed of the G domain (I) with the nucleotide binding site (arrow), domain II, domain III and domain IV.
Stable interactions of domain III with other domains are restricted to the area surrounding the N-terminal half of helix α9 (Figs1 and 3A and B). Van der Waals contacts are formed with domain II and the linker-helix α8 with a buried surface area of ˜700 Å2. Helix α9 also interacts with switch 2 as the only stable contact partner for domain III within the G domain. Further contacts are formed between the N-terminus of helix α12 and α4; however, these do not seem to be functionally relevant, as they differ between the various apo structures. Consequently, the orientation of domain III relative to the G domain differs considerably in Sc-eIF5B, Ct-eIF5B and aIF5B, which amounts to a displacement of the C-terminus of α12 by 12–22 Å between the three apo structures (Supplementary Fig S2A).
In a direct superposition of domain III from aIF5B, Ct-eIF5B(517C) and Ct-eIF5B(870C) the C-terminal ends of α12 lie only 3–3.5 Å apart. However, domain IV adopts significantly different orientations relative to α12 and domain III, indicating a high degree of flexibility (Supplementary Fig S2B).
Conformational changes in the G domain of eIF5B upon GTP binding
GTP is bound by eIF5B in a way common for G proteins involving five conserved sequence motifs termed G1–G5 (Bourne et al, 1991) (Fig2 and Supplementary Fig S3). The base is in contact with the 530NKID533 (G4) and 598SAx600 (G5) motifs (unless stated otherwise, S. cerevisiae sequence numbering will be used throughout), and the phosphates of the nucleotide are stabilized by main- and side-chain interactions with the P loop (G1).
Figure 2. The nucleotide-dependent conformational switch in the G domain of eIF5B.

A–C Structural transition of the G domain from its apo form (A) to the GTP- (B) and the GDP-bound (C) states. P loop, switch 1 and switch 2 are colored pink in the apo state, yellow in the GTP-bound state and cyan in the GDP-bound state. Thr439, Asp476, Gly479 and His480 (Saccharomyces cerevisiae numbering) are shown as sticks; the Mg2+ ion, Na+ ion and water molecules are shown as spheres in magenta, blue and gray, respectively; nucleotides are shown as balls and sticks.
D, E Network of interactions in the nucleotide binding pocket of the GTP- (D) and GDP-bound (E) factor. Direct interactions are indicated by dashed lines.
The most severe conformational changes are observed for the two switch regions which contain the 437GIT439 (G2) and 476DTPG479 (G3) motifs that function as sensors for the presence of the γ-phosphate (Fig2). In the apo state, most of switch 1 (residues 427–443) forms an unstructured loop, which points away from the nucleotide binding pocket. Only within its last third, the inactive switch 1 forms a short β strand (β2) oriented antiparallel to β3 as part of the interface with domain II. Upon GTP binding, switch 1 flips over by ∼180° using Gln427 and Gly443 as hinges (Fig2A and B and Supplementary S4A). As a result, its N-terminal part is oriented antiparallel to helix α1 toward the nucleotide binding pocket where it forms a one-turn α-helix (α1′) above the α-phosphate, followed by a turn toward the β- and γ-phosphates that continues into β3. The critical Thr439 is thereby displaced from its position in β2 of the apo state by nearly 20 Å to form direct contacts with the Mg2+ ion and the γ-phosphate. In this new position, switch 1 has almost no contacts outside the nucleotide binding pocket with the exceptions of a salt bridge between Glu434 and Arg688 of the β13-β14 loop and a hydrogen bond of Gln441 to the conserved Glu636 in domain II (Fig2D).
Switch 2 (476–492) undergoes a substantial rearrangement as well. In the apo state, the G3 motif in switch 2 runs parallel to β4 as far as Gly479, where the peptide backbone makes a sharp turn of > 90° and continues through the inter-domain cleft formed by domains I-III toward the back of the protein (Figs2A and 3B). Here, switch 2 turns a second time toward the dorsal side of the G domain forming the two-turn helix α3. In this conformation, switch 2 makes van der Waals contacts with domain II but mainly interacts with domain III: The backbone CO of Ser484 accepts a hydrogen bond from Gly763, Arg487 forms a hydrogen bond and salt bridges to Glu766 and Asp770, and Arg489 forms a strong salt bridge to Asp740 (Fig3A and B).
Figure 3. Nucleotide-dependent conformational changes in eIF5B.

- G domain-based superposition of domains I (G), II and III from Ct-eIF5B in the apo and GTP-bound state. Both G domains are shown in gray; otherwise the same color code was used as in Fig2. The GTP-induced rearrangements result in the loss of interactions between switch 2 and helices α8 and α9 (circle) and ultimately in the release of domain III from the G domain. Domain II rotates by ∼30° relative to the G domain and is stabilized in its new orientation by the newly formed contact between β13 and β14 loop and domain I.
- In apo eIF5B, the inactive switch 2 (pink) forms stable contacts with helices α8 and α9 of domain III which are broken upon the GTP-induced transition of switch 2 to its active state (yellow).
- Comparison of the molecular switch mechanisms in EF-Tu (left) and eIF5B (right). Both trGTPases are shown in their inactive apo or GDP-bound (top) and GTP-bound (bottom) states, respectively. Functionally relevant interactions between the switch regions (yellow) of the G domain and downstream functional domains (blue) are indicated by dashed circles.
- The contact surface (pink) found between domain III (blue) and domains I and II in apo eIF5B (top) is entirely lost in ribosome-bound eIF5B·GDPCP (bottom; PDB: 4BVX (Fernandez et al, 2013)), where domains III and IV become stabilized between SRL and Met-tRNAiMet (not shown).
Upon GTP binding, Asp476 moves 3.1 Å toward the γ-phosphate and forms a hydrogen bond to one of the water molecules coordinating the Mg2+ ion (Fig2B–D and Supplementary Fig S4B). The universally conserved Gly479 of switch 2 moves ∼8 Å toward the γ-phosphate; concomitantly, the peptide bond between Pro478 and Gly479 flips by ∼160°. In its new position, Gly479 interacts directly with the γ-phosphate and the putative catalytic water molecule (Wcat). The movement of the G3 motif has a profound impact on the rest of switch 2: His480 moves by 12 Å and forms part of a loop at the front of the G domain that is stabilized by Arg487 (Supplementary Fig S4B). In its new position, His480 lies next to Val414 (P loop) with the imidazole moiety pointing outward, away from Wcat (Fig2D). The rest of switch 2 (484–493) forms the extended helix α3 next to α4, with some residues up to 15 Å relocated from their original position in apo eIF5B; the axis through α3 is thereby rotated by more than 90° (Supplementary Fig S4A). In order to achieve this conformational change in switch 2, all its interactions to domain III in the apo state are necessarily broken (Fig3A and B).
Switch 1 and switch 2 are stabilized in their active GTP-bound conformations through a network of interactions surrounding the Mg2+ ion and γ-phosphate (Fig2D). The Mg2+ ion is coordinated by six oxygen ligands with octahedral coordination geometry; two of the ligands are water molecules, two come from the β- and γ-phosphates, and two are provided by the side chains of Thr419 and Thr439. The γ-phosphate is further stabilized by Lys418 (P loop), Thr439 and Gly479. Wcat lies 2.8 Å from the outward pointing γ-phosphate oxygen in position for an in-line attack on the γ-phosphate, stabilized by the backbone amides of Gly479 and His480 and the backbone CO of Thr439. Next to the γ-phosphate, an additional strong electron density was observed, which was assigned to a Na+ ion. Its pentagonal coordination shell with the typical bond lengths of 2.3–2.5 Å (Harding, 2002) is constituted by two oxygens from the α- and γ-phosphates, the β-γ-bridging oxygen, the caboxylate of Asp415 (P loop) and the CO from Gly437 in switch 1. Together, P loop, switch 1 and switch 2 form a closed ∼10 Å deep pocket that accommodates the Mg2+ and Na+ ions as well as all three phosphates with the γ-phosphate and Wcat at its bottom. His480 and bulk solvent are excluded from this pocket by a gate formed by Val414 and Ile438.
A comparison with the structure of EF-Tu·GDPNP (Hilgenfeld et al, 2000) shows a strikingly high degree of similarity between the catalytic centers in GTP/GDPNP-bound EF-Tu and eIF5B with nearly identical positions for all conserved residues with a pairwise Cα rmsd of 1.1 Å over 99 residues as well as for Wcat and the Mg2+ ion (Fig4A and Supplementary Fig S5A). However, the position occupied by the Na+ ion in eIF5B is vacant in the EF-Tu·GDPNP structure, and a perfect agreement between eIF5B and EF-Tu is restricted to residues that are directly involved in nucleotide binding or implicated in GTPase activity. Switch 1 in eIF5B lacks the second helix (A′′) that serves factor-specific functions in EF-Tu (Nissen et al, 1995).
Figure 4. Structural model of eIF5B·GTP on the ribosome.

- Superposition of the catalytic centers of eIF5B·GTP (yellow) and free EF-Tu·GDPNP (cyan; PDB: 1EXM). Conserved residues are shown as sticks; GDPNP is omitted for clarity.
- Superposition of the catalytic centers of eIF5B·GTP (yellow) and ribosome-bound EF-Tu·GDPCP (PDB: 2XQD, 2XQE) with the sarcin-ricin loop (SRL) in pink. Structural alterations relative to free eIF5B·GTP and EF-Tu·GDPNP are limited to Hiscat of EF-Tu, which is reoriented (arrow) into its active position between A2662 and Wcat.
- Model of domains I and II of eIF5B (orange) on the ribosome, based on the superposition with EF-Tu·GDPCP. Similar to eIF5B·GDPCP in the cryo-EM model of the 80S IC (see Supplementary Fig S5B), the G domain is associated with the SRL of the large subunit (LSU; green), while domain II interacts with the body of the small subunit (SSU; light pink).
- Putative interactions between the G domain and the SRL/H95 (green). Direct interactions are indicated by black dashed lines; red dashed lines indicate the positions in H95 that are cleaved by Fe(II)-BABE introduced in the position of Lys540 (Unbehaun et al, 2007). His505 lies only 3.5 Å from H95, explaining why the H505Y mutation results in a reduced affinity for the ribosome and GTPase deficiency in eIF5B (Shin et al, 2002) (see also Supplementary Table S1). The conserved Arg534 likely contributes to the interactions with H95.
Domain rearrangements in eIF5B upon GTP binding
Upon GTP binding, domain II performs two main movements that result from the activation of the G domain (Fig3A): On the one hand, the dorsal portion of domain II tilts inward, following the movement of β3 and β4 that was induced by the 3.1 Å shift of Asp476 toward the Mg2+ ion (Supplementary Fig S4); on the other hand, domain II rotates by ∼30° causing the front portion to move upward and the ventral side to move parallel to switch 2 toward the front. The rotated orientation of domain II is stabilized by a newly formed interaction of the β13-β14 loop (residues 684–689) with the G domain, which takes over the position next to β3 that was originally occupied by β2 and left vacant after the GTP-induced rearrangement of switch 1 (Figs2 and 3A).
As described above, all residues of switch 2 that are involved in contacts with domains II and III in the apo structure are rearranged and move by an average of ˜14 Å upon GTP binding (Fig3B and Supplementary Fig S4). Since switch 2 is the main contact area for domain III in the G domain, either a completely new set of interactions has to be formed or the interaction of domain III to the G domain is entirely lost. As domain III is not present in the structure of Ct-eIF5B·GTP, direct information about its position in free eIF5B·GTP is not available. However, our ITC experiments described below point toward a scenario in which domain III is released from the G domain without forming stable new contacts with the reorganized G domain in the free form of eIF5B·GTP (see below).
Conformational changes in eIF5B during the transition from the GTP- to the GDP-bound state
Subsequent to GTP hydrolysis and release of Pi, most conformational rearrangements that followed GTP binding are reversed in eIF5B·GDP (Fig2). Switch 1 flips back to its original position, thereby displacing the β13-β14 loop. Switch 2 loses part of its α-helical structure and retracts toward the back of the protein to adopt a conformation nearly identical to that found in the apo form (Supplementary Fig S4). However, Asp476 in the G3 motif of Ct-eIF5B·GDP remains in its activated position in contact with the Mg2+ ion. Likewise, Thr477 and Pro478 retain their activated positions, whereas Gly479 is rotated back. As a consequence, the entire switch 2 is still shifted ˜3 Å relative to its apo position but already forms the interactions to domains II and III found in apo eIF5B (Supplementary Fig S4). Accordingly, α9 and therewith domain III are shifted 3–4 Å relative to their apo state position. Finally, also the ventral side of domain II is still shifted forward; however, no upward movement of its frontal face occurs, indicating that the loss of interactions between domains I and III is a prerequisite for this domain rearrangement (Supplementary Fig S4C).
In summary, the transition from the GTP to the GDP state allows domain III to reassociate with the core domains of eIF5B and reverses the rotation of domain II. Interestingly, binding of GDP and Mg2+ seems to be able to partially activate switch 2 (including the peptide flip of Gly479), which induces conformational strain on the switch 2-domain III interactions.
Thermodynamics of the interactions between eIF5B and guanine nucleotides
In order to gain further insight into the domain rearrangements during the nucleotide cycle of eIF5B, we performed ITC experiments to determine the thermodynamic parameters of eIF5B binding to GTP and GDP in the temperature interval of 5–30°C (a summary of the data is given in Tables2 and 3; Fig5 and Supplementary Fig S6).
Table 2.
Thermodynamic parameters of eIF5B binding to GDP and GTP at different temperatures and in presence or absence of Mg2+ ions
| Construct | Ligand | MgCl2 (mM) | T (°C) | Kd (µM) | ΔH (cal/mol) | ΔG (kcal/mol) | TΔS (kcal/mol) |
|---|---|---|---|---|---|---|---|
| Ct-eIF5B(517C) | GDP | 2.5 | 5 | 2.09 | −6286 | −7.23 | 1.0 |
| GDP | 2.5 | 10 | 1.92 | −5057 | −7.4 | 2.3 | |
| GDP | 2.5 | 15 | 2.61 | −5240 | −7.36 | 2.0 | |
| GDP | 2.5 | 20 | 2.9 | −5520 | −7.43 | 1.9 | |
| GDP | 2.5 | 25 | 3.3 | −6058 | −7.48 | 1.4 | |
| GDP | 2.5 | 30 | 3.45 | −6650 | −7.58 | 0.9 | |
| GDP | 0 | 5 | 0.8 | −1855 | −7.76 | 5.9 | |
| GDP | 0 | 10 | 1.0 | −2385 | −7.77 | 5.4 | |
| GDP | 0 | 15 | 1.14 | −2805 | −7.84 | 5.0 | |
| GDP | 0 | 25 | 1.96 | −3912 | −7.79 | 3.9 | |
| GTP | 2.5 | 5 | 4.21 | −5346 | −6.84 | 1.5 | |
| GTP | 2.5 | 10 | 4.83 | −6192 | −6.89 | 0.7 | |
| GTP | 2.5 | 15 | 5.68 | −7033 | −6.92 | 0.12 | |
| GTP | 2.5 | 20 | 6.02 | −7423 | −7.0 | −0.6 | |
| GTP | 2.5 | 25 | 6.20 | −8426 | −7.1 | −1.3 | |
| GTP | 2.5 | 30 | 7.04 | −9344 | −7.03 | −2.2 | |
| Ct-eIF5B(517–858) | GTP | 2.5 | 5 | 1.34 | −4860 | −7.47 | 2.6 |
| GTP | 2.5 | 10 | 1.58 | −7822 | −7.52 | −0.3 | |
| GTP | 2.5 | 20 | 2.12 | −12,985 | −7.61 | −5.4 | |
| GTP | 2.5 | 30 | 4.07 | −18,810 | −7.48 | −11.3 |
All measurements were performed two to four times; for GTP binding to both constructs and for GDP binding in the presence of Mg2+ the experiments were done with two independent purifications of the respective construct; for GDP binding in the absence of Mg2+, the experiments were done with protein from one purification.
Kd, dissociation equilibrium constant; calculated as 1/Ka.
Ka, association equilibrium constant; standard deviation did not exceed ± 15%.
ΔH, standard enthalpy change; standard deviation did not exceed ± 10%.
ΔG, Gibbs energy; calculated from equation ΔG = −RTlnKa.
TΔS, standard entropy change; calculated from equation ΔG = ΔH − TΔS.
Table 3.
Changes in heat capacity and solvent-accessible surface area for eIF5B binding to GDP and GTP
| Construct | Ligand | ΔCp (cal/mol·K) | ΔASAmin (Å2) | ΔASAmax (Å2) | ΔASAcalc (Å2) |
|---|---|---|---|---|---|
| Ct-eIF5B(517C) | GDP+Mg | −140 ± 23 | −311 | −583 | −400 |
| GDP-Mg | −102 ± 5 | −227 | −425 | ||
| GTP+Mg | −155 ± 8 | −344 | −646 | ||
| Ct-eIF5B(517–858) | GTP+Mg | −553 ± 11 | −1229 | −2304 | −1800 |
ΔCp, heat capacity change; obtained from ΔH/dT.
ΔASAmin and ΔASAmax, changes in solvent-accessible surface areas assuming that all changes were conferred by either apolar or 70% apolar and 30% polar surfaces, respectively.
Calculated for 30°C from the first derivative of the second-order polynomial fit to ΔH measured at five different temperatures between 10 and 30°C.
Obtained from the slope of the linear fit to ΔH measured at different temperatures between 5 and 30°C.
Calculated from the crystal structures of GDP- and GTP-bound eIF5B relative to the apo state.
Figure 5. eIF5B interactions with guanine nucleotides measured by ITC.
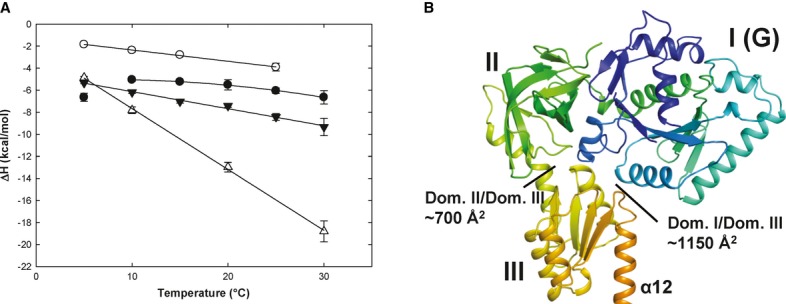
- Heat capacity changes upon eIF5B interaction with GDP or GTP. Temperature dependency of binding enthalpy changes (ΔH) upon Ct-eIF5B(517C) interactions with GDP in the presence (•) or absence (○) of MgCl2 and of Ct-eIF5B(517C) (▾) and Ct-eIF5B(517–852) (Δ) with GTP in the presence of MgCl2. Standard deviations are given by error bars (in some cases not visible because they are smaller than the symbol size).
- Domains I–III of apo Ct-eIF5B. Indicated are the contact areas of domain III to domains I and II, respectively.
To probe the conformational changes in eIF5B upon GTP binding and to test the particular influence of domain III, we performed ITC experiments with two different constructs: one comprising domains I–IV [Ct-eIF5B(517C)] and the other comprising only domains I and II [Ct-eIF5B(517-858)]. In both cases, GTP binding was driven by favorable negative changes in binding enthalpy (ΔH = −9.34 kcal/mol for domains I–IV and -18.8 kcal·mol−1 for domains I–II at 30°C) and opposed by unfavorable entropic contributions (Table2).
For both constructs, ΔH plotted against the temperature results in a straight line with negative slope (Fig5A) representing the change in heat capacity (ΔCp) which can be used as estimate for the change in solvent-accessible surface area (ΔASA) upon complex formation (see Materials and Methods). For Ct-eIF5B(517C), a ΔCp of −155 cal·mol−1·K−1 is calculated, corresponding to 344–646 Å2 of surface area that become buried upon GTP binding (Table3). In contrast, GTP binding to Ct-eIF5B(517–858), the construct lacking domains III and IV, gives a ΔCp of −553 cal·mol−1·K−1, corresponding to a GTP-dependent surface burial of 1229–2304 Å2, in agreement with the ∼1800 Å2 that become buried by switch 1 and the β13–β14 loop in domains I and II according to the crystal structures (Table3). Thus, the presence of domains III and IV contributes to the overall ΔCp of −155 cal·mol−1·K−1 in Ct-eIF5B(517C)·GTP with +398 cal·mol−1·K−1 to compensate the contribution of −553 cal·mol−1·K−1 by domains I and II alone. This corresponds to a ΔASA of 884–1658 Å2 that are exposed upon GTP binding simultaneously to the burial of ∼1800 Å2 (or 1229–2304 Å2) in domains I and II. The only reasonable candidates that can account for this compensatory effect are the surface areas buried between domain III and the G domain (∼1150 Å2) and domain II (∼700 Å2), respectively, in apo eIF5B (Fig5B). Thus, these data indicate that domain III is released from most or all its contacts with the G domain and domain II in response to the GTP-induced rearrangement of switch 2.
GDP binding to Ct-eIF5B(517C) was driven by favorable contributions of both, binding enthalpy and entropy (ΔH = −6.65 kcal/mol and TΔS = 0.9 kcal/mol at 30°C) (Table2). In contrast to GTP binding, the temperature dependency of ΔH was not linear for GDP binding; instead, the data between 10 and 30°C fit better to a second-order polynomial function, indicating a strong temperature dependency of ΔCp (Fig5A). This suggests that the amount of contact surface within the formed complex changes over the used temperature range. At higher temperatures (30°C), GDP binding results in a ΔCp of −140 cal·mol−1·K−1, corresponding to a surface burial of 311–583 Å2 (Table3), which agrees well with a ΔASA of ˜400 Å2 for GDP binding according to the crystal structures. However, the negative value for ΔCp decreases with lower temperatures. Below 10°C, the second-order polynomial behavior of ΔH breaks down and ΔCp changes sign, indicating a net exposure of ASA upon GDP binding. Here, three observations based on the eIF5B·GDP structures are of particular interest: (i) GDP/Mg2+ is able to partially activate switch 2 and to induce conformational strain on its interactions to domains II and III (Supplementary Fig S4), (ii) domain III contacts switch 2 primarily through ionic interactions (Fig3B) which are destabilized at low temperatures (Elcock, 1998; Hendsch & Tidor, 1994), and iii) domain III is released from the G domain in molecule B of Sc-eIF5B·GDP for which crystals were obtained at 10°C (Table1; Supplementary Fig S1E and S7A). Since switch 1 and the β13–β14 loop remain flexible in this structure, the release of domain III upon GDP binding results in a positive ΔASA corresponding to a positive contribution to ΔCp as observed in the ITC experiments. The non-linear behavior of ΔCp above 10°C would thus indicate that domain III always has the propensity to be released in eIF5B·GDP due to the partial activation of switch 2 in the presence of Mg2+, however, with a reduced tendency to do so with increasing temperatures at which the ionic interactions to the G domain become increasingly stable (Elcock, 1998; Hendsch & Tidor, 1994). In line with this interpretation, we found that the temperature dependency of ΔH does not break down at low temperatures when the ITC experiments are repeated with GDP in the absence of Mg2+ (Fig5A). Instead, ΔH plotted against the temperature results in a straight line with a slope (ΔCp) of −102 cal·mol−1·K−1, which is comparable to that for the eIF5B·GDP complex in the presence of Mg2+ at higher temperatures (Table3).
Taken together, these data support the idea that the conserved Asp476 in the G3 motif plays a critical role in the reorganization of switch 2 in response to nucleotide binding and indicate a direct connection between the Mg2+ ion and the temperature dependency of ΔCp in the eIF5B·GDP complex. The fact that GTP binding in contrast to GDP binding shows no temperature dependency of ΔCp indicates that the interactions of domain III to domains I and II in apo eIF5B are broken upon GTP binding regardless of the temperature at which the reaction takes place. However, here the positive contribution to ΔCp is compensated by the large negative contribution due to the burial of ASA in the GTP-bound G domain.
Discussion
The molecular switch between the GDP- and GTP-bound states of eIF5B: the domain release mechanism
The structural and thermodynamic data presented here provide the first detailed picture of the entire nucleotide cycle of eIF5B. The structures of eIF5B in its apo, GTP- and GDP-bound states reveal that its G domain follows the classical molecular switch mechanism, oscillating between an inactive (apo and GDP-bound) and a structurally distinct active GTP-bound form (Fig2). The GTP-induced transition from the inactive to the active conformation of the G domain is characterized by marked rearrangements in the two switch regions, which allow the conserved Thr439 of switch 1 and Gly479 of switch 2 to directly contact the γ-phosphate and Wcat, resulting in the formation of the catalytic GTPase center (Fig2B and D). This observation is in line with previous findings that the mutagenesis of Thr439 or Gly479 to Ala results in severe functional defects in eIF5B, including GTPase deficiency (Shin et al, 2007, 2002) (see also Supplementary Table S1). In switch 2, the initial signal of GTP binding experiences a considerable amplification along its way through the inter-domain cleft toward the back of the protein with a movement of 3 Å at Asp476, 8 Å in Gly479, 12 Å in His480 and finally ∼14 Å in Arg487 and Arg489, which form the primary contact surface for domain III in apo eIF5B (Fig3A and B and Supplementary Fig S4A and B). As a result, domain III is released from the activated G domain, accompanied by the counter-clockwise rotation of domain II with respect to the G domain (Fig3A). As indicated by the ITC data, domain III remains released in free eIF5B·GTP and does not form stable new contacts with the reorganized switch regions. Thus, the signal of GTP binding is amplified from a relatively small conformational change in the nucleotide binding pocket into the release of domain III and thereby ultimately translated into a gain of conformational freedom and higher structural flexibility of domains III and IV relative to domains I and II.
This mechanism for the activation of eIF5B in solution contradicts earlier assumptions that GTP alone is insufficient to induce the conformational switch in free eIF5B in the absence of the ribosome as auxiliary cofactor (Hauryliuk et al, 2008). Moreover, the domain release mechanism is in stark contrast to the previously proposed non-classical articulated lever model for eIF5B/IF2 function, in which the GTP-induced conformational changes in the G domain are limited to a ∼2 Å shift in switch 2. This causes a rigid body movement of domains III and IV and a displacement of the latter by ∼5 Å as ultimate result of eIF5B activation (Roll-Mecak et al, 2000, 2001). In contrast to the release mechanism, this involves neither a conformational change in switch 1 or switch 2 to form the canonical catalytic GTPase center nor does it require the loss or the formation of contacts between G domain and domains II and III at any stage of the activation process (Supplementary Fig S8). The articulated lever model therefore does not explain why switch 1 and switch 2 are universally conserved among eIF5B orthologs and why the mutagenesis of conserved residues in both motifs results in severe functional defects in eIF5B (Lee et al, 2002; Shin & Dever, 2007; Shin et al, 2002).
eIF5B combines the classical GTP operated switch mechanism in the G domain with a novel mechanism of activation for a trGTPase
TrGTPases such as eIF5B and EF-Tu are multidomain proteins which consist of a universally conserved structural core composed of the G domain and domain II that is supplemented with additional functional domains related to the respective role of the GTPase during translation. The activation of trGTPases by GTP is therefore not merely restricted to the G domain but involves a reorganization of the overall domain arrangement, induced by a modulation of the interactions between G domain and the downstream functional domains.
This principle was first established for the elongation factor EF-Tu. Here, the GTP-induced transition from the inactive apo form to the GTP-bound state depends on the rearrangement of the switch regions in the G domain that follows the canonical switch mechanism of Ras-like GTPases (Berchtold et al, 1993). As a consequence, domain II, which is separated from the G domain in inactive EF-Tu, stably associates with the reorganized G domain involving the newly formed surface of the activated switch 2 (Berchtold et al, 1993) (Fig3C). Only in this more compact GTP-conformation, EF-Tu is able to form a stable ternary complex with aminoacyl-tRNA—involving also the reorganized switch 1—for delivery of the latter to the ribosome (Abrahamson et al, 1985; Delaria et al, 1991; Louie & Jurnak, 1985; Nissen et al, 1995; Romero et al, 1985). Thus, the presence of the defined active conformation of switch 1 and 2 functions as the critical signal and thereby is a necessity for the overall activation of EF-Tu (Fig3C).
The same basic principle of activation also applies to eIF5B. The G domain follows the classical molecular switch mechanism (Fig2), and the signal of G domain activation is propagated into domains II–IV through the reorganization of the switch regions. However, the mechanism by which this signal transduction is achieved appears to be different from that in EF-Tu and so far unprecedented in trGTPases: In eIF5B, the GTP-induced absence of the inactive conformation of switch 2 and the resulting release of domain III seems to be the decisive signal that renders eIF5B·GTP activated for productive interactions with the ribosome (Fig3). This does not imply that the GTP-bound conformation of the G domain is irrelevant for eIF5B function but is most likely required for tight interactions of the G domain with the large ribosomal subunit and GTPase activity (Fig4) as well as to prevent the reassociation of domain III before GTP hydrolysis. Instead, this means that the defined GTP-conformation is not as critical for productive interactions between eIF5B and its effector molecules as it is for EF-Tu.
This scenario is in line with earlier biochemical studies that identified mutations in the G domain and domain III that are able to partially activate eIF5B by destabilizing the interactions between domain III and switch 2 in inactive eIF5B. The mutation of Gly479 in switch 2 to Ala was found to reduce GTP binding and to impair subunit joining and ribosome-dependent GTP hydrolysis (Shin et al, 2007). Our structural analysis shows that this Gly residue undergoes a peptide flip of ∼160° during the transition of switch 2 from its inactive to the active state (Fig4B), a conformational change that is energetically not allowed for any other residue. G479A would thus stabilize the inactive switch 2 preventing the formation of the GTPase center and the release of domain III, ultimately causing the inability of the mutant to promote subunit joining and to hydrolyze GTP. A444V and D740R were identified as two independent intragenic suppressor mutants for G479A that restore nucleotide binding, GTP hydrolysis and subunit joining activities in eIF5B (Shin et al, 2007). Interestingly, Asp740 is located in domain III ∼30 Å apart from the nucleotide binding pocket and forms a direct salt bridge to the conserved Arg489 of the inactive switch 2 which moves ∼15 Å upon GTP binding (Fig3B and Supplementary Fig S7B and C). Consequently, D740R would result in a steric and electrostatic repulsion of the inactive switch 2 and thereby a destabilization of the domain III-switch 2 contact in apo eIF5B. A444V is located at the N-terminus of strand β3 close to Asp476 in the G3 motif and most likely causes the constitutive reduction of the energy barrier that has to be overcome by GDP and GTP to move Asp476 into a GTP-like position thereby facilitating the distortion of the interactions between switch 2 and domain III (Shin et al, 2007) (Supplementary Fig S7C). Particularly interesting is that A444V does not only restore GTP dependency in eIF5B but even allows GDP to activate eIF5B for stable interactions with the ribosome (Shin et al, 2007). This demonstrates that the full GTP-conformation in the G domain is not an absolute requirement for stable interactions between eIF5B and the ribosome. Instead, it seems to be critical that the suppressor mutations overcome the increased energy barrier introduced by G479A by destabilizing the inactive conformation and thereby the contact between switch 2 and domain III either directly in the case of D740R or indirectly in the case of A444V (see also Supplementary Table S1).
Implications of the domain release mechanism for ribosomal subunit joining
eIF5B interacts with the ribosomal subunits and catalyzes subunit joining in a GTP-dependent manner (Acker et al, 2009; Pestova et al, 2000). This indicates that eIF5B undergoes a structural transition from the inactive apo-conformation to a GTP-bound state that allows productive interactions with its ribosomal effector complexes. However, it was shown in kinetic experiments that even in its GTP-bound state, eIF5B is unable to catalyze subunit joining unless the Met-tRNAiMet has been positioned in the P site of the 40S subunit (Acker et al, 2009). This seemingly paradoxical situation is compatible with the domain release mechanism of eIF5B activation. On the one hand, the GTP-induced release of domain III allows domain II to stably interact with the 18S rRNA (without steric hindrance by domain III as discussed below; see also Supplementary Fig S5C). On the other hand, the conformational freedom of domains III and IV relative to each other (Supplementary Fig S2B), relative to domains I and II, as well as to the 40S subunit, prevents eIF5B from self-supporting a conformation that allows efficient subunit docking on the 40S·eIF1A complex without Met-tRNAiMet (Fig6). Consequently, for the domain release model, a distinction has to be drawn between the activated state of eIF5B·GTP in solution and its subunit-joining-competent conformation on the 48S pre-IC, which is only one of the many possible conformations accessible to the free eIF5B·GTP, and which requires the reduction of conformational freedom of domains III and IV and their stabilization in the correct orientation. This proposed dependency of eIF5B·GTP on the ribosomal effector complex resembles the hypothesis of conditional switching for eIF5B (Hauryliuk et al, 2008). However, the critical conceptual difference is that eIF5B does not require the ribosome as cofactor to induce the GTP-dependent conformational switch, but instead depends on the ribosomal effector complex to stabilize the sole conformation of GTP-bound eIF5B (among the many possible) capable to promote the association of the 60S subunit. The recent cryo-EM structures of eIF5B on the 80S ribosome demonstrate that this stability is primarily provided by the methionylated 3′-CCA end of Met-tRNAiMet (Fernandez et al, 2013). In agreement with the domain release mechanism, the cryo-EM structures show that the activation of the G domain by GDPCP is sufficient to induce the release of domain III from switch 2 and domain II, but insufficient to stabilize domains III and IV in a defined conformation, as they remain disordered in the absence of amino-acylated tRNA. Only through the interactions between domain IV and the amino-acylated tRNA, domain III and IV become stabilized in their subunit-joining-competent conformation, in which domain III is released from all its contacts with the G domain and domain II that are also found in the apo state of eIF5B and is reoriented relative to domain I and II by ∼65° (Fig3D and Supplementary Fig S5C) (Fernandez et al, 2013).
Figure 6. Schematic model of the of nucleotide cycle of eIF5B during subunit joining.
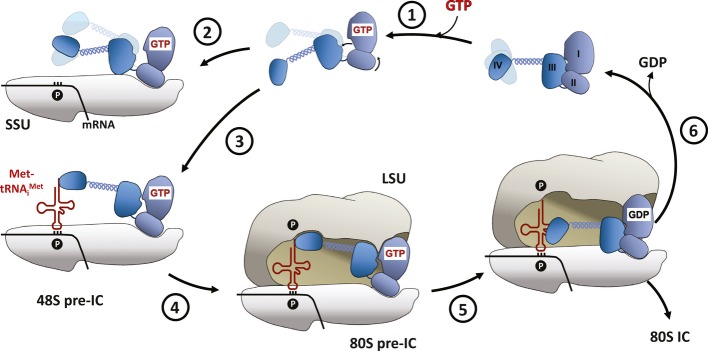
eIF5B with domains I (G) to IV is shown in blue. (1) GTP binding activates eIF5B by release of domain III and rotation of domain II relative to the G domain. (2) Binding of eIF5B·GTP to the small subunit (SSU) in the absence of Met-tRNAiMet results in a non-productive complex in which eIF5B is not able to stimulate subunit joining. (3) In the correctly preassembled 48S pre-IC, the subunit-joining-competent conformation of eIF5B·GTP is stabilized by the P site-bound initiator-tRNA, (4) resulting in the recruitment of the large ribosomal subunit (LSU). (5) Formation of the 80S pre-IC triggers GTP hydrolysis in eIF5B, which reverts back into its inactive conformation, (6) followed by the dissociation of eIF5B·GDP from the elongation-competent 80S ribosome.
Taken together, according to the presented domain release mechanism, the preparation of eIF5B for the catalysis of subunit joining involves a two-step process: i) activation of the G domain by GTP binding, resulting in the release of domain III and an increased intrinsic flexibility that allows productive binding to the effector complexes and ii) stabilization of eIF5B·GTP in the subunit-joining-competent conformation by the 48S pre-IC through a reduction of conformational entropy within the factor (Fig6). According to this scenario, eIF5B requires GTP as well as the correctly assembled 40S·Met-tRNAiMet effector complex for its function. Thus, the domain release mechanism seems evolved to ensure the formation of productive 80S ICs by discriminating against pre-ICs that do not contain Met-tRNAiMet.
Ribosome-induced GTP hydrolysis by eIF5B
Crystal structures of EF-G·GDPCP and EF-Tu·GDPCP on the ribosome suggest a common mechanism of GTPase activation for both elongation factors (Tourigny et al, 2013; Voorhees et al, 2010). According to the current model, the G domain of the translation factor binds to the sarcin-ricin loop (SRL) of the 50S ribosomal subunit and the GTPase activity is triggered as Hiscat (His84 in EF-Tu) rotates inward, where it is stabilized by hydrogen bond interactions with the phosphate of A2662 (SRL) and Wcat which becomes subsequently activated for the in-line attack on the γ-phosphate (Voorhees et al, 2010).
Our own structural investigations show that the catalytic centers in eIF5B·GTP and free EF-Tu·GDPNP exhibit nearly identical positions for residues implicated in ribosome binding and GTP hydrolysis (Fig4). In line with the recent cryo-EM model of ribosome-bound eIF5B (Fernandez et al, 2013), this suggests that domains I and II of eIF5B bind the 80S ribosome in the same way as the bacterial elongation factor (Fig4 and Supplementary Fig S5A and B). As for His84 in free EF-Tu·GDPNP, the imidazole moiety of His480 (Hiscat) in eIF5B points outward and therefore requires rearrangement in order to contact Wcat (Fig4B). In combination with earlier studies that highlight the importance of His480 for GTP hydrolysis (Lee et al, 2002; Shin & Dever, 2007), these observations strongly suggest that the ribosome-dependent GTPase activation of initiation factor eIF5B follows the same basic mechanism as employed for the elongation factors EF-Tu and EF-G.
Dissociation of eIF5B·GDP from the 80S ribosome
In accordance with the function of G proteins as molecular switches, the formation of the elongation-competent 80S ribosome by eIF5B·GTP results in GTP hydrolysis and Pi release, followed by the dissociation of eIF5B·GDP (Lee et al, 2002; Pestova et al, 2000). Thus, the reduced affinity for the ribosome and dissociation of eIF5B depends on its structural transition from the GTP-bound state to the inactive GDP-bound conformation.
Our data show that upon transition from the GTP- to the GDP-bound state, the G domain of eIF5B switches back into its inactive conformation (Fig2 and Supplementary Fig S4A). As a result, domain III is retrieved to interact with switch 2 and domain II, causing the latter to rotate clockwise relative to the G domain (Fig3A and Supplementary Fig S4C). This reorientation relative to each other necessarily disrupts the interactions of the G domain and domain II with the 60S and 40S subunits, respectively, which were formed by eIF5B·GTP in the rotated state (the rotation is also observable in the cryo-EM structure of eIF5B in the 80S IC (Fernandez et al, 2013)). Under the assumption that the interactions between domain II and the 18S rRNA are the last to be broken (as they do not directly depend on the presence of GTP as do those of the G domain with the SRL), domain III moves away from the SRL and domain IV moves away from the peptidyl-transferase center (PTC) toward the 40S subunit and helix H38 (60S) (Supplementary Fig S5C). This position of eIF5B·GDP is not stable as domains III and IV would clash with the ribosomal protein S23e and eIF1A, respectively, and the G domain is rotated into the SRL. As a result, eIF5B would have to retreat from the factor-binding site toward h6 of the 40S subunit as seen in the cryo-EM structure of IF2·GDP on the 70S ribosome (Myasnikov et al, 2005), followed by its dissociation (Fig6).
IF2 might function according to the domain release mechanism
Despite a large body of experimental data for IF2, its precise mode of function during subunit joining in bacteria is still unclear (Antoun et al, 2003; Eiler et al, 2013; Fabbretti et al, 2012; Rodnina et al, 2000; Tomsic et al, 2000). For the reasons discussed below, it is in our opinion reasonable to assume that the domain release mechanism also applies to IF2. Nearly all residues involved in the contacts between switch 2 and domain III in eIF5B, particularly also those in the N-terminus of helix α9, are highly conserved in a/eIF5B/IF2 orthologs (Supplementary Fig S3). In structures of the IF2 G domain, switch 2 is usually flexible with only partial helical character (Eiler et al, 2013; Simonetti et al, 2013; Wienk et al, 2012) and would thus be easily accessible for interactions with domain III. Moreover, from the high degree of structural and sequence homology, it can be inferred that the G domain of IF2, like in eIF5B, follows the classical switch mechanism, resulting in the formation of the canonical active site. The transition of IF2 between the GDP- and GTP-bound states would therefore result in the release of domain III and an increase in the overall flexibility of the factor, and the reduction of flexibility and retraction of domain III upon GTP hydrolysis. Indeed, this becomes apparent in cryo-EM structures of bacterial initiation complexes, where domain III is associated with domains I and II in IF2·GDP, whereas the GTP/GDPNP-bound forms adopt an elongated shape to contact the P site-bound fMet-tRNAfMet (Allen et al, 2005; Julian et al, 2011; Myasnikov et al, 2005; Simonetti et al, 2008).
The assumption that the domain release mechanism applies to IF2 seems to be contradicted by two recent structural studies (Eiler et al, 2013; Simonetti et al, 2013). The only so far available high-resolution structure of GTP-bound IF2 appears to indicate that its G domain does not follow the classical switch mechanism as switch 1 remains virtually unchanged upon GTP binding and switch 2 undergoes only a small local rearrangement without forming a contact with the γ-phosphate (Simonetti et al, 2013). However, it is important to note that this IF2·GTP structure was obtained by soaking GTP into crystals of apo IF2, in which both switch regions are fixed by extensive contacts with symmetry-related molecules. A reorganization of the G domain that would allow the G2 and G3 motifs to contact the GTP molecule in the classical way is thus most likely prevented by crystal contacts and not due to a non-classical behavior of IF2. The recent crystal structure of T. thermophilus IF2(3–467) in the apo and GDP-bound state as well seems to argue against the domain release mechanism for IF2 as domain III has no direct contact with either of the switch regions (Eiler et al, 2013). The authors propose that the increased length of helix α8 (the linker between domains II and III) compared to α8 in aIF5B accounts for the inability of domain III to contact switch 2 (Eiler et al, 2013). However, the lengths of helix α8 and the following flexible linker to domain III are actually compatible with a direct contact between the N-terminal half of helix α9 and switch 2 as observed in apo a/eIF5B (Fig3A and B). Instead, the crystal packing shows that the position occupied by domain III in a/eIF5B is occupied by symmetry-related molecules in the IF2 crystals. Crystallization would therefore be selective for the state in which domain III is released, irrespective of its fraction among the IF2 molecules in solution. As we show by means of ITC and observe in the Sc-eIF5B·GDP structure (Supplementary Fig S1E and S7A), domain III in eIF5B has the ability to dissociate from the G domain even in the absence of GTP. This, we suggest, also applies to IF2.
As for eIF5B, the domain release model can explain why IF2 requires GTP for efficient interactions with ribosomal complexes and the fMet-tRNAfMet in the context of the 30S pre-IC (Antoun et al, 2003, 2004), why IF2·GTP is unable to promote subunit docking in the absence of the initiator-tRNA (Antoun et al, 2006) and finally, why even GDP is able to partially activate IF2 for ribosome binding and subunit joining in in vitro studies despite its inability to stabilize the GTP-conformation of the G domain. With the critical conceptual difference that in the domain release mechanism, the GTP-dependent conformational switch in IF2 precedes and is therefore not a consequence of its interaction with the ribosome, this hypothesis of a common mechanism for eIF5B and IF2 is in agreement with previous proposals of a stepwise activation mechanism for IF2 by GTP and the 30S·fMet-tRNAfMet complex made on the basis of biochemical experiments (Antoun et al, 2003; Pavlov et al, 2011).
Materials and Methods
Protein preparation, crystallization and structure determination
The N-terminally His-tagged versions of eIF5B from C. thermophilum [Ct-eIF5B(517–1116), −(517–970) and −(517–858)] and S. cerevisiae [Sc-eIF5B(399–852)] were expressed in E. coli and purified using standard procedures. Crystals used for structure determination were obtained by sitting-drop vapor diffusion using standard screens either in the presence or absence of GDP or GTP (for details see Table1 and Supplementary Materials and Methods).
X-ray diffraction data were collected using synchrotron radiation. For all structures, the phase problem was solved by molecular replacement using the program PHASER (McCoy et al, 2007). Structures were refined to reasonable R-values and stereochemistry using the program PHENIX (Adams et al, 2010). Data collection and refinement statistics are summarized in Table1. See Supplementary Materials and Methods for details.
Isothermal Titration Calorimetry
The thermodynamic parameters of Ct-eIF5B binding to GDP or GTP were measured using a MicroCal VP-ITC instrument (GE Healthcare). Experiments were carried out essentially as described previously (Mitkevich et al, 2006). The obtained values for enthalpy changes (ΔH) at different temperatures were used to estimate the change in heat capacity (ΔCp) for the various protein–nucleotide complexes. These ΔCp values were then used to estimate the conformational changes occurring in Ct-eIF5B upon GDP or GTP binding, using the empirically determined relation ΔCp = Δcap · ΔASAap + Δcp · ΔASAp (where Δcap and Δcp are the area coefficients in cal·K−1·(mole·Å2)−1 for contributions of apolar or polar side chains to the change in solvent-accessible surface area (ΔASA), respectively) (Perozzo et al, 2004). For more details, see supplementary text.
Coordinates
Coordinates have been deposited in the PDB: Apo Ct-eIF5B(517C) (4N3N); Ct-eIF5B(870C) (4N3G); Ct-eIF5B(517-970)·GDP (4NCL); Ct-eIF5B(517-858)·GTP (4NCN); Apo Sc-eIF5B(399-852) (4N3S); Sc-eIF5B(399-852)·GDP (4NCF).
Acknowledgments
We thank the beam line scientists at BESSY (Berlin) and EMBL/DESY (Hamburg) as well as P. Neumann for support during X-ray diffraction data collection, M. Franke for technical assistance and L. K. Dörfel and A. Dickmanns for critical reading of the manuscript.
Author contributions
BK designed the experiments, prepared and crystallized proteins, collected X-ray data, solved structures, performed ITC experiments and created figures. BK and RF analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Abrahamson JK, Laue TM, Miller DL, Johnson AE. Direct determination of the association constant between elongation factor Tu X GTP and aminoacyl-tRNA using fluorescence. Biochemistry. 1985;24:692–700. doi: 10.1021/bi00324a023. [DOI] [PubMed] [Google Scholar]
- Acker MG, Shin BS, Nanda JS, Saini AK, Dever TE, Lorsch JR. Kinetic analysis of late steps of eukaryotic translation initiation. J Mol Biol. 2009;385:491–506. doi: 10.1016/j.jmb.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GS, Zavialov A, Gursky R, Ehrenberg M, Frank J. The cryo-EM structure of a translation initiation complex from Escherichia coli. Cell. 2005;121:703–712. doi: 10.1016/j.cell.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Antoun A, Pavlov MY, Andersson K, Tenson T, Ehrenberg M. The roles of initiation factor 2 and guanosine triphosphate in initiation of protein synthesis. EMBO J. 2003;22:5593–5601. doi: 10.1093/emboj/cdg525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoun A, Pavlov MY, Lovmar M, Ehrenberg M. How initiation factors tune the rate of initiation of protein synthesis in bacteria. EMBO J. 2006;25:2539–2550. doi: 10.1038/sj.emboj.7601140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoun A, Pavlov MY, Tenson T, Ehrenberg MM. Ribosome formation from subunits studied by stopped-flow and Rayleigh light scattering. Biol Proced Online. 2004;6:35–54. doi: 10.1251/bpo71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne R, Naaktgeboren N, Gubbens J, Voorma HO. Recycling of initiation factors IF-1, IF-2 and IF-3. Eur J Biochem. 1973;32:372–380. doi: 10.1111/j.1432-1033.1973.tb02619.x. [DOI] [PubMed] [Google Scholar]
- Berchtold H, Reshetnikova L, Reiser CO, Schirmer NK, Sprinzl M, Hilgenfeld R. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature. 1993;365:126–132. doi: 10.1038/365126a0. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Delaria K, Guillen M, Louie A, Jurnak F. Stabilization of the Escherichia coli elongation factor Tu-GTP-aminoacyl-tRNA complex. Arch Biochem Biophys. 1991;286:207–211. doi: 10.1016/0003-9861(91)90029-i. [DOI] [PubMed] [Google Scholar]
- Dubnoff JS, Lockwood AH, Maitra U. Studies on the role of guanosine triphosphate in polypeptide chain initiation in Escherichia coli. J Biol Chem. 1972;247:2884–2894. [PubMed] [Google Scholar]
- Eiler D, Lin J, Simonetti A, Klaholz BP, Steitz TA. Initiation factor 2 crystal structure reveals a different domain organization from eukaryotic initiation factor 5B and mechanism among translational GTPases. Proc Natl Acad Sci USA. 2013;110:15662–15667. doi: 10.1073/pnas.1309360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock AH. The stability of salt bridges at high temperatures: implications for hyperthermophilic proteins. J Mol Biol. 1998;284:489–502. doi: 10.1006/jmbi.1998.2159. [DOI] [PubMed] [Google Scholar]
- Fabbretti A, Brandi L, Milon P, Spurio R, Pon CL, Gualerzi CO. Translation initiation without IF2-dependent GTP hydrolysis. Nucleic Acids Res. 2012;40:7946–7955. doi: 10.1093/nar/gks569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez IS, Bai XC, Hussain T, Kelley AC, Lorsch JR, Ramakrishnan V, Scheres SH. Molecular architecture of a eukaryotic translational initiation complex. Science. 2013;342:1240585. doi: 10.1126/science.1240585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualerzi CO, Brandi L, Caserta E, Garofalo C, Lammi M, La Teana A, Petrelli D, Spurio R, Tomsic J, Pon CL. Initiation factors in the early events of mRNA translation in bacteria. Cold Spring Harb Symp Quant Biol. 2001;66:363–376. doi: 10.1101/sqb.2001.66.363. [DOI] [PubMed] [Google Scholar]
- Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]
- Hauryliuk V, Hansson S, Ehrenberg M. Cofactor dependent conformational switching of GTPases. Biophys J. 2008;95:1704–1715. doi: 10.1529/biophysj.107.127290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendsch ZS, Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R, Mesters JR, Hogg T. In: The Ribosome: Structure, Function, Antibiotics, and Cellular Interactions. Garrett RA, Douthwaite SR, Liljas A, Matheson AT, Moore PB, Noller AF, editors. Vol. 28. Washington, DC: American Society for Microbiology Press; 2000. pp. 347–357. [Google Scholar]
- Julian P, Milon P, Agirrezabala X, Lasso G, Gil D, Rodnina MV, Valle M. The Cryo-EM structure of a complete 30S translation initiation complex from Escherichia coli. PLoS Biol. 2011;9:e1001095. doi: 10.1371/journal.pbio.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurberg M, Kristensen O, Martemyanov K, Gudkov AT, Nagaev I, Hughes D, Liljas A. Structure of a mutant EF-G reveals domain III and possibly the fusidic acid binding site. J Mol Biol. 2000;303:593–603. doi: 10.1006/jmbi.2000.4168. [DOI] [PubMed] [Google Scholar]
- Lee JH, Pestova TV, Shin BS, Cao C, Choi SK, Dever TE. Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc Natl Acad Sci USA. 2002;99:16689–16694. doi: 10.1073/pnas.262569399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie A, Jurnak F. Kinetic studies of Escherichia coli elongation factor Tu-guanosine 5'-triphosphate-aminoacyl-tRNA complexes. Biochemistry. 1985;24:6433–6439. doi: 10.1021/bi00344a019. [DOI] [PubMed] [Google Scholar]
- Marintchev A, Wagner G. Translation initiation: structures, mechanisms and evolution. Q Rev Biophys. 2004;37:197–284. doi: 10.1017/S0033583505004026. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitkevich VA, Kononenko AV, Petrushanko IY, Yanvarev DV, Makarov AA, Kisselev LL. Termination of translation in eukaryotes is mediated by the quaternary eRF1*eRF3*GTP*Mg2+ complex. The biological roles of eRF3 and prokaryotic RF3 are profoundly distinct. Nucleic Acids Res. 2006;34:3947–3954. doi: 10.1093/nar/gkl549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myasnikov AG, Marzi S, Simonetti A, Giuliodori AM, Gualerzi CO, Yusupova G, Yusupov M, Klaholz BP. Conformational transition of initiation factor 2 from the GTP- to GDP-bound state visualized on the ribosome. Nat Struct Mol Biol. 2005;12:1145–1149. doi: 10.1038/nsmb1012. [DOI] [PubMed] [Google Scholar]
- Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- Pavlov MY, Zorzet A, Andersson DI, Ehrenberg M. Activation of initiation factor 2 by ligands and mutations for rapid docking of ribosomal subunits. EMBO J. 2011;30:289–301. doi: 10.1038/emboj.2010.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozzo R, Folkers G, Scapozza L. Thermodynamics of protein-ligand interactions: history, presence, and future aspects. J Recept Signal Transduct Res. 2004;24:1–52. doi: 10.1081/rrs-120037896. [DOI] [PubMed] [Google Scholar]
- Pestova TV, Lomakin IB, Lee JH, Choi SK, Dever TE, Hellen CU. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature. 2000;403:332–335. doi: 10.1038/35002118. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Stark H, Savelsbergh A, Wieden HJ, Mohr D, Matassova NB, Peske F, Daviter T, Gualerzi CO, Wintermeyer W. GTPases mechanisms and functions of translation factors on the ribosome. Biol Chem. 2000;381:377–387. doi: 10.1515/BC.2000.050. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A, Cao C, Dever TE, Burley SK. X-Ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Cell. 2000;103:781–792. doi: 10.1016/s0092-8674(00)00181-1. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A, Shin BS, Dever TE, Burley SK. Engaging the ribosome: universal IFs of translation. Trends Biochem Sci. 2001;26:705–709. doi: 10.1016/s0968-0004(01)02024-2. [DOI] [PubMed] [Google Scholar]
- Romero G, Chau V, Biltonen RL. Kinetics and thermodynamics of the interaction of elongation factor Tu with elongation factor Ts, guanine nucleotides, and aminoacyl-tRNA. J Biol Chem. 1985;260:6167–6174. [PubMed] [Google Scholar]
- Shin BS, Acker MG, Maag D, Kim JR, Lorsch JR, Dever TE. Intragenic suppressor mutations restore GTPase and translation functions of a eukaryotic initiation factor 5B switch II mutant. Mol Cell Biol. 2007;27:1677–1685. doi: 10.1128/MCB.01258-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin BS, Dever TE. Molecular genetic structure-function analysis of translation initiation factor eIF5B. Methods Enzymol. 2007;429:185–201. doi: 10.1016/S0076-6879(07)29009-3. [DOI] [PubMed] [Google Scholar]
- Shin BS, Maag D, Roll-Mecak A, Arefin MS, Burley SK, Lorsch JR, Dever TE. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell. 2002;111:1015–1025. doi: 10.1016/s0092-8674(02)01171-6. [DOI] [PubMed] [Google Scholar]
- Simonetti A, Marzi S, Fabbretti A, Hazemann I, Jenner L, Urzhumtsev A, Gualerzi CO, Klaholz BP. Structure of the protein core of translation initiation factor 2 in apo, GTP-bound and GDP-bound forms. Acta Crystallogr D Biol Crystallogr. 2013;69:925–933. doi: 10.1107/S0907444913006422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti A, Marzi S, Myasnikov AG, Fabbretti A, Yusupov M, Gualerzi CO, Klaholz BP. Structure of the 30S translation initiation complex. Nature. 2008;455:416–420. doi: 10.1038/nature07192. [DOI] [PubMed] [Google Scholar]
- Tomsic J, Vitali LA, Daviter T, Savelsbergh A, Spurio R, Striebeck P, Wintermeyer W, Rodnina MV, Gualerzi CO. Late events of translation initiation in bacteria: a kinetic analysis. EMBO J. 2000;19:2127–2136. doi: 10.1093/emboj/19.9.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourigny DS, Fernandez IS, Kelley AC, Ramakrishnan V. Elongation factor G bound to the ribosome in an intermediate state of translocation. Science. 2013;340:1235490. doi: 10.1126/science.1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unbehaun A, Marintchev A, Lomakin IB, Didenko T, Wagner G, Hellen CU, Pestova TV. Position of eukaryotic initiation factor eIF5B on the 80S ribosome mapped by directed hydroxyl radical probing. EMBO J. 2007;26:3109–3123. doi: 10.1038/sj.emboj.7601751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330:835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienk H, Tishchenko E, Belardinelli R, Tomaselli S, Dongre R, Spurio R, Folkers GE, Gualerzi CO, Boelens R. Structural dynamics of bacterial translation initiation factor IF2. J Biol Chem. 2012;287:10922–10932. doi: 10.1074/jbc.M111.333393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.