

Abstract
Background
Excessive levels of methylglyoxal (MG) encountered in diabetes foster enhanced leukocyte-endothelial cell interactions, mechanisms of which are incompletely understood. MG genomically upregulates endothelial serum- and glucocorticoid-inducible kinase 1 (SGK1) which orchestrates leukocyte recruitment by regulating the activation and expression of transcription factors and adhesion molecules. SGK1 regulates a myriad of ion channels and carriers including the Na+/H+ exchanger NHE1. Here, we explored the effect of MG on SGK1-dependent NHE1 activation and the putative role of NHE1 activation in MG-induced leukocyte recruitment and microvascular hyperpermeability.
Methods
Using RT-PCR and immunoblotting, we analyzed NHE1 mRNA and protein levels in murine microvascular SVEC4-10EE2 endothelial cells (EE2 ECs). NHE1 phosphorylation was detected using a specific antibody against the 14-3-3 binding motif at phospho-Ser703. SGK in EE2 ECs was silenced using targeted siRNA. ROS production was determined using DCF-dependent fluorescence. Leukocyte recruitment and microvascular permeability in murine cremasteric microvasculature were measured using intravital microscopy. The expression of endothelial adhesion molecules was determined by immunoblotting and confocal imaging analysis.
Results
MG treatment significantly upregulated NHE1 mRNA and dose-dependently increased total- and phospho-NHE1. Treatment with SGK1 inhibitor GSK650394, antioxidant Tempol and silencing SGK all blunted MG-triggered phospho-NHE1 upregulation in EE2 ECs. NHE1 inhibitor cariporide attenuated MG-triggered ROS production, leukocyte adhesion and emigration and microvascular hyperpermeability, without affecting leukocyte rolling. Cariporide treatment did not alter MG-triggered upregulation of P- and E-selectins, but reduced endothelial ICAM-1 expression.
Conclusion
MG elicits SGK1-dependent activation of endothelial Na+/H+ exchanger NHE1 which participates in MG-induced ROS production, upregulation of endothelial ICAM-1, leukocyte recruitment and microvascular hyperpermeability. Pharmacological inhibition of NHE1 attenuates the proinflammatory effects of excessive MG and may, thus, be beneficial in diabetes-associated inflammation.
Keywords: Methylglyoxal, NHE1, Oxidative stress, SGK1, Leukocyte recruitment
Introduction
Increased interaction of leukocytes with activated vascular endothelium participates in the inflammatory sequelae of diabetes [1-3]. Excessive levels of the glycolysis metabolite methylglyoxal (MG) in vivo, that contribute to increased carbonyl stress, are associated with conditions such as diabetes, renal failure, obesity and metabolic syndrome [4-9]. In diabetes, increased levels of MG are implicated in the pathogenesis of vascular complications such as hypertension [10], impaired microcirculation [11], and thrombosis [12]. Ramifications of pathological MG concentrations include modulation of immune cell functions by stimulation of cytokine induction [13], activation of macrophages [14], and suppression of T-cell functions [15]. MG alters cellular functions by influencing energy and redox balance [16], modulating cytosolic Ca2+ [17] and by triggering apoptotic or necrotic cell death [11,18].
MG was shown to influence innate immunity in diabetes by enhancing neutrophil apoptosis and by eliciting the expression of integrins on neutrophils [12]. Recently, MG was shown to stimulate leukocyte-endothelial cell interactions by reducing intravascular rolling velocity and by potentiating adhesion and transendothelial migration of leukocytes through the upregulated endothelial adhesion molecules [19]. MG was shown to induce eNOS uncoupling [20] which supports redox-sensitive leukocyte recruitment and enhanced microvascular permeability during inflammation [21]. However, putative mechanisms of MG-induced leukocyte recruitment are largely unknown.
MG-induced leukocyte recruitment was recently reported to be mediated by signal transduction downstream of endothelial phosphoinositide 3-kinases (PI3K) [22]. MG was shown to temporally activate glycogen synthase kinase 3 (GSK3) and serum- and glucocorticoid-inducible kinase 1 (SGK1) which, in turn, stimulate endothelial nuclear factor-κB (NF-κB) and cyclic AMP response element-binding protein (CREB), two transcription factors that are important in mediating inflammatory responses [22]. The kinase SGK1, ubiquitously expressed and regulated by a myriad of cell stressors [23], is genomically upregulated in endothelial cells in response to MG treatment [22]. SGK1 regulates a wide array of ion channels and carriers including the Na+/H+ exchangers NHE1 and NHE3 [24-29]. Phosphorylation of NHE1 Ser703 by SGK1 is essential for the binding of 14-3-3 protein to NHE1 [25,30] which, in turn, is critical in the activation of this Na+/H+ exchanger [25,31].
The plasma membrane transport protein NHE1 regulates cellular pH and volume and, thus, participates in a multitude of physiological functions such as proliferation, migration and apoptosis [32]. Endothelial cells express the Na+/H+ exchanger isoforms NHE1 and NHE2 that regulate functions such as endocytosis [33], apoptosis [34] and blood brain barrier [35-38]. NHE1 is constitutively phosphorylated and additional phosphorylation enhances its activity [39]. Several studies have documented that reactive oxygen species (ROS)-mediated signaling activates NHE1 [24,40]. Na+/H+ exchange is pharmacologically inhibitable by NHE1-selective acylguanidine-derived compounds such as cariporide (HOE-642) [39,41,42]. Cariporide blunts the phosphorylation of NHE1 [31] and has been widely used to study putative functions of NHE1 in vitro and in vivo [24,41,43,44]. The influence of MG on NHE1 expression and functions remains elusive.
The present study explores the mechanisms involved in the activation of endothelial NHE1 by MG. Using intravital microscopy, which enables us to directly visualize and determine leukocyte-endothelial interactions with high imaging quality in the microvasculature of anaesthetised mice, we elucidate the effects of pharmacological suppression of NHE1 on leukocyte recruitment and microvascular hyperpermeability elicited by MG.
Materials and methods
Mice and intravital microscopy
Male C57BL/6 mice (Charles River, Saint-Constant, QC, Canada) aged 8–12 wk-old were used in this study with the approval of animal protocols from University Committee on Animal Care and Supply (#20070028) at the University of Saskatchewan. Mice were anaesthetised using an i.p. injection of 10 mg/kg xylazine (Bayer, Toronto, ON, Canada) and 200 mg/kg ketamine hydrochloride (Rogar, Montreal, QC, Canada). The mouse cremaster muscle preparation was used to study dynamic leukocyte-endothelial interactions in microvasculature as described [21,22,45,46]. The cremaster muscle microvasculature is considered to be the goldstandard in vivo model for intravital microscopy where leukocyte-endothelial cell interactions in the postcapillary venules are readily visualized [47]. Leukocyte rolling flux (cells/min), velocity of rolling leukocytes (μm/sec), and the number of adherent (cells/100-μm venule) and emigrated leukocytes (cells/443 × 286 μm2 field) were determined in the cremasteric postcapillary venule (25–40 μm diameter) using video playback analysis [21,22,45,46]. MG-triggered microvascular leakage was determined in postcapillary venules of the cremaster muscle by intravital microscopy measuring fluorescence intensity of FITC-labelled BSA (25 mg/kg i.v.; Sigma) inside and outside the vessel for calculating permeability index as described [21,45]. Where indicated, MG (50 mg/kg; Sigma, Oakville, ON, Canada) and cariporide (20 mg/kg, Sigma) were administered by separate intrascrotal injections or superfusion of exposed cremaster muscle with MG and cariporide at 100 μM and 50 μM, respectively.
Cell culture and gene silencing
Murine microvascular SVEC4-10EE2 endothelial cell line cells (EE2 ECs; ATCC, Manassas, VA) were cultured as described earlier [22]. Previously, MG-sensitive SGK1 signaling was studied in EE2 ECs [22] and in the present study we elucidate the role of NHE1 in MG-induced leukocyte recruitment in the context of SGK1-dependent activation of NHE1. We, therefore, used EE2 ECs to corroborate our in vivo findings. Where indicated, Tempol (300 μM; Sigma), cariporide (50 μM) or GSK650394 (20 μM; Sigma) was added at the specified concentrations. Targeted gene silencing was accomplished by a 48-h transfection of EE2 ECs with siRNA specifically targeting SGK (Santa Cruz) and with siRNA transfection medium and reagent (Santa Cruz) as described previously [48]. The control cells were transfected with negative control scrambled siRNA (Santa Cruz) having no homology to any known RNA sequence.
RT-PCR
RT-PCR was performed to determine NHE1 and β-actin mRNA expression as described previously [22]. Briefly, RNA was isolated from the cells using RNA isolation kit (Qiagen) and reverse-transcribed using reverse transcription kit (Qiagen). RT-PCR was carried out by SYBR green PCR kit (Qiagen) in an iCycler iQ apparatus (Bio-Rad, Hercules, CA) with primers targeting NHE1 (QT00105413; Qiagen) and β-actin (QT00095242; Qiagen). All PCRs were performed in triplicate and ran for 30 cycles at 95°C for 30 sec, 55°C for 30 sec, and 72°C for 40 sec.
Immunoprecipitation and immunoblotting
Phosphorylation of NHE1 was determined in NHE1-immunoprecipitated samples using an anti-phospho-(Ser) 14-3-3 protein binding motif antibody [25]. Briefly, EE2 ECs or excised cremaster muscles were harvested using ice-cold IP lysis buffer containing complete protease and phosphatase inhibitor cocktail (Cell Signaling) and the samples were sonicated on ice three times for 5 seconds each. The protein concentration was determined by BCA assay (Sigma). The samples were then aliquoted into two equal portions of 500 μg proteins each. In the first portion, β-actin (Santa Cruz) was detected as described previously [22]. In the second portion, lysates containing 500 μg proteins were incubated overnight at 4°C with 5 μl mouse monoclonal NHE1 antibody (Abcam). Then, immune complexes were mixed with protein G agarose (20 μl of 50% bead slurry, Thermo Fisher Scientific) for 3 h at 4°C and then washed five times with ice-cold IP lysis buffer. The immune complexes were dissociated by adding 20 μl 3× SDS sample buffer and heating for 5 min at 95°C, and then centrifuged for 1 min at 14,000× g for removing the agarose beads. The resulting supernatant was separated into two equal portions. One portion of the protein lysate was separated on 10% SDS-PAGE gels and electrotransferred to a nitrocellulose membrane. The membrane was incubated overnight at 4°C with rabbit polyclonal phospho-(Ser) 14-3-3 binding-motif protein antibody (1:1000, Cell Signaling), followed by 2-h incubation with HRP-conjugated goat anti-rabbit secondary antibody (1:1000, Santa Cruz) at room temperature. In the other portion, total NHE1 (1:1000, Abcam) was determined respectively using routine immunoblotting [45]. Where indicated, expression of adhesion molecules was detected in cremaster muscle homogenates as described previously [19,22] using primary antibodies against ICAM-1 (Abcam), P-selectin (LifeSpan Biosciences) and E-selectin (Abcam). Intensity values for the proteins were normalized to β-actin and densitometric quantification of the detected bands was performed using Quantity One® Software (Bio-Rad).
Determination of ROS production
The levels of ROS production in EE2 ECs were measured as previously described [21]. Following the indicated treatments, EE2 ECs were loaded (30 min, 37°C in the dark) with a membrane-permeable and non-fluorescent 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA; 30 μM, Invitrogen) which was oxidized to fluorescent 2’,7’-dichlorofluorescein (DCF) in the presence of ROS. To remove excess probe, the cells were washed with PBS and fluorescence intensity in EE2 ECs was excited at 495 nm and measured at 527 nm emission wavelengths. Data are normalized to the control.
Confocal imaging
EE2 ECs treated under different conditions were fixed using 4% paraformaldehyde for 30 min, washed twice with PBS, blocked for 1 h with 5% goat serum (Abcam) and then incubated overnight at 4°C with antibodies against ICAM-1 (1:500; eBioscience). After washing, the slides were incubated for 1 h with fluorescence goat anti-rat antibody (Alexa Fluor 488; Invitrogen) and the cells were then washed, permeabilized with 0.1% Triton X-100 and stained with Hoechst 33342 (Invitrogen). The slides were mounted in ProLong® Gold Antifade Reagent (Invitrogen) and observed under a laser scanning confocal microscope (Zeiss, LSM 700). As the antibodies recognize extracellular epitopes of target proteins, the labelled proteins are considered to have surface localization.
Statistical analysis
Data are shown as arithmetic mean ± SEM. Statistical analysis was made using ANOVA with Tukey’s post-hoc comparison test. n denotes the number of different mice, different batches of cremaster muscle or endothelial cells studied in each group. Values of p < 0.05 were considered statistically significant.
Results
Firstly, we tested whether MG treatment influences endothelial NHE1 protein levels. We found that treatment of EE2 ECs with MG significantly enhanced NHE1 and phospho-NHE1 in a dose-dependent manner, an effect reaching statistical significance at 100 μM MG (NHE1) and 50 μM (phospho-NHE1) respectively (Figure 1A). Next, we determined whether MG influences NHE1 transcription in EE2 ECs. As shown in Figure 1B, treatment of EE2 ECs with MG for 0–8 h significantly enhanced NHE1 mRNA levels, an effect reaching statistical significance 2 h after MG treatment. To corroborate our in vitro observations, we demonstrate that an intrascrotal injection of MG for 4 h significantly upregulated total- and phospho-NHE1 levels in cremaster muscle (Figure 1C).
Figure 1.
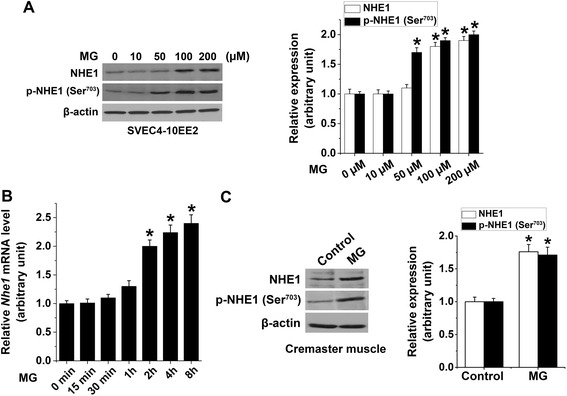
Methylglyoxal-induced upregulation of NHE1 expression. (A) Representative original Western blot and means ± SEM (n = 4) showing total NHE1 and phospho-NHE1 (14-3-3 binding motif at p-Ser703) determined in MG-treated (0–200 μM for 4 h) EE2 ECs (relative to β-actin). * indicates significant difference (p < 0.05) from 0 μM MG. (B) Mean ± SEM of NHE1 mRNA levels (n = 4) determined in MG-treated (100 μM for 0–8 h) EE2 ECs (relative to β-actin). * indicates significant difference (p < 0.05) from 0 h. (C) Representative original Western blot and means ± SEM (n = 4) showing total NHE1 and phospho-NHE1 (14-3-3 binding motif at p-Ser703; relative to β-actin) determined in excised cremaster muscle 4 h after an intrascrotal injection with saline (Control) or MG (50 mg/kg). * indicates significant difference (p < 0.05) from the absence of MG (Control).
We then sought to elucidate the participation of SGK1 in the activation of endothelial NHE1 elicited by MG treatment. As shown in Figure 2A and B, MG treatment (100 μM) of EE2 ECs significantly upregulated total NHE1 and phospho-NHE1 as compared to the control. Treatment with the SGK1 inhibitor GSK650394 significantly attenuated MG-induced upregulation of phospho-NHE, but not total NHE1 (Figure 2A and B). To confirm the role of SGK1 in NHE1 activation, an additional series of experiments were performed using SGK-targeted siRNA silencing. As illustrated in Figure 2C and D, MG treatment significantly enhanced NHE1 protein expression in EE2 ECs at 4–8 h after MG treatment, an effect that was not significantly altered by SGK silencing. Furthermore, phospho-NHE1 levels in EE2 ECs were significantly enhanced at 1–8 h after MG treatment, an effect that was abolished by SGK silencing (Figure 2C and E). These data suggest that endothelial SGK1 activates NHE1 in response to MG treatment.
Figure 2.
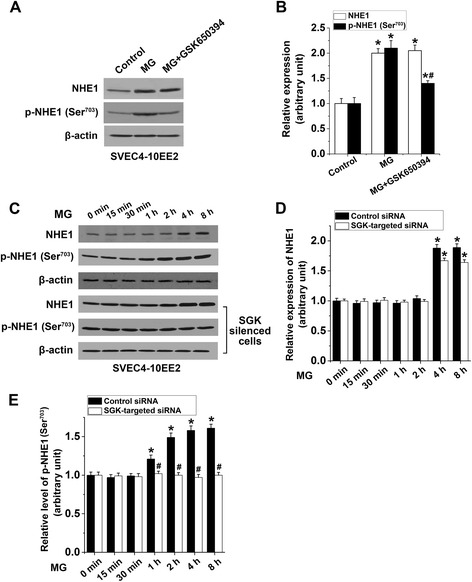
Methylglyoxal-triggered endothelial SGK-dependent NHE1 activation. A − B. Representative original Western blot (A) and means ± SEM (B; n = 4) showing total NHE1 (white bars) and phospho-NHE1 (14-3-3 binding motif at p-Ser703; black bars; relative to β-actin) determined in the absence (Control) or in the presence of MG-treatment (100 μM for 4 h) in EE2 ECs without (MG) or with the SGK1 inhibitor GSK650394 (20 μM, 1 h prior to MG). * indicates significant difference (p < 0.05) from the Control (0 μM MG). # indicates significant difference (p < 0.05) from MG alone. C − E. Representative original Western blot (C) and means ± SEM (n = 4) showing total NHE1 (D) and phospho-NHE1 (14-3-3 binding motif at p-Ser703; relative to β-actin; E) determined in MG-treated (100 μM for 0–8 h) EE2 ECs with scrambled siRNA (Control siRNA; black bars) or with SGK-targeted siRNA silencing (white bars). * indicates significant difference (p < 0.05) from 0 h. # indicates significant difference (p < 0.05) from scrambled control siRNA.
To address the role of oxidative stress in MG-induced NHE1 activation, we analyzed ROS generation. Figure 3A shows that MG treatment significantly enhanced DCF-dependent fluorescence in a time-dependent manner, an effect that was significantly curtailed by treatment with cariporide or the antioxidant Tempol. These data indicate that NHE1 activation is required for MG-triggered ROS production. An additional series of experiments explored whether MG-induced ROS production [20] is essential as an upstream activator of endothelial NHE1. As shown in Figure 3B, total NHE1 expression in EE2 ECs was significantly upregulated after treatment with MG, an effect that was significantly blunted by the antioxidant Tempol but not by cariporide. In contrast, MG-triggered enhanced phospho-NHE1 expression was significantly reduced by both Tempol and cariporide treatment suggesting that MG-induced ROS production is crucial for NHE1 activation and both oxidative burst and NHE1 activation depend on each other in response to MG treatment (Figure 3B).
Figure 3.
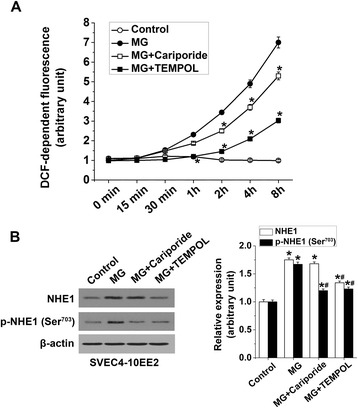
Contribution of oxidative stress in methylglyoxal-triggered endothelial NHE1 activation. (A) Means ± SEM (n = 6) of DCF-dependent fluorescence in EE2 ECs treated with MG (100 μM for 0–8 h) alone (MG) or co-treated with MG and cariporide (50 μM; 1 h prior to MG) or Tempol (300 μM; 1 h prior to MG). * indicates significant difference (p < 0.05) from MG alone. (B) Representative original Western blot and means ± SEM (n = 4) showing total NHE1 and phospho-NHE1 (14-3-3 binding motif at p-Ser703; relative to β-actin) determined in EE2 ECs treated with MG (100 μM for 1 h) alone or co-treated with MG and cariporide (50 μM; 1 h prior to MG) or Tempol (300 μM; 1 h prior to MG). * indicates significant difference (p < 0.05) from the absence of MG (Control). # indicates significant difference (p < 0.05) from MG alone.
Next, we explored whether SGK1-dependent NHE1 activation participates in MG-induced leukocyte recruitment. Intravital microscopy of murine cremasteric microvasculature revealed increased adhesion to endothelium and emigration of leukocytes following MG treatment, an effect that was thwarted by cariporide treatment (Figure 4). Analysis of leukocyte recruitment 4.0 − 5.5 h after an intrascrotal injection of MG showed decreased leukocyte rolling velocity and increased leukocyte rolling flux, adhesion and emigration as compared to saline control (Figure 4B-E). Pretreatment with cariporide, however, significantly blunted MG-induced increased leukocyte adhesion and emigration but did not modify leukocyte rolling flux and velocity (Figure 4B-E) indicating that NHE1 activation participates in leukocyte recruitment in response to excessive MG. Cariporide treatment reduced leukocyte adhesion by ~25% and emigration by ~34% at 5.5 h after MG treatment.
Figure 4.
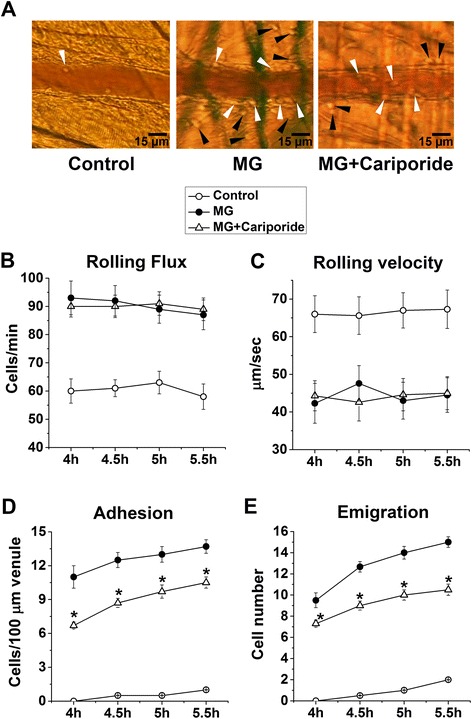
Effect of cariporide on methylglyoxal-induced leukocyte recruitment . (A) Representative intravital microscopy images showing adherent (white arrowheads) and emigrated (black arrowheads) leukocytes. B − E. Means ± SEM (n = 6) of leukocyte rolling flux (cells/min; B), rolling velocity (μm/sec; C), adhesion (cells/100-μm venule; D) and emigration (cells/443 × 286 μm2 field; E) determined 4.0 − 5.5 h after an intrascrotal injection of saline (Control) or MG (50 mg/kg) alone or co-treated with MG and cariporide (20 mg/kg, 1 h prior to MG). * indicates significant difference (p < 0.05) from MG alone.
Impaired microvascular barrier function during leukocyte transendothelial migration fosters increased microvascular permeability. We, therefore, measured fluorescence changes of FITC-conjugated albumin inside and outside cremasteric postcapillary venules to examine the effect of cariporide treatment on MG-elicited microvascular leakage. As depicted in Figure 5, permeability index analysis revealed that an intrascrotal injection of MG increased microvascular permeability, an effect that was significantly inhibited by treatment with cariporide. These data indicate that NHE1 activation participates in MG-triggered increases in microvascular permeability.
Figure 5.
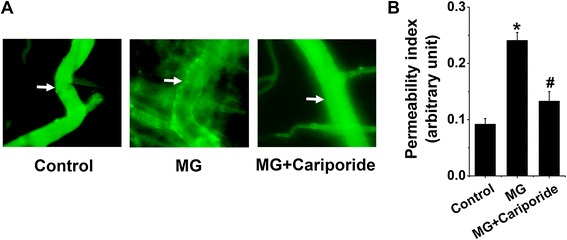
Effect of cariporide on methylglyoxal-induced microvascular leakage. (A) Representative fluorescence intravital microscopy images (arrows point to the segment of postcapillary venule where permeability index was determined) and (B) means ± SEM (n = 4) of permeability index analysis of mouse cremasteric postcapillary venules showing the leakage of FITC-conjugated BSA post 1-h i.v. injection and after an intrascrotal injection for 4 h with saline (Control), MG (50 mg/kg) alone or co-treatment with MG and cariporide (20 mg/kg, 1 h prior to MG). * indicates significant difference (p < 0.05) from the absence of MG (Control). # indicates significant difference (p < 0.05) from MG alone.
To identify the mechanisms that regulate sensitivity of MG-induced leukocyte recruitment to cariporide, we analyzed the expression of endothelial adhesion molecules. Murine cremaster muscles treated with MG had enhanced expression of P- and E-selectins and ICAM-1. Cariporide treatment attenuated MG-induced ICAM-1 expression, but not P- and E-selectin expression in vivo (Figure 6A). To confirm these observations, we used confocal microscopy to visualize the surface expression of ICAM-1 on murine EE2 ECs. As shown in Figure 6B, MG treatment enhanced the expression of ICAM-1 on EE2 ECs, an effect that was blunted by cariporide treatment. These results confirm that inhibition of NHE1 in endothelial cells blunted MG-induced leukocyte adhesion and transmigration through the suppression of ICAM-1 upregulation indicating that MG-triggered activation of NHE1 regulates ICAM-1 upregulation and ICAM-1-mediated endothelial functions.
Figure 6.
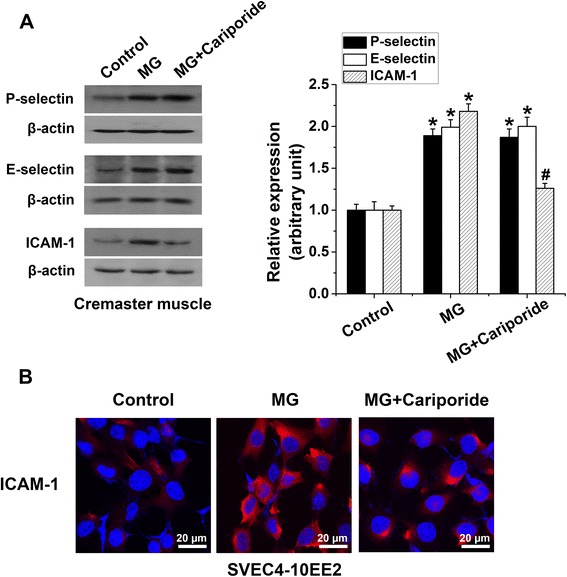
Effect of cariporide on methylglyoxal-induced upregulation of endothelial adhesion molecule expression. (A) Representative original Western blot and means ± SEM (n = 4) showing the expression of P-selectin, E-selectin and ICAM-1 (relative to β-actin) determined in whole cremaster muscle 4 h after an intrascrotal injection of saline (Control), MG (50 mg/kg) alone or co-treated with MG and cariporide (20 mg/kg, 1 h prior to MG). * indicates significant difference (p < 0.05) from absence of MG (Control). # indicates significant difference (p < 0.05) from MG alone. (B) Representative confocal micrographs of cell surface ICAM-1 immunostaining (Red) in EE2 ECs in the absence (Control) or in the presence of MG (100 μM, 4 h) alone or co-treatment with MG and cariporide (50 μM, 1 h prior to MG). Nuclear staining is shown in blue (Hoechst 33342).
Discussion
The present study reveals the role of SGK1-dependent activation of endothelial NHE1 in regulating leukocyte recruitment through the upregulation of ICAM-1 in response to MG. We observed that MG treatment potentiated dose- and time-dependent endothelial NHE1 mRNA and protein levels and triggered SGK1- and ROS-dependent NHE1 phosphorylation. By using intravital microscopy of cremasteric microvasculature in a previously described murine model of MG-triggered acute inflammation [19,21,22], we demonstrate that pharmacological inhibition of NHE1 using cariporide ameliorated leukocyte adhesion, transendothelial migration and microvascular leakage elicited by MG. Although, the decrease may not appear dramatic in terms of absolute recruited cell numbers presented in the data, it turns out to be a significant and stable difference in terms of inhibition of leukocyte recruitment in vivo which was determined in a small segment of the postcapillary venule. The physiological relevance of MG concentrations used in this study is corroborated by previous reports [49,50]. Intrascrotal injection of MG (50 mg/kg) was shown to yield MG concentrations of 1.7 μM and 3.9 nmol/mg protein in plasma and tissue respectively [19]. MG concentrations detected in cremaster muscle are similar to those detected in other organs [51,52]. Moreover, the concentration of MG (100 μM) used in vitro in the present study is based on previous reports [53-55].
Here, we show that MG treatment increased the levels of both total and phosphorylated endothelial NHE1. Mounting evidence suggests that NHE1 participates in the pathogenesis of various complications of diabetes. Recently, high glucose concentrations were demonstrated to stimulate p38 MAPK- and ERK1/2-dependent NHE1 activation in distal nephron cells [56]. Strikingly, hyperglycemia was shown to induce endothelial dysfunction via Ca2+-dependent calpain signaling mediated by the Na+/H+ exchanger [57]. In diabetic rats, vascular hypertrophy was accompanied by elevated NHE1 activity, an effect thwarted by cariporide administration [58]. Cariporide treatment was also found to ameliorate retinal microangiopathy in diabetic rats [59] and to curtail hyperglycemia-induced hypertrophy of rat cardiomyocytes [60]. Chronic cariporide administration was further shown to attenuate left ventricular hypertrophy in diabetic rats [61]. Intriguingly, cariporide treatment was shown to counteract the formation of methylglyoxal-derived advanced glycation end products (AGEs) in type 1 diabetic rats [62]. AGEs, in turn, are known to amplify inflammatory responses [63]. It is, therefore, tempting to speculate that enhanced MG levels may contribute to or even account for NHE1-mediated cardiovascular dysfunctions encountered in diabetic complications.
Mechanistically, PI3K signaling is decisive in NHE1-mediated cellular functions [64,65]. SGK1 is activated through PI3K and phosphoinositide-dependent kinase PDK1 [23,66]. The PI3K downstream kinase AKT was previously shown to phosphorylate and inhibit NHE1 [67]. Along these lines, SGK1 may play a decisive role in PI3K-dependent NHE1 regulation. It was reported that MG treatment did not alter SGK1 expression in neutrophils [22]. Accordingly, it is unlikely that cariporide treatment inhibits MG-induced leukocyte recruitment in vivo by mitigating neutrophil NHE1 activity. MG-induced leukocyte recruitment may, therefore, be effectively accomplished by activation of SGK1-dependent NHE1 activation in endothelial cells.
Redox imbalance is an essential pathological element of the diabetic milieu [68]. MG is known to stimulate ROS and superoxide production [16,20-22]. Furthermore, MG modulates the expression and functions of antioxidant cytoprotective molecules such as superoxide dismutase and H2S [69,70]. Compelling evidence links ROS-dependent signaling to NHE1 activation. Inhibition of NHE1 by cariporide was shown to blunt ROS formation in dendritic cells [40]. Conversely, antioxidant treatment is known to attenuate NHE activity [71]. Interestingly, ROS was previously shown to enhance NHE1 gene expression [72]. Thus, it is apparent from our data that ROS production and NHE1 activation upon stimulation with MG are dependent on each other.
Previous studies have shown a crucial role of NHE-dependent functions in the brain microvascular endothelium [36-38]. These studies, however, preclude us to draw mechanistic parallels of NHE-dependent functions in brain microvascular endothelial cells to peripheral endothelial cells due to their distinct features. Upregulation of adhesion molecules in diabetes is fostered by hyperglycemia [73-75] and increased levels of MG [19]. We observed that cariporide treatment did not alter selectin-dependent leukocyte rolling during MG-induced leukocyte recruitment in vivo but mitigated endothelial ICAM-1-dependent leukocyte adhesion. Surprisingly, thrombin-induced P-selectin upregulation and leukocyte rolling were reported to be inhibited by cariporide [76]. Cariporide treatment was shown to mitigate increased ICAM-1 mRNA levels in microvascular endothelial cells following ischemia-reperfusion [77] and to foster increased shedding of L-selectin on leukocytes [78]. Remarkably, cariporide treatment was found to decrease monocyte adhesion to endothelial cell, an effect that was attributed to decreased ICAM-1 expression [79]. Pharmacological blockade of NHE was previously reported to suppress NF-κB activation [80], an effect that contributes to reduced ICAM-1 expression [81]. Cariporide may, in addition, counteract inflammation by abating cytokine production [82].
MG was previously shown to trigger increased microvascular permeability [21], an effect mediated by oxidative stress [21,83]. Our data reveal that NHE1 inhibition attenuated MG-triggered microvascular hyperpermeability. However, whether NHE1 activation directly regulates microvascular barrier function is not known. It is possible that reduction in MG-triggered microvascular hyperpermeability after cariporide treatment results from the suppression of NHE1 activation which regulates both oxidative stress and transendothelial migration of leukocytes, key elements that are vital to the microvascular permeability regulation [83,84]. By the same token, leukocyte recruitment is also triggered by oxidative stress [21,85] and our data suggests that inhibition of MG-induced leukocyte recruitment by cariporide involves a redox-sensitive mechanism.
Conclusions
In conclusion, MG elicits SGK1-dependent activation of endothelial Na+/H+ exchanger NHE1 which participates in MG-induced ROS production, upregulation of endothelial ICAM-1, leukocyte recruitment and microvascular hyperpermeability (Figure 7). Pharmacological inhibition of NHE1 attenuates the proinflammatory effects of excessive MG and may, thus, be beneficial in diabetes-associated inflammation.
Figure 7.
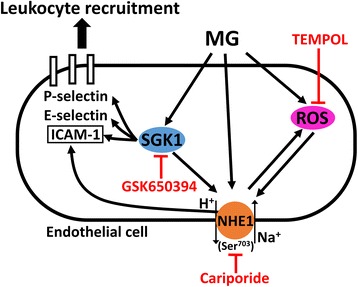
Scheme of the role of Na + /H + exchanger NHE1 in methylglyoxal-induced leukocyte recruitment.
Acknowledgments
This work is supported in part by research grants to L. Liu from Canadian Institutes of Health Research (CIHR, MOP-86749) and Natural Sciences and Engineering Research Council of Canada (NSERC, #386732-2010). Y. Su is supported by a scholarship from China Scholarship Council. L. Liu is a recipient of CIHR New Investigator Award. S.M. Qadri is supported by a fellowship from the Saskatchewan Health Research Foundation (SHRF).
Abbreviations
- MG
Methylglyoxal
- Tempol
1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine
- ROS
Reactive oxygen species
- PI3K
Phosphoinositide 3-kinases
- SGK1
Serum- and glucocorticoid-inducible kinase 1
- EE2 ECs
SVEC4-10EE2 endothelial cells
- CREB
Cyclic AMP response element-binding protein
- NF-κB
Nuclear factor-κB
- NHE1
Na+/H+ exchanger 1
- p38 MAPK
p38 mitogen-activated protein kinases
- ERK1/2
Extracellular-signal-regulated kinases 1/2
- ICAM-1
Intercellular adhesion molecule-1
- AGEs
Advanced glycation end products
Footnotes
Syed M Qadri and Yang Su contributed equally.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
Participated in research design: SMQ and LL; conducted experiments: YS and FSC; all authors performed data analysis and interpretation; contributed to the writing of the manuscript and critically revised the manuscript: SMQ and LL; all authors read and approved the final manuscript.
Authors’ information
Syed M Qadri and Yang Su share the first authorship.
Contributor Information
Syed M Qadri, Email: syed.qadri@usask.ca.
Yang Su, Email: yas467@mail.usask.ca.
Francisco S Cayabyab, Email: frank.cayabyab@usask.ca.
Lixin Liu, Email: lixin.liu@usask.ca.
References
- 1.Fortes ZB, Farsky SP, Oliveira MA, Garcia-Leme J. Direct vital microscopic study of defective leukocyte-endothelial interaction in diabetes mellitus. Diabetes. 1991;40:1267–1273. doi: 10.2337/diab.40.10.1267. [DOI] [PubMed] [Google Scholar]
- 2.Pettersson US, Christoffersson G, Massena S, Ahl D, Jansson L, Henriksnas J, Phillipson M. Increased recruitment but impaired function of leukocytes during inflammation in mouse models of type 1 and type 2 diabetes. PLoS One. 2011;6:e22480. doi: 10.1371/journal.pone.0022480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haidari M, Zhang W, Willerson JT, Dixon RA. Disruption of endothelial adherens junctions by high glucose is mediated by protein kinase C-β-dependent vascular endothelial cadherin tyrosine phosphorylation. Cardiovasc Diabetol. 2014;13:112. doi: 10.1186/1475-2840-13-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karg E, Papp F, Tassi N, Janaky T, Wittmann G, Turi S. Enhanced methylglyoxal formation in the erythrocytes of hemodialyzed patients. Metabolism. 2009;58:976–982. doi: 10.1016/j.metabol.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 5.Lapolla A, Flamini R, Lupo A, Arico NC, Rugiu C, Reitano R, Tubaro M, Ragazzi E, Seraglia R, Traldi P. Evaluation of glyoxal and methylglyoxal levels in uremic patients under peritoneal dialysis. Ann N Y Acad Sci. 2005;1043:217–224. doi: 10.1196/annals.1333.027. [DOI] [PubMed] [Google Scholar]
- 6.Miyata T, Van Ypersele de SC, Kurokawa K, Baynes JW. Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int. 1999;55:389–399. doi: 10.1046/j.1523-1755.1999.00302.x. [DOI] [PubMed] [Google Scholar]
- 7.McLellan AC, Thornalley PJ, Benn J, Sonksen PH. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin Sci (Lond) 1994;87:21–29. doi: 10.1042/cs0870021. [DOI] [PubMed] [Google Scholar]
- 8.Atkins TW, Thornally PJ. Erythrocyte glyoxalase activity in genetically obese (ob/ob) and streptozotocin diabetic mice. Diabetes Res. 1989;11:125–129. [PubMed] [Google Scholar]
- 9.Liu J, Wang R, Desai K, Wu L. Upregulation of aldolase B and overproduction of methylglyoxal in vascular tissues from rats with metabolic syndrome. Cardiovasc Res. 2011;92:494–503. doi: 10.1093/cvr/cvr239. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Mori T, Guo Q, Hu C, Ohsaki Y, Yoneki Y, Zhu W, Jiang Y, Endo S, Nakayama K, Ogawa S, Nakayama M, Miyata T, Ito S. Carbonyl stress induces hypertension and cardio-renal vascular injury in Dahl salt-sensitive rats. Hypertens Res. 2013;36:361–367. doi: 10.1038/hr.2012.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicolay JP, Schneider J, Niemoeller OM, Artunc F, Portero-Otin M, Haik G, Jr, Thornalley PJ, Schleicher E, Wieder T, Lang F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell Physiol Biochem. 2006;18:223–232. doi: 10.1159/000097669. [DOI] [PubMed] [Google Scholar]
- 12.Gawlowski T, Stratmann B, Stirban AO, Negrean M, Tschoepe D. AGEs and methylglyoxal induce apoptosis and expression of Mac-1 on neutrophils resulting in platelet-neutrophil aggregation. Thromb Res. 2007;121:117–126. doi: 10.1016/j.thromres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Kuntz S, Kunz C, Rudloff S. Carbonyl compounds methylglyoxal and glyoxal affect interleukin-8 secretion in intestinal cells by superoxide anion generation and activation of MAPK p38. Mol Nutr Food Res. 2010;54:1458–1467. doi: 10.1002/mnfr.200900408. [DOI] [PubMed] [Google Scholar]
- 14.Pal A, Bhattacharya I, Bhattacharya K, Mandal C, Ray M. Methylglyoxal induced activation of murine peritoneal macrophages and surface markers of T lymphocytes in sarcoma-180 bearing mice: involvement of MAP kinase, NF-kappa beta signal transduction pathway. Mol Immunol. 2009;46:2039–2044. doi: 10.1016/j.molimm.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Price CL, Hassi HO, English NR, Blakemore AI, Stagg AJ, Knight SC. Methylglyoxal modulates immune responses: relevance to diabetes. J Cell Mol Med. 2010;14:1806–1815. doi: 10.1111/j.1582-4934.2009.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Liu J, Wu L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem Pharmacol. 2009;77:1709–1716. doi: 10.1016/j.bcp.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 17.Jan CR, Chen CH, Wang SC, Kuo SY. Effect of methylglyoxal on intracellular calcium levels and viability in renal tubular cells. Cell Signal. 2005;17:847–855. doi: 10.1016/j.cellsig.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Chan WH, Wu HJ. Methylglyoxal and high glucose co-treatment induces apoptosis or necrosis in human umbilical vein endothelial cells. J Cell Biochem. 2008;103:1144–1157. doi: 10.1002/jcb.21489. [DOI] [PubMed] [Google Scholar]
- 19.Su Y, Lei X, Wu L, Liu L. The role of endothelial cell adhesion molecules P-selectin, E-selectin and intercellular adhesion molecule-1 in leucocyte recruitment induced by exogenous methylglyoxal. Immunology. 2012;137:65–79. doi: 10.1111/j.1365-2567.2012.03608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su Y, Qadri SM, Wu L, Liu L. Methylglyoxal modulates endothelial nitric oxide synthase-associated functions in EA.hy926 endothelial cells. Cardiovasc Diabetol. 2013;12:134. doi: 10.1186/1475-2840-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Y, Qadri SM, Hossain M, Wu L, Liu L. Uncoupling of eNOS contributes to redox-sensitive leukocyte recruitment and microvascular leakage elicited by methylglyoxal. Biochem Pharmacol. 2013;86:1762–1774. doi: 10.1016/j.bcp.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 22.Su Y, Qadri SM, Cayabyab FS, Wu L, Liu L. Regulation of methylglyoxal-elicited leukocyte recruitment by endothelial SGK1/GSK3 signalling. Biochim Biophys Acta. 1843;2014:2481–2491. doi: 10.1016/j.bbamcr.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Lang F, Shumilina E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J. 2013;27:3–12. doi: 10.1096/fj.12-218230. [DOI] [PubMed] [Google Scholar]
- 24.Rotte A, Pasham V, Eichenmuller M, Yang W, Bhandaru M, Lang F. Influence of dexamethasone on Na+/H+ exchanger activity in dendritic cells. Cell Physiol Biochem. 2011;28:305–314. doi: 10.1159/000331746. [DOI] [PubMed] [Google Scholar]
- 25.Voelkl J, Pasham V, Ahmed MS, Walker B, Szteyn K, Kuhl D, Metzler B, Alesutan I, Lang F. Sgk1-dependent stimulation of cardiac Na+/H+ exchanger Nhe1 by dexamethasone. Cell Physiol Biochem. 2013;32:25–38. doi: 10.1159/000350120. [DOI] [PubMed] [Google Scholar]
- 26.Wang D, Sun H, Lang F, Yun CC. Activation of NHE3 by dexamethasone requires phosphorylation of NHE3 at Ser663 by SGK1. Am J Physiol Cell Physiol. 2005;289:C802–C810. doi: 10.1152/ajpcell.00597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasham V, Rotte A, Gu S, Yang W, Bhandaru M, Rexhepaj R, Pathare G, Lang F. Upregulation of intestinal NHE3 following saline ingestion. Kidney Blood Press Res. 2013;37:48–57. doi: 10.1159/000343401. [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee S, Schmidt S, Pouli S, Honisch S, Alkahtani S, Stournaras C, Lang F. Membrane androgen receptor sensitive Na+/H+ exchanger activity in prostate cancer cells. FEBS Lett. 2014;588:1571–1579. doi: 10.1016/j.febslet.2014.02.040. [DOI] [PubMed] [Google Scholar]
- 29.Voelkl J, Lin Y, Alesutan I, Ahmed MS, Pasham V, Mia S, Gu S, Feger M, Saxena A, Metzler B, Kuhl D, Pichler BJ, Lang F. Sgk1 sensitivity of Na+/H+ exchanger activity and cardiac remodeling following pressure overload. Basic Res Cardiol. 2012;107:236. doi: 10.1007/s00395-011-0236-2. [DOI] [PubMed] [Google Scholar]
- 30.Lehoux S, Abe J, Florian JA, Berk BC. 14-3-3 Binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. J Biol Chem. 2001;276:15794–15800. doi: 10.1074/jbc.M100410200. [DOI] [PubMed] [Google Scholar]
- 31.Garciarena CD, Fantinelli JC, Caldiz CI, de CG C, Ennis IL, Perez NG, Cingolani HE, Mosca SM. Myocardial reperfusion injury: reactive oxygen species vs. NHE-1 reactivation. Cell Physiol Biochem. 2011;27:13–22. doi: 10.1159/000325201. [DOI] [PubMed] [Google Scholar]
- 32.Wakabayashi S, Hisamitsu T, Nakamura TY. Regulation of the cardiac Na+/H+ exchanger in health and disease. J Mol Cell Cardiol. 2013;61:68–76. doi: 10.1016/j.yjmcc.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S. Intercellular adhesion molecule 1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler Thromb Vasc Biol. 2012;32:1178–1185. doi: 10.1161/ATVBAHA.111.244186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Cui G, Zhang N, Liu Z, Sun W, Peng Q. Lipopolysaccharide induces endothelial cell apoptosis via activation of Na+/H+ exchanger 1 and calpain-dependent degradation of Bcl-2. Biochem Biophys Res Commun. 2012;427:125–132. doi: 10.1016/j.bbrc.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 35.Mokgokong R, Wang S, Taylor CJ, Barrand MA, Hladky SB. Ion transporters in brain endothelial cells that contribute to formation of brain interstitial fluid. Pflugers Arch. 2014;466:887–901. doi: 10.1007/s00424-013-1342-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam TI, Wise PM, O’Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Physiol Cell Physiol. 2009;297:C278–C289. doi: 10.1152/ajpcell.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood–brain barrier Na/H exchanger. J Cereb Blood Flow Metab. 2013;33:225–234. doi: 10.1038/jcbfm.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuen N, Lam TI, Wallace BK, Klug NR, Anderson SE, O’Donnell ME. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: evidence for involvement of ERK1/2 MAP kinase. Am J Physiol Cell Physiol. 2014;306:C931–C942. doi: 10.1152/ajpcell.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cingolani HE, Ennis IL. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation. 2007;115:1090–1100. doi: 10.1161/CIRCULATIONAHA.106.626929. [DOI] [PubMed] [Google Scholar]
- 40.Rotte A, Pasham V, Bhandaru M, Bobbala D, Zelenak C, Lang F. Rapamycin sensitive ROS formation and Na+/H+ exchanger activity in dendritic cells. Cell Physiol Biochem. 2012;29:543–550. doi: 10.1159/000338508. [DOI] [PubMed] [Google Scholar]
- 41.Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, Weichert A, Schölkens BA. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–268. doi: 10.1016/0008-6363(96)88579-8. [DOI] [PubMed] [Google Scholar]
- 42.Weichert A, Faber S, Jansen HW, Scholz W, Lang HJ. Synthesis of the highly selective Na+/H+ exchange inhibitors cariporide mesilate and (3-methanesulfonyl-4-piperidino-benzoyl) guanidine methanesulfonate. Arzneimittelforschung. 1997;47:1204–1207. [PubMed] [Google Scholar]
- 43.Anzawa R, Seki S, Nagoshi T, Taniguchi I, Feuvray D, Yoshimura M. The role of Na+/H+ exchanger in Ca2+ overload and ischemic myocardial damage in hearts from type 2 diabetic db/db mice. Cardiovasc Diabetol. 2012;11:33. doi: 10.1186/1475-2840-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russ U, Balser C, Scholz W, Albus U, Lang HJ, Weichert A, Schölkens BA, Gögelein H. Effects of the Na+/H+-exchange inhibitor Hoe 642 on intracellular pH, calcium and sodium in isolated rat ventricular myocytes. Pflugers Arch. 1996;433:26–34. doi: 10.1007/s004240050244. [DOI] [PubMed] [Google Scholar]
- 45.Lei X, Hossain M, Qadri SM, Liu L. Different microvascular permeability responses elicited by the CXC chemokines MIP-2 and KC during leukocyte recruitment: role of LSP1. Biochem Biophys Res Commun. 2012;423:484–489. doi: 10.1016/j.bbrc.2012.05.146. [DOI] [PubMed] [Google Scholar]
- 46.Liu L, Puri KD, Penninger JM, Kubes P. Leukocyte PI3Kγ and PI3Kδ have temporally distinct roles for leukocyte recruitment in vivo. Blood. 2007;110:1191–1198. doi: 10.1182/blood-2006-11-060103. [DOI] [PubMed] [Google Scholar]
- 47.Hickey MJ, Westhorpe CL. Imaging inflammatory leukocyte recruitment in kidney, lung and liver–challenges to the multi-step paradigm. Immunol Cell Biol. 2013;91:281–289. doi: 10.1038/icb.2012.83. [DOI] [PubMed] [Google Scholar]
- 48.Scoditti E, Massaro M, Carluccio MA, Distante A, Storelli C, De CR. PPARγ agonists inhibit angiogenesis by suppressing PKCα- and CREB-mediated COX-2 expression in the human endothelium. Cardiovasc Res. 2010;86:302–310. doi: 10.1093/cvr/cvp400. [DOI] [PubMed] [Google Scholar]
- 49.Nagaraj RH, Sarkar P, Mally A, Biemel KM, Lederer MO, Padayatti PS. Effect of pyridoxamine on chemical modification of proteins by carbonyls in diabetic rats: characterization of a major product from the reaction of pyridoxamine and methylglyoxal. Arch Biochem Biophys. 2002;402:110–119. doi: 10.1016/S0003-9861(02)00067-X. [DOI] [PubMed] [Google Scholar]
- 50.Thornalley PJ, Hooper NI, Jennings PE, Florkowski CM, Jones AF, Lunec J, Barnett AH. The human red blood cell glyoxalase system in diabetes mellitus. Diabetes Res Clin Pract. 1989;7:115–120. doi: 10.1016/0168-8227(89)90101-0. [DOI] [PubMed] [Google Scholar]
- 51.Dhar A, Desai KM, Wu L. Alagebrium attenuates acute methylglyoxal-induced glucose intolerance in Sprague–Dawley rats. Br J Pharmacol. 2010;159:166–175. doi: 10.1111/j.1476-5381.2009.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhar A, Dhar I, Jiang B, Desai KM, Wu L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague–Dawley rats. Diabetes. 2011;60:899–908. doi: 10.2337/db10-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheader EA, Benson RS, Best L. Cytotoxic action of methylglyoxal on insulin-secreting cells. Biochem Pharmacol. 2001;61:1381–1386. doi: 10.1016/S0006-2952(01)00603-7. [DOI] [PubMed] [Google Scholar]
- 54.Du J, Suzuki H, Nagase F, Akhand AA, Ma XY, Yokoyama T, Miyata T, Nakashima I. Superoxide-mediated early oxidation and activation of ASK1 are important for initiating methylglyoxal-induced apoptosis process. Free Radic Biol Med. 2001;31:469–478. doi: 10.1016/S0891-5849(01)00611-6. [DOI] [PubMed] [Google Scholar]
- 55.Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101:1142–1147. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.da Costa-Pessoa JM, Damasceno RS, Machado UF, Beloto-Silva O, Oliveira-Souza M. High glucose concentration stimulates NHE-1 activity in distal nephron cells: the role of the Mek/Erk1/2/p90RSK and p38MAPK signaling pathways. Cell Physiol Biochem. 2014;33:333–343. doi: 10.1159/000356673. [DOI] [PubMed] [Google Scholar]
- 57.Wang S, Peng Q, Zhang J, Liu L. Na+/H+ exchanger is required for hyperglycaemia-induced endothelial dysfunction via calcium-dependent calpain. Cardiovasc Res. 2008;80:255–262. doi: 10.1093/cvr/cvn179. [DOI] [PubMed] [Google Scholar]
- 58.Jandeleit-Dahm K, Hannan KM, Farrelly CA, Allen TJ, Rumble JR, Gilbert RE, Cooper ME, Little PJ. Diabetes-induced vascular hypertrophy is accompanied by activation of Na+/H+ exchange and prevented by Na+/H+ exchange inhibition. Circ Res. 2000;87:1133–1140. doi: 10.1161/01.RES.87.12.1133. [DOI] [PubMed] [Google Scholar]
- 59.Cukiernik M, Hileeto D, Downey D, Evans T, Khan ZA, Karmazyn M, Chakrabarti S. The role of the sodium hydrogen exchanger-1 in mediating diabetes-induced changes in the retina. Diabetes Metab Res Rev. 2004;20:61–71. doi: 10.1002/dmrr.421. [DOI] [PubMed] [Google Scholar]
- 60.Chen S, Khan ZA, Karmazyn M, Chakrabarti S. Role of endothelin-1, sodium hydrogen exchanger-1 and mitogen activated protein kinase (MAPK) activation in glucose-induced cardiomyocyte hypertrophy. Diabetes Metab Res Rev. 2007;23:356–367. doi: 10.1002/dmrr.689. [DOI] [PubMed] [Google Scholar]
- 61.Darmellah A, Baetz D, Prunier F, Tamareille S, Rucker-Martin C, Feuvray D. Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia. 2007;50:1335–1344. doi: 10.1007/s00125-007-0628-x. [DOI] [PubMed] [Google Scholar]
- 62.Lupachyk S, Watcho P, Shevalye H, Vareniuk I, Obrosov A, Obrosova IG, Yorek MA. Na+/H+ exchanger 1 inhibition reverses manifestation of peripheral diabetic neuropathy in type 1 diabetic rats. Am J Physiol Endocrinol Metab. 2013;305:E396–E404. doi: 10.1152/ajpendo.00186.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, Kislinger T, Stern DM, Schmidt AM, De Caterina R. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation. 2002;105:816–822. doi: 10.1161/hc0702.104183. [DOI] [PubMed] [Google Scholar]
- 64.Lawrence SP, Holman GD, Koumanov F. Translocation of the Na+/H+ exchanger 1 (NHE1) in cardiomyocyte responses to insulin and energy-status signalling. Biochem J. 2010;432:515–523. doi: 10.1042/BJ20100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rotte A, Pasham V, Yang W, Eichenmuller M, Bhandaru M, Shumilina E, Lang F. Phosphoinositide 3-kinase-dependent regulation of Na+/H+ exchanger in dendritic cells. Pflugers Arch. 2010;460:1087–1096. doi: 10.1007/s00424-010-0879-0. [DOI] [PubMed] [Google Scholar]
- 66.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Snabaitis AK, Cuello F, Avkiran M. Protein kinase B/Akt phosphorylates and inhibits the cardiac Na+/H+ exchanger NHE1. Circ Res. 2008;103:881–890. doi: 10.1161/CIRCRESAHA.108.175877. [DOI] [PubMed] [Google Scholar]
- 68.Patel H, Chen J, Das KC, Kavdia M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc Diabetol. 2013;12:142. doi: 10.1186/1475-2840-12-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang T, Untereiner A, Liu J, Wu L. Interaction of methylglyoxal and hydrogen sulfide in rat vascular smooth muscle cells. Antioxid Redox Signal. 2010;12:1093–1100. doi: 10.1089/ars.2009.2918. [DOI] [PubMed] [Google Scholar]
- 70.Kang JH. Modification and inactivation of human Cu, Zn-superoxide dismutase by methylglyoxal. Mol Cells. 2003;15:194–199. [PubMed] [Google Scholar]
- 71.Rotte A, Pasham V, Eichenmuller M, Mahmud H, Xuan NT, Shumilina E, Götz F, Lang F. Effect of bacterial lipopolysaccharide on Na+/H+ exchanger activity in dendritic cells. Cell Physiol Biochem. 2010;26:553–562. doi: 10.1159/000322323. [DOI] [PubMed] [Google Scholar]
- 72.Akram S, Teong HF, Fliegel L, Pervaiz S, Clement MV. Reactive oxygen species-mediated regulation of the Na+/H+ exchanger 1 gene expression connects intracellular redox status with cells’ sensitivity to death triggers. Cell Death Differ. 2006;13:628–641. doi: 10.1038/sj.cdd.4401775. [DOI] [PubMed] [Google Scholar]
- 73.Puente N, Chettab K, Duhault J, Koenig-Berard E, McGregor JL. Glucose and insulin modulate the capacity of endothelial cells (HUVEC) to express P-selectin and bind a monocytic cell line (U937) Thromb Haemost. 2001;86:680–685. [PubMed] [Google Scholar]
- 74.Zhu M, Chen J, Jiang H, Miao C. Propofol protects against high glucose-induced endothelial adhesion molecules expression in human umbilical vein endothelial cells. Cardiovasc Diabetol. 2013;12:13. doi: 10.1186/1475-2840-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen JS, Chen YH, Huang PH, Tsai HY, Chen YL, Lin SJ, Chen JW. Ginkgo biloba extract reduces high-glucose-induced endothelial adhesion by inhibiting the redox-dependent interleukin-6 pathways. Cardiovasc Diabetol. 2012;11:49. doi: 10.1186/1475-2840-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buerke U, Pruefer D, Carter JM, Russ M, Schlitt A, Prondzinsky R, Makowski J, Rohrbach S, Niemann B, Schulze C, Dahm M, Vahl CF, Werdan K, Buerke M. Sodium/hydrogen exchange inhibition with cariporide reduces leukocyte adhesion via P-selectin suppression during inflammation. Br J Pharmacol. 2008;153:1678–1685. doi: 10.1038/sj.bjp.0707647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hattori R, Otani H, Moriguchi Y, Matsubara H, Yamamura T, Nakao Y, Omiya H, Osako M, Imamura H. NHE and ICAM-1 expression in hypoxic/reoxygenated coronary microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2001;280:H2796–H2803. doi: 10.1152/ajpheart.2001.280.6.H2796. [DOI] [PubMed] [Google Scholar]
- 78.Redlin M, Werner J, Habazettl H, Griethe W, Kuppe H, Pries AR. Cariporide (HOE 642) attenuates leukocyte activation in ischemia and reperfusion. Anesth Analg. 2001;93:1472–1479. doi: 10.1097/00000539-200112000-00024. [DOI] [PubMed] [Google Scholar]
- 79.Wang SX, Sun XY, Zhang XH, Chen SX, Liu YH, Liu LY. Cariporide inhibits high glucose-mediated adhesion of monocyte-endothelial cell and expression of intercellular adhesion molecule-1. Life Sci. 2006;79:1399–1404. doi: 10.1016/j.lfs.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 80.Nemeth ZH, Deitch EA, Lu Q, Szabo C, Hasko G. NHE blockade inhibits chemokine production and NF-κB activation in immunostimulated endothelial cells. Am J Physiol Cell Physiol. 2002;283:C396–C403. doi: 10.1152/ajpcell.00491.2001. [DOI] [PubMed] [Google Scholar]
- 81.Wu S, Gao X, Yang S, Liu L, Ge B, Yang Q. Protective effects of cariporide on endothelial dysfunction induced by homocysteine. Pharmacology. 2013;92:303–309. doi: 10.1159/000356318. [DOI] [PubMed] [Google Scholar]
- 82.Yang X, Bai H, Cai W, Liu J, Wang Y, Xu Y, Li J, Zhou Q, Han J, Zhu X, Dong M, Hu D. Inhibition of Na+/H+ exchanger 1 by cariporide alleviates burn-induced multiple organ injury. J Surg Res. 2013;185:797–804. doi: 10.1016/j.jss.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 83.Jin BY, Lin AJ, Golan DE, Michel T. MARCKS protein mediates hydrogen peroxide regulation of endothelial permeability. Proc Natl Acad Sci U S A. 2012;109:14864–14869. doi: 10.1073/pnas.1204974109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Petri B, Kaur J, Long EM, Li H, Parsons SA, Butz S, Phillipson M, Vestweber D, Patel KD, Robbins SM, Kubes P. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. 2011;117:942–952. doi: 10.1182/blood-2010-02-270561. [DOI] [PubMed] [Google Scholar]
- 85.Lo SK, Janakidevi K, Lai L, Malik AB. Hydrogen peroxide-induced increase in endothelial adhesiveness is dependent on ICAM-1 activation. Am J Physiol. 1993;264:L406–L412. doi: 10.1152/ajplung.1993.264.4.L406. [DOI] [PubMed] [Google Scholar]