

Abstract
Mice overexpressing the mitotic checkpoint kinase gene BubR1 live longer, whereas mice hypomorphic for BubR1 (BubR1H/H) live shorter and show signs of accelerated aging. As wild-type mice age, BubR1 levels decline in many tissues, a process that is proposed to underlie normal aging and age-related diseases. Understanding why BubR1 declines with age and how to slow this process is therefore of considerable interest. The sirtuins (SIRT1-7) are a family of NAD+-dependent deacetylases that can delay age-related diseases. Here, we show that the loss of BubR1 levels with age is due to a decline in NAD+ and the ability of SIRT2 to maintain lysine-668 of BubR1 in a deacetylated state, which is counteracted by the acetyltransferase CBP. Overexpression of SIRT2 or treatment of mice with the NAD+ precursor nicotinamide mononucleotide (NMN) increases BubR1 abundance in vivo. Overexpression of SIRT2 in BubR1H/H animals increases median lifespan, with a greater effect in male mice. Together, these data indicate that further exploration of the potential of SIRT2 and NAD+ to delay diseases of aging in mammals is warranted.
Keywords: acetylation, aging, BubR1, NAD +, sirtuin
See also: C Cosentino & R Mostoslavsky (July 2014)
Introduction
BubR1 is a component of the spindle assembly checkpoint that has emerged as a key regulator of aging and longevity in mice (Baker et al, 2004, 2011). BubR1 was first characterized as an inhibitor of the anaphase-promoting complex (APC/C), an E3 ligase complex that triggers the transition from metaphase to anaphase by targeting cell cycle regulators for degradation (Elowe, 2011). The evolutionarily conserved amino-terminal domain of BubR1 acts as a pseudosubstrate inhibitor of the APC/C (Oberg et al, 2001; Wei et al, 2005), and the C-terminal domain is thought to act as a pseudokinase that regulates conformational stability of BubR1 (Nateri et al, 2004). Whether it is the cell cycle role of BubR1 or another downstream target that regulates lifespan is not yet known.
In wild-type mice, levels of BubR1 significantly decline with age (Baker et al, 2004). BubR1 hypomorphic mice (BubR1H/H) that constitutively express low levels of BubR1 are normal in appearance and size at birth, but undergo slow postnatal growth and have a shortened lifespan exhibiting premature aging phenotypes, including cachectic dwarfism, lordokyphosis, cataracts, loss of subcutaneous fat, and impaired wound healing. Conversely, sustained expression of BubR1 extends lifespan and delays many age-associated changes including cardiac stress, muscle loss, retinal atrophy, and renal interstitial fibrosis and glomerulosclerosis (Baker et al, 2013). In humans, mutations in BubR1 have been associated with mosaic variegated aneuploidy (MVA) syndrome. MVA patients have various progeroid traits including a shortened lifespan, short stature, facial dysmorphisms, and cataracts (Jacquemont et al, 2002; Lane et al, 2002; Garcia-Castillo et al, 2008). BubR1H/H mice exhibit widespread senescence, and although deletion of senescent cells from these mice delays the onset of numerous age-related phenotypes, their lifespan is not extended (Baker et al, 2011). A major obstacle to extending the lifespan of BubR1 hypomorphic mice is their small heart size and altered electrophysiology. Together, these studies indicate that BubR1 regulates longevity in mammals and that the decline in BubR1 levels over time is an important contributor to normal aging. However, why BubR1 levels decrease over time, and how this may be counteracted remains unknown.
Sirtuins enzymes are evolutionarily conserved NAD+-dependent deacetylases that can delay a variety of age-related diseases (Haigis & Sinclair, 2010). In mammals, there are seven sirtuins, SIRT1-7. SIRT1 is the most extensively characterized sirtuin and protects mice from age-related metabolic dysfunction, liver steatosis, neurodegeneration, cardiovascular disease, and various types of cancer (Haigis & Sinclair, 2010). Many of the beneficial effects attributed to CR have been shown to require sirtuins. For example, the effects of CR on spontaneous activity, cell survival, and lifespan extension require SIRT1 (Cohen et al, 2004b; Chen et al, 2005; Boily et al, 2008) and the suppression of age-related hearing loss requires SIRT3 (Someya et al, 2010).
Despite intensive research on the biology of sirtuins, the cytoplasmic SIRT2 protein remains the least understood. In cell culture, SIRT2 is involved in cell cycle regulation by deacetylating tubulin and histone H4 (North et al, 2003; Vaquero et al, 2006; North & Verdin, 2007b). Through the regulation of histone H4 K16 acetylation, SIRT2 controls the methylation status of histone H4 K20, preventing genomic instability during mitosis (Serrano et al, 2013). SIRT2 also inhibits adipocyte differentiation in vitro by enhancing insulin-stimulated phosphorylation of FOXO1 (Jing et al, 2007). Mice deficient in SIRT2 are tumor prone, which is believed to be through negative regulation of the anaphase-promoting complex by SIRT2 (Kim et al, 2011). SIRT2 and its cofactor NAD+ are upregulated in a variety of tissues during CR (Wang et al, 2007; Haigis & Sinclair, 2010). Recently, SIRT2 has also been implicated in Listeria monocytogenes-mediated reprograming of host cell gene expression (Eskandarian et al, 2013).
Here, we report that BubR1 stability is under the control of SIRT2 in vitro and in vivo. Deacetylation of lysine 668 (K668) of BubR1 inhibits ubiquitylation and the targeting of BubR1 to the proteasome, in a manner independent of altering the cell cycle. Interestingly, SIRT2 overexpression extends both mean and maximum lifespan of BubR1H/H mice. Pharmacologically inducing NAD+ synthesis in mice reverses the age-related decline in NAD+ and, in turn, restores BubR1 protein abundance. Together, our results show that raising NAD+ levels and stimulating SIRT2 activity warrants further investigation as a means to counteract the age-related decline in BubR1 and improving healthspan in mammals.
Results
The ability of BubR1 and the sirtuins to forestall diseases of aging led us to hypothesize that they might be in the same pathway. To test this, we examined the effects of the sirtuin inhibitors nicotinamide and sirtinol on BubR1 levels. Treatment with either of these inhibitors resulted in a reduction in BubR1 protein levels (Fig1A and B). BubR1 expression fluctuates dramatically during the cell cycle, exhibiting low levels during the G1 phase, followed by transcriptional up-regulation during G2 (Davenport et al, 1999). To determine whether sirtuin inhibition was simply reducing BubR1 levels altering the cell cycle, we assessed cell cycle profiles by staining with propidium iodide and did not observe a significant difference in cell cycle profiles following treatment of cells with either nicotinamide or sirtinol (Fig1C, Supplementary Fig S1A). Furthermore, the reduction in BubR1 protein abundance due to sirtuin inhibition was not a result of transcriptional down-regulation as neither treatment affected BubR1 mRNA levels (Fig1D). In addition, treatment of MEFs with nicotinamide resulted in a decrease in BubR1 but had minimal to no effect on other cell cycle regulators such as Aurora A or Aurora B (Supplementary Fig S1B). These results indicated that one or more of the sirtuins control BubR1 abundance without affecting the cell cycle.
Figure 1. BubR1 abundance is modulated by SIRT2.
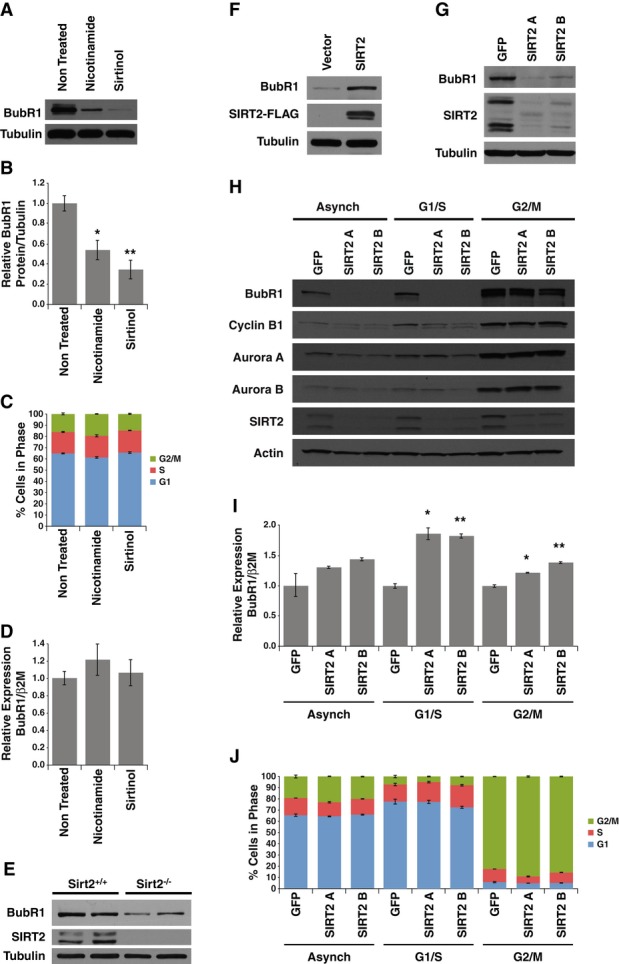
- HeLa cells untreated or treated with nicotinamide or sirtinol were Western blotted for BubR1 and tubulin.
- Quantification of BubR1 protein levels from (A).
- Cell cycle stages of cells from (A).
- Gene expression analysis of BubR1 normalized to β2 microglobulin in cells from (A).
- MEFs from wild-type and Sirt2−/− embryos were Western blotted for BubR1, SIRT2, and tubulin.
- Wild-type MEFs infected with control virus or virus expressing SIRT2-FLAG were Western blotted for BubR1, FLAG, and tubulin.
- HeLa cells expressing control shRNA or two SIRT2 shRNAs were Western blotted for BubR1, SIRT2, and tubulin.
- HeLa cells expressing control shRNA or two SIRT2 shRNAs were either left asynchronous or arrested at G1/S phase or M phase of the cell cycle were Western blotted for BubR1, cyclin B1, Aurora A, Aurora B, SIRT2, and actin.
- Gene expression analysis of BubR1 normalized to β2 microglobulin in cells from (H).
- Cell cycle stages of cells from (H).
Data information: Error bars represent SEM. P-values calculated using Student’s t-test (n = 3), *P < 0.05, **P < 0.005.
Of the seven sirtuins inhibited by these molecules, we speculated SIRT2 would be likely candidate based because it is the predominant sirtuin in the cytoplasm where BubR1 resides (Inoue et al, 2007; North & Verdin, 2007a; Izumi et al, 2009). To test this, we assessed BubR1 protein levels in mouse embryonic fibroblasts (MEFs) isolated from Sirt2−/− embryos (Vaquero et al, 2006). Consistent with our results using sirtuin inhibitors, we found that Sirt2−/− MEFs had reduced BubR1 abundance (Fig1E). Furthermore, overexpression of SIRT2 in wild-type MEFs resulted in an increase in BubR1 protein abundance (Fig1F). Similar to what we observed in Sirt2−/− MEFs, we found a decrease in BubR1 protein levels in cells when SIRT2 was knocked down by shRNA (Fig1G).
These results indicated that BubR1 protein levels are regulated by SIRT2. As with our previous studies utilizing sirtuin inhibitors, we tested whether SIRT2 increases BubR1 abundance by altering the cell cycle distribution or gene transcription. We first assessed the abundance of BubR1 in cells expressing SIRT2 shRNA during various stages of the cell cycle. Cells were either maintained in an asynchronous state or arrested in either the G1/S phase of the cell cycle by double thymidine block or at M phase of the cell cycle by nocodazole treatment. In asynchronous cultures, knockdown of SIRT2 by shRNA did not alter the proportion of cells undergoing mitosis, as the levels of the mitotic regulatory proteins cyclin B1, Aurora A, and Aurora B were not significantly altered (Fig1H). In addition, these cells did not have altered cell cycle profiles (Supplementary Fig S1C). Furthermore, BubR1 abundance was reduced in cells with SIRT2 knocked down, irrespective of whether they were asynchronous, or arrested at either G1/S or M phases of the cell cycle, although the extent that SIRT2 knockdown reduced BubR1 was greater during interphase (asynchronous and G1/S) compared to those arrested in M phase (Fig1H). Interestingly, the degree to which BubR1 levels were reduced by SIRT2 knockdown correlated with the extent of degraded BubR1 in each sample (Supplementary Fig S1D). In addition, knockdown of SIRT2 did not reduce BubR1 mRNA levels, or block the ability of BubR1 to be transcriptionally upregulated during the G2 phase of the cell cycle (Fig1I, Supplementary Fig S1E). Surprisingly, knockdown of SIRT2 appeared to increase levels of BubR1 mRNA (Fig1I), perhaps as a compensation mechanism for reduced BubR1 protein levels. Finally, reduction in SIRT2 did not dramatically influence either the cell cycle profiles of asynchronous, G1/S or M phase-arrested cells, or the ability of cells to be arrested in the G1/S or M phases of the cell cycle (Fig1J, Supplementary Fig S1C). Taken together, these results indicate that SIRT2 modulates the abundance of BubR1 in a manner independent of altering the cell cycle.
These results suggested that SIRT2 might regulate BubR1 stability via a post-translational mechanism. We speculated that SIRT2 could directly modulate BubR1 abundance by deacetylating specific lysine residues on BubR1. First, to determine whether acetylation is involved in regulating BubR1, we tested whether any of the major acetyltransferases affected endogenous BubR1 protein levels. While GCN5, PCAF, and p300 had no effect on endogenous BubR1 protein abundance, CBP decreased BubR1 abundance (Fig2A), an effect that was reversed by SIRT2 (Fig2B). BubR1 physically interacted with both CBP (Fig2C) and SIRT2 (Fig2D), although the interactions appear transient in nature due to the small fraction of proteins interacting at any given time. The interaction between SIRT2 and BubR1 was confirmed at the endogenous level (Fig2E). These data indicate that direct acetylation and deacetylation by CBP and SIRT2 may regulate BubR1 protein levels. Consistent with this, endogenous BubR1 was robustly acetylated by CBP in vivo, as assessed using a pan-anti-acetyl-lysine antibody (Fig2F).
Figure 2. Reversible acetylation of BubR1 by CBP and SIRT2.

- 293T cells were transfected with vector, GCN5-FLAG, PCAF-FLAG, p300-HA, or CBP-HA and Western blotted for BubR1, FLAG, HA, and tubulin.
- 293T cells transfected with vector or CBP-HA with or without SIRT2-FLAG and Western blotted for BubR1, FLAG, HA, and tubulin.
- 293T cells were co-transfected with Myc-BubR1 and CBP-HA. Extracts were immunoprecipitated with anti-HA antibodies and Western blotted for Myc and HA.
- 293T cells were co-transfected with Myc-BubR1 and SIRT2-FLAG. Extracts were immunoprecipitated with anti-FLAG antibodies and Western blotted for Myc and FLAG.
- SIRT2 was immunoprecipitated from 293T cells with anti-SIRT2 antibodies and Western blotted for BubR1 and SIRT2.
- 293T cells were transfected with vector or CBP-HA. BubR1 was immunoprecipitated with anti-BubR1 antibodies and Western blotted for Ac-lysine and BubR1.
- Sirtuin enzymes and acetylated BubR1 purified from 293T cells were incubated in vitro with NAD+. Reactions were Western blotted for Ac-lysine, Myc, and FLAG.
- 293T cells were transfected with Myc-BubR1, CBP-HA, and either vector, wild-type, or catalytically inactive mutations of SIRT2-FLAG and SIRT3-FLAG. Cells extracts were immunoprecipitated with anti-Myc antibodies, and Western blotted for Ac-lysine, Myc, and FLAG.
Next, the ability of SIRT2 and the other six sirtuins (SIRT1, SIRT3-7) to deacetylate BubR1 was tested. We found that SIRT2 and the closely related sirtuin, SIRT3 (North & Verdin, 2004), were the only sirtuins capable of deacetylating BubR1 in vitro (Fig2G). However, when SIRT2 or SIRT3 was transfected into cells, only SIRT2 expression led to a decrease in BubR1 acetylation, in an activity-dependent manner (Fig2H). This is consistent with the fact that SIRT3 is targeted to the mitochondria and would not routinely come into contact with BubR1 (Schwer et al, 2002). To confirm that SIRT2 was the sole deacetylase activity within the immunoprecipitated material, we tested both the sirtuin-specific inhibitor nicotinamide, as well as a class I and II deacetylase specific inhibitor trichostatin A (TSA), on either wild-type or catalytically inactive SIRT2, in an in vitro deacetylation reaction. In this assay, only wild-type SIRT2 was capable to deacetylate BubR1, in both an NAD+-dependent and nicotinamide-sensitive manner (Supplementary Fig S2A). In addition, recombinant SIRT2 purified from bacteria was also capable of deacetylating BubR1 in vitro, indicating that SIRT2 can act directly on BubR1, rather than influencing another deacetylase or acetyltransferase to modulate BubR1 acetylation (Supplementary Fig S2B). Together, these results indicate that BubR1 is subjected to direct and reversible lysine acetylation and deacetylation by CBP and SIRT2, respectively.
To further elucidate the mechanism of regulation of BubR1 abundance by reversible acetylation, we utilized mass spectrometry to determine which lysine residues are acetylated on BubR1. We purified BubR1 acetylated in vivo by CBP and incubated it in vitro with or without rSIRT2, to determine sites specifically acetylated by CBP and deacetylated by SIRT2 (Supplementary Fig S3A and B). We detected acetylation on lysine-668 (K668; Fig3A), a residue that lies in a highly conserved region among vertebrates, between the amino-terminal domain, which is involved in inhibition of the APC, and the kinase domain of BubR1 (Fig3B). To confirm our mass spectrometry analysis, we generated an acetylation-specific antibody for BubR1-K668. The antibody recognized acetylated BubR1 but not a version of BubR1 lacking K668, demonstrating specificity of the antibody for this acetylated site (Supplementary Fig S3C). Using this antibody, we showed that SIRT2 deacetylates BubR1-K668 in cells (Fig3C). Furthermore, of the acetyltransferases that were tested, CBP preferentially acetylated BubR1 at K668 (Fig3D). These results indicate that CBP acetylates BubR1 on K668, regulating BubR1 abundance, which is reversed by SIRT2.
Figure 3. Acetylation of BubR1 at K668 regulates proteasome-mediated degradation of BubR1.

- Myc-BubR1 protein was analyzed by tandem mass spectrometry. Fragmentation spectrum of acetylated K668 peptide showing y and b series fragment ions.
- Schematic diagram and alignment of region surrounding K668 from BubR1 of vertebrate species.
- 293T cells were transfected with Myc-BubR1, CBP-HA, with or without wild-type or catalytically inactive mutant of SIRT2. Extracts were immunoprecipitated with anti-Myc antibodies and Western blotted for Ac-lysine, Ac-BubR1 K668, Myc, and FLAG.
- 293T cells were transfected with vector, GCN5-FLAG, PCAF-FLAG, p300-HA, or CBP-HA, and Myc-BubR1. Extracts were immunoprecipitated with anti-Myc antibodies and Western blotted for Ac-BubR1 K668, Ac-lysine, Myc, HA, FLAG, and tubulin.
- 293T cells were transfected with CBP and treated with MG132. Cell extracts were immunoprecipitated with anti-BubR1 antibodies and Western blotted for ubiquitin, BubR1, and HA.
- 293T cells were co-transfected with HA-Ubiquitin, and wild-type, K668Q, or K668R BubR1-FLAG. Cells were treated with MG132, and cell extracts were immunoprecipitated with anti-FLAG antibodies and Western blotted for HA and FLAG.
- 293T cells were transfected with wild-type, K668Q, or K668R BubR1-FLAG. Cells were treated with cycloheximide for indicated time points were Western blotted for FLAG and tubulin.
- >Quantification of BubR1-FLAG protein levels in (G).
Data information: Error bars represent SEM. P-values calculated using Student’s t-test (n = 3), *P < 0.05, **P < 0.005.
We next explored how acetylation regulates BubR1 abundance. To assess targeting of BubR1 to the proteasome by ubiquitination, cells were transfected with CBP in the presence or absence of SIRT2 and subsequently treated with the proteasome inhibitor MG132. Expression of CBP increased the amount of BubR1 targeted for ubiquitination, which was reversed by SIRT2 expression (Fig3E, Supplementary Fig S3D). To determine whether the acetylation status of the single site, K668, was sufficient to regulate BubR1 degradation, we replaced K668 with glutamine (K668Q) to mimic a constitutively acetylated state or with arginine (K668R) to mimic the non-acetylated state. The K668Q enhanced ubiquitinated BubR1, and, conversely, the K668R mutation reduced it (Fig3F, Supplementary Fig S3E). Furthermore, mutation of additional putative acetylated lysines on BubR1, identified during our mass spectrometry analysis, did not influence ubiquitination of BubR1 as observed for K668 (Supplementary Fig S3F). We next tested whether acetylation mimetic or non-acetylated mutants at K668 altered stability of BubR1 by assessing the rate of degradation following blocking de novo protein synthesis by cycloheximide treatment. Consistent with our previous results, we found that the degradation rate of the K668R mutant was slower than wild-type BubR1, whereas the rate of the K668Q mutant was faster than the wild-type BubR1 (Fig3G). These results indicate that CBP-mediated acetylation at K668 drives BubR1 toward proteasome-mediated protein degradation, and this pathway is counteracted by SIRT2 (Fig7A).
Figure 7. Models for the regulation of BubR1 by SIRT2 and its relevance to age-related decline.
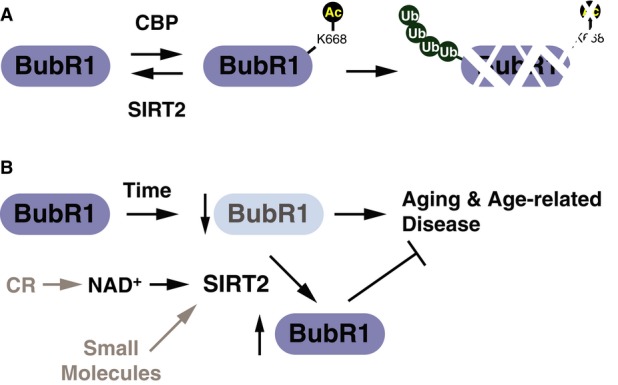
- Model for regulation of BubR1 stability by reversible acetylation mediated by CBP and SIRT2.
- Working model for the stabilization of BubR1 protein abundance by SIRT2 during aging and the effects of increasing NAD+ levels.
To determine whether SIRT2 could influence the levels of BubR1 in vivo and increase the health and lifespan of BubR1H/H mice, we generated a mouse constitutively overexpressing SIRT2. The mice contain a floxed transcriptional stop cassette between the promoter and SIRT2 cDNA (Supplementary Fig S4A). When crossed to a mouse expressing a CMV-driven Cre recombinase, F2 offspring overexpress SIRT2 in all tissues tested (Supplementary Fig S4B). To address whether SIRT2 could extend BubR1-dependent longevity in vivo, SIRT2 transgenic (SIRT2tg) mice were crossed to BubR1H/H mice. Interestingly, although the SIRT2tg/BubR1H/H mice remained small (Supplementary Fig S4C–E), they experienced a 58%increase in median lifespan and 21%increase in maximal lifespan compared to the BubR1H/H control mice (Fig4A). Lifespan extension was observed preferentially in male mice, with a 123%increase in median lifespan (Fig4B). There was no change in median lifespan for females (Fig4C). These results indicate that increasing SIRT2 activity can extend the lifespan of male BubR1H/H mice.
Figure 4. Lifespan extension of BubR1H/H mice by SIRT2 overexpression.

A Lifespan of BubR1H/H (n = 31) and SIRT2tg/BubR1H/H (n = 33) mice. P = 0.0384 determined by log-rank test.
B Lifespan of males BubR1H/H (NT, n = 16) and SIRT2tg/BubR1H/H (SIRT2tg, n = 17) mice, P = 0.004.
C Lifespan of females BubR1H/H (NT, n = 15) and SIRT2tg/BubR1H/H (SIRT2tg, n = 16) mice, N.S. (P = 0.7915).
D–F Lysates from testes (D), spleen (E), and hearts (F) isolated from wild-type, BubR1H/H, and SIRT2tg/BubR1H/H mice were Western blotted for BubR1, SIRT2, and tubulin.
Based on our previous results, we suspected that overexpression of SIRT2 in BubR1H/H mice would stabilize BubR1 in these animals. In initial studies, we purified MEFs from both wild-type and BubR1H/H mice and infected BubR1H/H MEFs with retrovirus expressing wild-type or catalytically inactive SIRT2. We found that expression of wild-type SIRT2 led to stabilization of the BubR1 protein levels, whereas expression of the catalytically inactive SIRT2 had no effect (Supplementary Fig S5A). Following birth, BubR1 levels decline quite dramatically in most tissues. We isolated spleen, lung, and testes tissues from animals obtained from crossing SIRT2tg with BubR1H/H mice as we were readily able to see BubR1 protein within these tissues. We found that, as observed in the isolated MEFs, SIRT2 overexpression resulted in an increase in BubR1 protein in tissues where we were able to detect BubR1 (Fig4D–F). Of note, we were not able to see a restoration of BubR1 levels observed in the BubR1H/H mice back to wild-type levels, which may be due to the fact that the BubR1 protein levels in these mice are due to altered gene transcription, and we would expect that SIRT2-mediated deacetylation is only able to stabilize the protein that is present but not induce a greater level of mRNA driven from the BubR1 promoter. This inability to restore BubR1 back to wild-type levels might also explain why we were able to see a difference in lifespan whereas many of the age-related phenotypes were not significantly altered, suggesting that SIRT2 overexpression may not have achieved a level of BubR1 able to reverse the senescence effects of BubR1 depletion.
Previous studies identified that deletion of senescence cells within the BubR1H/H mice reversed many of their aging-associated phenotypes (Baker et al, 2011). However, removing senescence cells from these mice was not sufficient to reverse their shortened lifespan, due to their defects in cardiac electrophysiology (Baker et al, 2011). During the course of the longevity study, we noted that BubR1H/H mice tended to lose weight only within their last week of life and would die rather suddenly (Supplementary Fig S4F). These observations, combined with the fact that male mice are more susceptible to cardiac abnormalities than females (Du, 2004), led us to hypothesize that BubR1H/H mice were dying from a cardiac-related event that was relieved by SIRT2 overexpression. Consistent with this, the heart weight relative to tibia length of male BubR1H/H animals was less than wild-type animals, and this reduction was partially reversed by SIRT2 (Fig5A). In contrast, reduced kidney size in BubR1H/H animals was not affected by SIRT2 (Fig5B). These results indicated the potential that SIRT2 overexpression in the heart was able to reverse the cardiac abnormalities observed in the BubR1H/H animals.
Figure 5. Reversal of cardiac defects in BubR1H/H mice by SIRT2.

- Hearts from male 3-month-old wild-type (n = 6), BubR1H/H (n = 5), and SIRT2tg/BubR1H/H (n = 6) mice were weighed and normalized to tibia length.
- Kidneys analyzed as in (A).
- LVID;d measured by echocardiography.
- Left ventricle mass calculated with echocardiography measurements.
- Representative ECG tracings from 3-month-old wild-type, BubR1H/H, and SIRT2tg/BubR1H/H mice with inset highlighting J-point. Electrocardiography measurements from male 3-month-old wild-type (n = 12), BubR1H/H (n = 14), and SIRT2tg/BubR1H/H (n = 12) mice representing (F) J-point amplitudes and (G) normalization of J-point to R-wave amplitude.
Data information: Error bars represent SEM. P-values calculated using Student’s t-test (n as specified), *P < 0.05, **P < 0.005.
To explore the possibility that SIRT2 overexpression improved cardiac function in male BubR1H/H mice, we performed echocardiography and discovered the BubR1H/H mice had a reduction in left ventricular (LV) chamber dimension and a corresponding reduction in LV mass (Fig5C and D). Consistent with reversal of heart weight, SIRT2 counteracted the reduced heart size as measured by echocardiography (Fig5C and D). Consequently, we observed that reversal of the abnormally small heart size and dimensions in the BubR1H/H mice overexpressing SIRT2 coincided with increased BubR1 protein abundance in heart tissue (Fig4F).
We next tested whether there was altered cardiac electrocardiography (ECG) in male BubR1H/H mice that was influenced by SIRT2. We found that electrophysiology in BubR1H/H mice was altered including a lengthened duration of the QTc interval, the QRS complex, and an increase in amplitude of the R- and P-wave, and a decrease in the PR duration (Supplementary Fig S5B–F). However, the most prominent effect was depression of the J-point, which represents the approximate end of depolarization and beginning of repolarization (Gussak et al, 1995). We observed a suppression of the J-point in ECG tracings (Fig5E). Interestingly, SIRT2 overexpression prevented the depression in the J-point, both in the measured J-point amplitude (Fig5F) as well as when the J-point amplitude was corrected for overall change in size of the QRS complex (Fig5G) demonstrating that SIRT2 suppresses the repolarization defect of BubR1H/H mice. Together, these data demonstrate that cardiac irregularities influence the lifespan of BubR1H/H mice and that repolarization defects can be reversed by expression of SIRT2.
Our results indicate that BubR1 protein stability is under the control of SIRT2, an NAD+-dependent deacetylase. This raised the intriguing possibility that the age-related loss of BubR1 (Baker et al, 2004) might be correlated with a decrease in levels of the cofactor NAD+ with age. We measured total NAD+ levels in hearts from 6-month- and 30-month-old mice using quantitative HPLC/mass spectrometry and detected significantly lower levels of NAD+ in hearts of older mice, compared to young mice (Fig6A). We next wanted to test whether raising NAD+ levels would increase the levels of BubR1 in both young and old mice. Mice were treated with nicotinamide mononucleotide (NMN), an intermediate of the NAD+ salvage pathway that was shown to raise NAD+ levels in vivo (Supplementary Fig S6A; Yoshino et al, 2011). In addition to the heart, we examined testes, a tissue that expresses high levels of BubR1 (Baker et al, 2004). Intraperitoneal injection of NMN for 7 days (500 mg/kg/day) lead to an increase in NAD+ within the heart and testes in young and in aged mice (Fig6B and C). Although BubR1 protein levels were difficult to determine in the heart tissue of aged animals, BubR1 protein levels were clearly increased in testes following NMN treatment (Fig6D). This increase in BubR1 was observed in both young and aged mice, and more remarkable, BubR1 levels in aged NMN-treated animals were brought back to the levels of the 3-month-old animals (Fig6E).
Figure 6. Decline in NAD+ levels with age and reversal by nicotinamide mononucleotide (NMN).

- NAD+ measured from hearts excised from male 6-month- (n = 4), and 30-month-old (n = 4) mice.
- Relative NAD+ levels in hearts of young and aged mice treated with either PBS or NMN (n = 3 for each group).
- Relative NAD+ levels in testes from same mice as in (B).
- Lysates were prepared from testes of young and aged mice treated with either PBS or NMN and Western blotted for BubR1 and actin.
- Quantification of BubR1 protein levels from (D).
- Wild-type and Sirt2−/− MEFs treated with or without 1 mM NMN for 24 h and measured for relative NAD+ levels.
- Lysates from MEFs in (F) were Western blotted for BubR1, SIRT2, and tubulin.
Data information: Error bars represent SEM. P-values calculated using Student’s t-test (n = 3), *P < 0.05, **P < 0.005.
To determine whether the increase in BubR1 following treatment with NMN was dependent on SIRT2, we utilized MEFs isolated from Sirt2−/− embryos (Vaquero et al, 2006). NMN raised NAD+ levels in MEFs, irrespective of their SIRT2 status (Fig6F) and did not alter cell cycle profiles (Supplementary Fig S6B and C). NMN treatment of wild-type MEFs induced BubR1 protein abundance, and this induction was almost completely absent in the Sirt2−/− MEFs (Fig6G). Together, these results are consistent with a model in which NAD+ levels dictate the level of BubR1 via SIRT2 and indicate that pharmacological modulation of the NAD+/SIRT2 pathway can reverse the age-related decline of BubR1.
Discussion
In this study, we show that SIRT2 controls BubR1 abundance and identify the first intervention to increase the lifespan of BubR1H/H mice. The study provides the first link between SIRT2 and a longevity pathway and provides a mechanistic understanding of how SIRT2 regulates BubR1 abundance in vivo. Specifically, we demonstrate that BubR1 is acetylated at K668, that acetylation at this site promotes ubiquitination and degradation of BubR1, and that SIRT2 stabilizes BubR1 in cell culture and in vivo (Fig7A). We also show that BubR1H/H mice overexpressing SIRT2 have increased BubR1 protein levels and a concomitant lifespan extension.
We were initially surprised that the ability of SIRT2 to extend the lifespan of BubR1H/H mice was gender specific, with a bias toward male animals. However, the recently published lifespan extension of BubR1 transgenic mice also showed a bias toward male mice (Baker et al, 2013), suggesting a shared etiology of lifespan extension between these two studies. Baker et al (2011) recently demonstrated that deletion of senescent cells from BubR1H/H mice improves health but does not extend their lifespan, likely due to early mortality caused by cardiac defects. Our data are consistent with this possibility, based on the improvements in cardiac physiology and lifespan extension provided by SIRT2. In our electrophysiology analysis, we discovered that BubR1 deficiency results in abnormalities in a variety of ECG components, including suppression of the J-point. Furthermore, overexpression of SIRT2 reversed this j-point suppression. The J-point represents the approximate end of depolarization and beginning of repolarization (Gussak et al, 1995). Comparable with the effects we observed in BubR1H/H mice, repolarization of the heart declines with aging in humans (Bachman et al, 1981) and the J-point becomes suppressed with age, specifically in males (Surawicz & Parikh, 2003). Given that both age and male gender are two of the most important risk factors for sudden death in humans (Zheng et al, 2001), our findings suggest that modulation of BubR1 by SIRT2 could mitigate the decline in cardiac function with age and reduce sudden death.
Recent results involving sirtuins and longevity have also shown that overexpression of SIRT6 extends lifespan in mice (Kanfi et al, 2012). Interestingly, the ability of SIRT6 to extend lifespan was also biased toward male mice, where female mice did not respond in a similar fashion in SIRT6 overexpressing mice (Kanfi et al, 2012). The fact that we also observe a gender bias might indicate that the ability of sirtuins to be active when overexpressed might differ between genders, warranting further studies to elucidate mechanisms regulating sirtuin activity or NAD synthesis pathways, and differences that might exist in a gender-specific manner.
In our study, we did not observe a reversal of many of the premature aging phenotypes associated in BubR1H/H mice with SIRT2 overexpression, including lordokyphosis, cataract formation, or the small size. Baker and colleagues demonstrated that by deletion of senescence cells in this model of BubR1 depletion, many of the premature aging phenotypes can be reversed, while this is not sufficient to mitigate the shortened lifespan associated with reduced BubR1 expression. Whereas in this current study, we find that SIRT2 can extend the lifespan, although it was unable to reverse many of the aging-associated phenotypes. We believe that this may be due to the fact that expression of SIRT2 is not able to restore BubR1 levels completely back to wild-type levels in the hypomorphic setting, suggesting that there may be a critical level of BubR1 required to prevent progression to senescence. Furthermore, the amount of BubR1 necessary to preserve tissue homeostasis may vary between tissues or that some of the phenotypes of the germline hypomorphic mouse, which has low levels of BubR1 due to reduced gene expression of the conditional allele, may be irreversible on account of the fact that BubR1 expression is reduced during development and throughout its entire lifespan. In wild-type mice, however, BubR1 levels decline progressively with age (Baker et al, 2004). The data in this study indicate that the age-related decline in BubR1 levels is mediated in part through a decline in NAD+ and SIRT2 activity (Fig7). In this model, NAD+ and SIRT2 maintain BubR1 protein levels and can extend the duration of time in which BubR1 is maintained above the threshold level required for maintenance of tissue homeostasis. This model is also consistent with the observations that aging is associated with a substantial reduction in NAD+ in multiple tissues including heart, muscle, lung, liver, and kidney (Fig5A; Braidy et al, 2011; Price et al, 2012; Yoshino et al, 2011) and that treatment of 30-month-old mice with NMN, a precursor of NAD+, increases NAD+ and restores BubR1 to levels similar to those of 3-month-old mice.
We have shown here that targeting the SIRT2 pathway by inducing NAD+ through treatment with NMN may be a potential mechanism to mitigate the decline of BubR1 with age as we were able to restore BubR1 in aged animals back to that observed in young animals. Although our data suggest that SIRT2 is the primary factor mediated this increase in BubR1, we did see a slight increase of BubR1 following NMN treatment in Sirt2−/− MEFs, suggesting that another sirtuin, or other pathways regulated by NMN, may also be able to influence BubR1 abundance.
Our results also indicate that inducing SIRT2 activity can counteract the aging effects caused by BubR1 depletion in the heart. Although our data suggest that SIRT2-mediated deacetylation and stabilization of BubR1 can reverse the effect of BubR1 depletion in BubR1H/H mice, there remains a strong possibility that SIRT2 has additional targets through which it might exert its lifespan extension effect under BubR1-depleted circumstances. Given that CR is associated with increases in NAD+ and SIRT2 levels (Wang et al, 2007; Haigis & Sinclair, 2010), and that the effects of CR are blunted by preventing the increase in NAD+ (Song et al, 2013), our findings also raise the possibility that SIRT2-mediated increases in BubR1 abundance could underlie some of the health benefits associated with CR. Taken together, these data indicate that increasing NAD+ levels or SIRT2 activity, by genetic or pharmacological means, could have beneficial effects on healthspan and lifespan in mammals.
Materials and Methods
Mice generation and breeding
BubR1H/H mice were previously described (Baker et al, 2004). Sirt2−/− mice for generation of MEFs were kindly provided by Dr. Frederick W. Alt, HHMI, Children’s Hospital Boston (Vaquero et al, 2006). To generate a Cre-inducible SIRT2 transgenic mouse, a transcriptional STOP element was flanked with loxP sites and inserted between a CAGGS promoter and the murine SIRT2 cDNA. This construct was targeted into the mouse Collagen A1 locus using flp recombinase-mediated genomic integration into C10 ES cells (derived from V6.5; BL/6 × 129Sv/Jae F1 ES cells) as described previously (Beard et al, 2006). Mouse embryonic stem cells carrying a single copy of the SIRT2STOP construct were identified by resistance to the antibiotic marker hygromycin and Southern blotting. Clones were injected into blastocysts and pups genotyped for positive recombination. Subsequently, mice were backcrossed to C57BL/6 to 10 generations. To generate constitutive SIRT2 transgenic animals (SIRT2tg), SIRT2STOP mice were crossed with CMV-Cre transgenic mice strains obtained in the C57BL/6J background from Jackson Labs (Bar Harbor, ME, USA). SIRT2tg;CMV-Cre double transgenics were then backcrossed to C57BL/6J to outcross the CMV-Cre allele. SIRT2tg mice were then crossed to BubR1H/H mice to generate SIRT2tg;BubR1H/+ males and females, which were interbred to generate cohorts of mice for longevity study. Mice were weighed twice per week for the duration of the aging study. 3-month- and 30-month-old C57BL/6 mice were obtained from the NIA Aged Rodent Colony. Mice were injected with 500 mg/kg/day nicotinamide mononucleotide (NMN; Sigma Aldrich, St. Louis, MO, USA) diluted in PBS or vehicle for 7 days. Injections were performed prior to dark cycle. On the final day, mice were injected at beginning of light cycle and sacrificed 5 h following injection. Animals were maintained at Harvard Medical School, and experiments were approved by the Animal Care Committee of Harvard Medical School.
Constructs
Sirtuin constructs were previously described (Schwer et al, 2002; North et al, 2003). Myc-BubR1 was kindly provided by Dr. Hongtao Yu, UT-Southwestern (Tang et al, 2004). BubR1 cDNA was cloned to generate C-terminal FLAG-tagged fusions in a derivative of the pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA) backbone (FLAG vector), human SIRT2 was cloned into pMSCVpuro (Invitrogen), and mouse SIRT2 was cloned into pMSCVhygro (Invitrogen), by standard PCR-based strategies. Vectors for expressing SIRT2 shRNA in pLKO.1 were obtained from OpenBiosystems. Sequences for shRNAs used SIRT2 shRNA A CCTGTGGCTAAGTAAACCATA and SIRT2 shRNA B GCCATCTTTGAGATCAGCTAT. CBP-HA (Addgene plasmid 16701) was obtained from Dr. Richard Goodam (Chrivia et al, 1993), p300-HA (Addgene plasmid 10718) from Dr. William Sellers, GCN5-FLAG (Addgene plasmid 14424) from Dr. Puigserver (Lerin et al, 2006), PCAF-FLAG (Addgene plasmid 8941) from Dr. Yoshihiro Nakatani (Yang et al, 1996), and pMD2.G VSV G (Addgene plasmid 12259) and psPAX2 (Addgene plasmid 12260) from Dr. Didier Trono. Mutagenesis of BubR1 was performed with QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA).
Cell culture, transfections, infections, and treatments
293T and HeLa cells were obtained from American Type Culture Collection (ATCC), grown in Dulbecco’s modified Eagle’s medium (DMEM, Mediatech, Inc., Herndon, VA, USA) supplemented with 10%fetal bovine serum (Gemini Bio-products, Woodland, CA, USA) in the presence of penicillin, streptomycin, and 2 mM l-glutamine (Invitrogen). MEFs were prepared from crosses between male and female Sirt2+/−, or between male and female BubR1H/+ mice. On day E14.5, embryos were isolated from pregnant female mice and embryonic fibroblasts prepared as previously described (Xu, 2005). Lentivirus was produced by transfecting 293T cells pMD2.G VSV G, psPAX2, and shRNA encoding transfer vector by the calcium phosphate DNA precipitation method. Media were changed 16 h post transfection, and virus-containing media were harvested 36 h later and filtered through a 0.45-μm filter (Corning) fitted to a 10-ml syringe (BD Biosciences, San Jose, CA). Retrovirus expressing SIRT2 was produced by transfection of Phx-ampho cells (Orbigen) with pMD2.G VSV G and pMSCVpuro-SIRT2 by the calcium phosphate DNA precipitation method, and virus was produced and harvested as described for lentivirus. Target cells were incubated with virus-containing media for 24 h. Media were changed, and 24 h later, infected cells were selected with 2 μg/ml puromycin (Invivogen). Selected cells were harvested and lysed in IPLS lysis buffer (50 mM Tris–HCl, pH 7.5, 0.5 mM EDTA, 0.5%NP-40, 150 mM NaCl) in the presence of protease inhibitor cocktail (Complete, Roche), protein concentrations normalized, and samples diluted in SDS-PAGE buffer. HeLa cells were treated with either 20 mM nicotinamide (Sigma Aldrich) or 25 μM sirtinol (Enzo Life Sciences) for 16 h and harvested as described above. Wild-type and Sirt2−/− MEFs were treated with 1 mM NMN for 24 h and harvested as described above. For cell cycle arrests, cells were treated for 16 h with 2 mM thymidine (Sigma Aldrich), washed, and released into fresh media for 8 h. Subsequently, cells were retreated for 16 h with either 2 mM thymidine for G1/S phase arrest or 200 nM nocodazole (Sigma Aldrich) for G2/M phase arrest. For ubiquitination assays, cells were transfected as described above and treated prior to harvest with 10 μM MG132 (Sigma Aldrich) for 16 h (CBP/SIRT2 assay) or 4 h (BubR1 acetylation mutants assay). Cells were harvested in RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1.0%Triton X-100 (or NP-40), 0.5%sodium deoxycholate, 0.1%SDS) in the presence of protease inhibitor cocktail (Complete, Roche) and immunoprecipitated as described below. For cycloheximide time course assays, transfected cells were treated with 100 μg/ml cycloheximide (Sigma) for indicated time and harvested as described above. Tissue samples were lysed in RIPA buffer following tissue disruption with a Potter-Elvehjem homogenizer. Lysates were cleared by centrifugation, and protein concentrations normalized, and samples diluted in SDS-PAGE buffer.
Cell cycle analysis
Cells were harvested by trypsinization, washed in PBS, and fixed in ice-cold 70%ethanol, and stored at −20°C for 2 h. Fixed cells were pelleted, washed in PBS, and stained in PI Buffer (0.1%sodium citrate, 0.3%Triton X-100, 0.01%propidium iodide, and 0.02 mg/ml RNase A: modified as described previously; Krishan, 1975) for 30 min. Stained cells were subjected to flow cytometric analysis using a FACSCalibur (BD Biosciences) and analyzed by FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Gene expression analysis
Cells were harvested by trypsinization and washed in PBS. RNA was isolated from pelleted cells using RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and resulting RNA was quantified using a NanoDrop 1000 spectrophotometer (Thermo Scientific). cDNA synthesis was performed with Illustra Ready-To-Go RT-PCR Beads (GE Healthcare) using 1 µg RNA and random hexamer primers. Quantitative RT-PCR was performed with 1 µM of each primer and SYBR Green Master Mix (Roche) on a LightCycler 480 detection system (Roche). Relative gene expression was calculated using the delta-delta Ct method using β2-microglobulin as internal standard. Primers used for qPCR: BubR1 CTCGTGGCAATACAGCTTCA and CTGGTCAATAGCTCGGCTTC, β2-microglobulin CAAGATAGTTAAGTGGGATCGAG and GTGCATAAAGTGTAAGTGTATAAGC.
Mass spectrometry analysis
Excised gel bands were cut into approximately 1 mm3 pieces. The samples were reduced with 1 mM DTT for 30 min at 60°C and then alkylated with 5 mM iodoacetamide for 15 min in the dark at room temperature. For modified in-gel trypsin digestion procedure, gel pieces were washed and dehydrated with acetonitrile for 10 min followed by removal of acetonitrile. Pieces were then completely dried in a speed-vac and rehydrated with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega) at 4°C. Samples were then placed at 37°C overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50%acetonitrile and 5%acetic acid. The extracts were dried in a speed-vac (˜1 h) and stored at 4°C until analysis. For mass spectrometry analysis, the samples were reconstituted in 5 μl HPLC solvent A (2.5%acetonitrile, 0.1%formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 5 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter × 12 cm length) with a flame-drawn tip. After equilibrating the column, each sample was pressure-loaded off-line onto the column. The column was then reattached to the HPLC system. A gradient was formed, and peptides were eluted with increasing concentrations of solvent B (97.5%acetonitrile, 0.1%formic acid). As each peptide was eluted, they were subjected to electrospray ionization and then they entered into an LTQ-Orbitrap mass spectrometer (ThermoFinnigan). Eluting peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern by Sequest (ThermoFinnigan) software. The modification of 42.0106 mass units to lysine was included in the database searches to determine acetylated peptides. Each acetylated peptide that was determined by the Sequest program was also manually inspected to ensure confidence (Shevchenko et al, 1996; Peng & Gygi, 2001).
Recombinant proteins
DH5α were transformed with pHEX vector containing the human SIRT2 cDNA and induced with 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 37°C for 2 h. Bacteria were sonicated in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole, 1 mg/ml lysozyme), and the resulting 6×His-tagged protein was purified under native conditions at 4°C by Ni-NTA (Qiagen). Captured protein was washed with lysis buffer containing 20 mM imidazole and eluted with lysis buffer containing 250 mM imidazole. Eluted protein was dialyzed against storage buffer (50 mM Tris–HCl, pH 8.0, 4 mM MgCl2, 0.2 mM DTT, 150 mM NaCl, 10%glycerol), aliquoted, and stored at −20°C.
Immunoprecipitation
293T cells were transfected by the calcium phosphate DNA precipitation method and lysed 48 h after transfection in IPLS lysis buffer (50 mM Tris–HCl, pH 7.5, 0.5 mM EDTA, 0.5%NP-40, 150 mM NaCl) in the presence of protease inhibitor cocktail (Complete, Roche). FLAG-tagged, HA-tagged, and Myc-tagged proteins were immunoprecipitated for 2 h at 4°C with anti-FLAG M2 agarose affinity gel (Sigma), anti-HA agarose (Roche), and anti-Myc (9E10, Santa Cruz Biotechnology), respectively. Myc-tagged proteins were absorbed with Protein A/G Plus agarose (Santa Cruz Biotechnology) for an additional 1 h. Immunoprecipitated material was washed three times for 15 min each in lysis buffer and either used for subsequent in vitro activity assays or resuspended in SDS-PAGE buffer. Endogenous immunoprecipitations were performed from 293T cells using anti-SIRT2 (Epitomics) as described above.
Immunoblotting
A rabbit polyclonal antibody was raised against a peptide of BubR1, CQTLSIK-AcK-LSPII-amide, and purified over non-acetylated and acetylated peptides (YenZym Antibodies). Samples were separated on 10%SDS-polyacrylamide gels and transferred to Hybond ECL nitrocellulose membrane (Amersham Pharmacia Biotech, Inc.). Membranes were blocked with 5%non-fat dry milk in TBS-Tween (10 mM Tris, pH 7.5, 150 mM NaCl, and 0.1%Tween-20) and were probed with antibodies to BubR1 (BD Biosciences) at 1:1,000, tubulin (Sigma Aldrich) at 1:5,000, SIRT2 (Epitomics or Bethyl Laboratories) at 1:1,000, FLAG (Sigma Aldrich) at 1:5,000, HA (Sigma Aldrich) at 1:5,000, Myc (Santa Cruz Biotechnology) at 1:5,000, Ac-lysine (Cohen et al, 2004a) at 1:2,000, ubiquitin (Santa Cruz Biotechnology) at 1:1,000, and BubR1 Acetylated K668 at 0.05 μg/ml. Secondary detection was performed using horseradish peroxidase-coupled sheep anti-mouse IgG (Amersham Pharmacia Biotech, Inc.), goat anti-rabbit IgG (Pierce Chemical Co., Rockford, IL, USA) diluted 1:5,000, and ECL Western blotting detection system (Amersham Pharmacia Biotech, Inc.). Quantifications of Western blot films were performed using ImageJ (National Institutes of Health, Bethesda, MD, USA).
In vitro activity assay
Immunoprecipitates for FLAG-tagged sirtuins and for Myc-tagged BubR1 were washed two times for 15 min each in SIRT deacetylase buffer (50 mM Tris–HCl, pH 9.0, 4 mM MgCl2, 0.2 mM DTT). Myc-BubR1 immunoprecipitates were resuspended in 100 μl of SIRT deacetylase buffer containing 1 mM NAD+ (Sigma) and added to each FLAG-tagged sirtuin immunoprecipitated, or purified SIRT2, and incubated for 2 h at 37°C. Reactions containing deacetylase inhibitors were pre-incubated for 10 min with either 20 mM nicotinamide or 400 nM trichostatin A (TSA) prior to reaction being started by addition of NAD+. Reactions were stopped by addition of 20 μl of 6× SDS-PAGE buffer. Beads were pelleted by centrifugation at 16,000 g for 10 min, and 10 μl of each supernatant was separated on 10%SDS-PAGE gels and Western blotted as described above.
NAD+ quantification
NAD+ from young and aged hearts were determined as described previously (Sauve & Schramm, 2003), with the following modifications. The reference standard 18O-NAD+ (typically 640 pmol) was added to the sample. NAD+ was extracted from heart tissue by adding ice-cold 7%perchloric acid, followed by vortexing for 30 s, and sonicating for 5 min, on ice. The vortex–sonication cycle was repeated three times. Samples were then centrifuged at room temperature for 3 min at 16,800 g. Cleared supernatant was neutralized by 3 M NaOH and 1 M phosphate buffer (pH ˜9). Neutralized sample was centrifuged again at room temperature for 3 min at 16,800 g. Cleared supernatant was injected into HPLC to separate NAD+ from other cellular components; NAD+ peaks were collected according to the retention time of authentic standard. Collections were lyophilized to dryness and subjected to MALDI-TOF analysis. Ratio of intensities for m/z = 664 and 666 peaks, corresponding to 16O- and 18O-NAD+ isotopomers, was multiplied by 640 pmol and then divided by tissue weight to determine NAD+ concentration in the sample. Standards containing only 16O and 18O-NAD+ (600 pmol each) were also run to determine corrections for isotopic purity and for procedure calibration.
For NAD+ quantifications from testes following NMN treatment, NAD+ was measured with NAD/NADH Quantification Kit (Biovision, Milpitas, CA, USA) and normalized to total protein content.
Echocardiography
Mice were anesthetized under an isoflurane vaporizer (VetEquip), body temperature was maintained at 37°C, and paws were secured to the ECG leads on Vevo Mouse Handling Table (VisualSonics Inc.). Chest hair was removed with Nair cream, and ultrasound transmission gel was applied. Echocardiography was conducted using a Vevo 770 High-Resolution In Vivo Micro-Imaging System and RMV 707B scanhead (VisualSonics Inc.) with heart rate of 500–550 beats per minute. M-mode imaging was obtained with parasternal short axis view. Three consecutive cardiac cycles were measured and averaged accordingly. LV mass was calculated from diastolic M-mode measurements as has been previously described (Liu & Rigel, 2009).
Electrocardiography
Mice were lightly anesthetized with isofluorane (1–1.5%in O2 at 3–5 cc/h), and 4-unipolar subcutaneous leads (PowerLab, AD Instruments) were placed on the proximal limbs. Baseline cardiac cycle intervals were measured, including the heart rate, cycle length, PR, QRS, and QT intervals. P, R, and J-point amplitudes were measured. Any arrhythmia, if present, was documented. Data were recorded and analyzed using Chart 5 (version 5.5.3, AD Instruments) software.
Statistical analysis
Log-rank test were used to evaluate significance between groups for Kaplan–Meier survival curves, and Student’s t-test was used to evaluate significance between groups for all other data and P-values indicated.
Acknowledgments
DS was supported by grants from NIH/NIA (R01 AG028730 and RO1 AG019719), the Glenn Foundation for Medical Research, the United Mitochondrial Disease Foundation, The Juvenile Diabetes foundation, and a gift from the Schulak family. B.J.N was supported by BIDMC/Harvard Translational Research in Aging Training Program (T32 AG023480). A.R., M.R., and J.D. are supported in part by grants from the NIH and a Leducq Foundation Network of Research Excellence.
Author contributions
BN, MR, KJ, AH, SM, JD, DB, and YC performed the experiments. BN, MR, KJ, AH, DB, AS, JD, AR, and DS analyzed the results and participated in experimental design. LW supplied reagents. BN and DS wrote the manuscript.
Conflict of interest
D.S. is a consultant to and inventor on patents licensed to GlaxoSmithKline, OvaScience, MetroBiotech, companies working on NAD+ and sirtuin modulation.
Supporting information for this article is available online: http://emboj.embopress.org
References
- Bachman S, Sparrow D, Smith LK. Effect of aging on the electrocardiogram. Am J Cardiol. 1981;48:513–516. doi: 10.1016/0002-9149(81)90081-3. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol. 2013;15:96–102. doi: 10.1038/ncb2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis. 2006;44:23–28. doi: 10.1002/gene.20180. [DOI] [PubMed] [Google Scholar]
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, Xuan J, Evans M, Harper ME, McBurney MW. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004a;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004b;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Davenport JW, Fernandes ER, Harris LD, Neale GA, Goorha R. The mouse mitotic checkpoint gene bub1b, a novel bub1 family member, is expressed in a cell cycle-dependent manner. Genomics. 1999;55:113–117. doi: 10.1006/geno.1998.5629. [DOI] [PubMed] [Google Scholar]
- Du XJ. Gender modulates cardiac phenotype development in genetically modified mice. Cardiovasc Res. 2004;63:510–519. doi: 10.1016/j.cardiores.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Elowe S. Bub1 and BubR1: at the interface between chromosome attachment and the spindle checkpoint. Mol Cell Biol. 2011;31:3085–3093. doi: 10.1128/MCB.05326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandarian HA, Impens F, Nahori MA, Soubigou G, Coppee JY, Cossart P, Hamon MA. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science. 2013;341:1238858. doi: 10.1126/science.1238858. [DOI] [PubMed] [Google Scholar]
- Garcia-Castillo H, Vasquez-Velasquez AI, Rivera H, Barros-Nunez P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am J Med Genet A. 2008;146A:1687–1695. doi: 10.1002/ajmg.a.32315. [DOI] [PubMed] [Google Scholar]
- Gussak I, Bjerregaard P, Egan TM, Chaitman BR. ECG phenomenon called the J wave. History, pathophysiology, and clinical significance. J Electrocardiol. 1995;28:49–58. doi: 10.1016/s0022-0736(05)80007-x. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Hiratsuka M, Osaki M, Oshimura M. The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle. 2007;6:1011–1018. doi: 10.4161/cc.6.9.4219. [DOI] [PubMed] [Google Scholar]
- Izumi H, Matsumoto Y, Ikeuchi T, Saya H, Kajii T, Matsuura S. BubR1 localizes to centrosomes and suppresses centrosome amplification via regulating Plk1 activity in interphase cells. Oncogene. 2009;28:2806–2820. doi: 10.1038/onc.2009.141. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Boceno M, Rival JM, Mechinaud F, David A. High risk of malignancy in mosaic variegated aneuploidy syndrome. Am J Med Genet. 2002;109:17–21. doi: 10.1002/ajmg.10281. ; discussion 16. [DOI] [PubMed] [Google Scholar]
- Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66:188–193. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AH, Aijaz N, Galvin-Parton P, Lanman J, Mangano R, Wilson TA. Mosaic variegated aneuploidy with growth hormone deficiency and congenital heart defects. Am J Med Genet. 2002;110:273–277. doi: 10.1002/ajmg.10462. [DOI] [PubMed] [Google Scholar]
- Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Liu J, Rigel DF. Echocardiographic examination in rats and mice. Methods Mol Biol. 2009;573:139–155. doi: 10.1007/978-1-60761-247-6_8. [DOI] [PubMed] [Google Scholar]
- Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004;5:224. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS One. 2007a;2:e784. doi: 10.1371/journal.pone.0000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Verdin E. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J Biol Chem. 2007b;282:19546–19555. doi: 10.1074/jbc.M702990200. [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem. 2001;276:35847–35853. doi: 10.1074/jbc.M103992200. [DOI] [PubMed] [Google Scholar]
- Peng J, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Schramm VL. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry. 2003;42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano L, Martinez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB, Kane-Goldsmith N, Tong Q, Rabanal RM, Fondevila D, Munoz P, Kruger M, Tischfield JA, Vaquero A. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013;27:639–653. doi: 10.1101/gad.211342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Ke SF, Zhou CC, Zhang SL, Guan YF, Xu TY, Sheng CQ, Wang P, Miao CY. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J Gerontol A Biol Sci Med Sci. 2013;69:44–57. doi: 10.1093/gerona/glt122. [DOI] [PubMed] [Google Scholar]
- Surawicz B, Parikh SR. Differences between ventricular repolarization in men and women: description, mechanism and implications. Ann Noninvasive Electrocardiol. 2003;8:333–340. doi: 10.1046/j.1542-474X.2003.08411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Shu H, Oncel D, Chen S, Yu H. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004;16:387–397. doi: 10.1016/j.molcel.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–1261. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. In: Ausubel FM, et al., editors. Current Protocols in Molecular Biology. New York: Wiley and Sons; 2005. (eds), Chapter 28: Unit 28 21. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–2163. doi: 10.1161/hc4301.098254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.