

Abstract
Rad17 is best known as a checkpoint clamp loader in the activation of ATR kinase signaling. A new study in The EMBO Journal suggests that it also plays a role in initial recruitment of the MRN complex to sites of DNA double-strand breaks, thereby promoting early ATM checkpoint responses and homologous recombination repair.
See also: Q Wang et al (April 2014)
The cellular response to DNA double-strand breaks (DSBs) involves immediate binding of break sites by early-acting DNA repair and checkpoint proteins (Goodarzi & Jeggo, 2013). Coordinated loading of DNA end processing factors promotes repair through either homologous or non-homologous mechanisms, and signaling of breaks through post-translational modifications that emanate from the break site ensures rapid cell cycle arrest or apoptosis. One of the first complexes arriving at a break to mediate these events is the Mre11-Rad50-Nbs1 (MRN) complex, which promotes repair of breaks through 5′ strand resection and homologous recombination and also initiates a signaling cascade through the ATM protein kinase to trigger checkpoint activation.
It has been understood for some time that MRN is recruited to sites of DNA breaks via at least two different mechanisms. Over a time course of DNA damage (ionizing radiation in most cases), MRN is observed by indirect immunofluorescence to arrive within seconds of double-strand break induction (early recruitment) and also remains at a subset of these sites in distinct foci at later time points (retention) (Lukas et al, 2004). The retention of MRN at regions of chromatin adjacent to break sites is much better understood: MRN binds directly to the Mdc1 protein through phospho-specific interactions between the Nbs1 component of MRN and the constitutively phosphorylated Mdc1 protein, which is recruited to chromatin near DSBs through recognition of the phosphorylated form of the histone H2AX (γ-H2AX) (Goodarzi & Jeggo, 2013). Cells deleted for H2AX still exhibit early recruitment of MRN but are deficient in forming the large foci at later time points that are dependent on Mdc1 (Celeste et al, 2003).
This distinction between two kinds of binding patterns and kinetics was also observed in the physical size of the γ-H2AX-independent Nbs1 foci which were limited to a smaller region compared to γ-H2AX foci and thus termed “microfoci” in the absence of the Mdc1-dependent late retention pathway (Lukas et al, 2004). We have assumed that early recruitment of MRN to the actual break site (as opposed to the mega-base-sized region of the chromatin surrounding the break site) is mediated by MRN itself, since the complex exhibits picomolar affinity for DNA ends, and in vitro reconsitituted assays for DNA binding, 5′ resection, end tethering, and ATM activation do not require any other factors (Lee & Paull, 2007). However, a new study in this issue of The EMBO Journal shows that the Rad17 protein, previously implicated in ATR signaling through its role as part of the Rad17-Rfc2-5 clamp loader for the Rad1-Rad9-Hus1 (9-1-1) complex, is necessary for the early recruitment of MRN to double-strand break sites (Wang et al, 2014).
In this article, Wang and colleagues show that depletion of Rad17 causes a dramatic loss of ionizing radiation (IR)-induced foci including γ-H2AX, Nbs1, Mdc1, and RPA as well as defects in ATM-dependent phosphorylation and homologous recombination, suggesting that Rad17 has a role in the early stages of DNA DSB response (Wang et al, 2014). Previous studies using genetic deletions of Rad17 had suggested a role for this factor in homologous recombination (Nishino et al, 2008; Saberi et al, 2008), although the function of Rad17 in this process was not clear. Here, the authors show that Rad17 is required for the early recruitment of the MRN complex to DSB sites but is not required for Mdc1/γ-H2AX-dependent recruitment of the MRN complex at late time points after IR. A direct interaction between Nbs1 and Rad17 was demonstrated, which requires ATM phosphorylation of Rad17 on Thr622. They also show that expression of a T622A Rad17 mutant causes severe reduction in MRN complex levels on chromatin after IR treatment and loss of ATM, Nbs1, and Chk2 phosphorylation at early time points after IR treatment. Taken together, these data suggest that Rad17 has a role in the recruitment of the MRN complex to DSBs independent of Mdc1/γ-H2AX (Fig 1).
Figure 1. Tentative model of how Rad17 may help to promote initial MRN recruitment and early damages response signaling at DNA double-strand break sites.
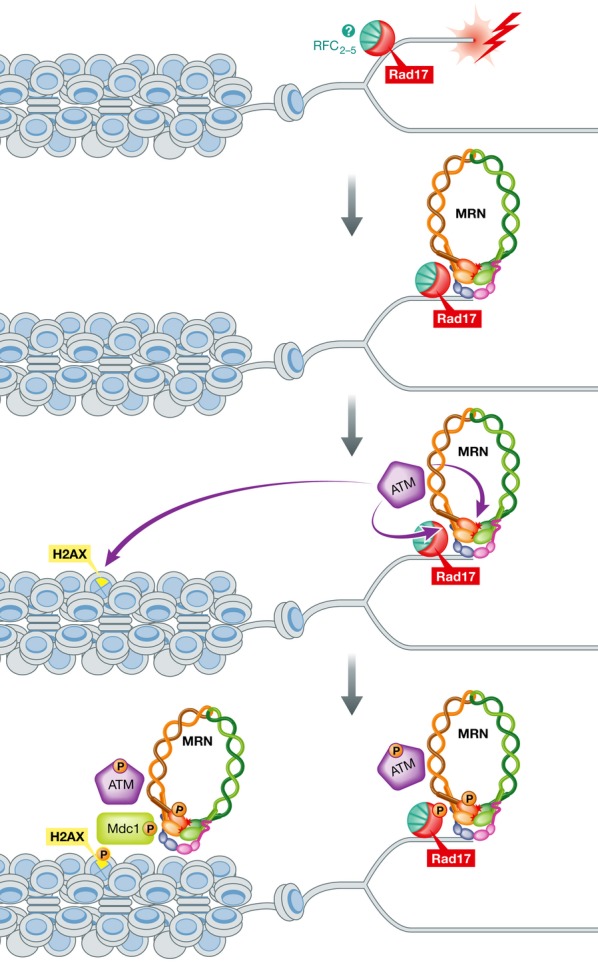
See text for details. Please note that it remains to be determined whether Rad17 acts together with Rfc2-5 as part of an RFC complex in this instance.
The MRN complex has two important roles after DNA damage: initiation of DNA repair and activation of ATM. The MRN complex exhibits endo- and exo-nuclease activity on double-stranded DNA and interacts with the CtIP endonuclease, promoting homologous recombination by initiating DSB end resection (Goodarzi & Jeggo, 2013). In the article by Wang et al, the authors show that Rad17 recruits the MRN complex to DSBs, promoting the initiation of DNA resection, resulting in more efficient DNA repair by homologous recombination.
The MRN complex also acts as a DNA break sensor and activator of the ATM protein kinase, which phosphorylates many proteins that collectively promote checkpoint activation and DNA repair (Goodarzi & Jeggo, 2013). Many groups have shown that the MRN complex is essential for ATM activation after DSBs in vivo and in vitro (Lee & Paull, 2007). One of the most interesting findings by Wang et al is that Rad17 is required for ATM phosphorylation of DNA damage targets, including Chk2 and H2AX itself. Thus, the Rad17 pathway directly promotes the subsequent γ-H2AX-dependent pathway that occurs later and promotes retention of MRN to sites of Mdc1/γ-H2AX. The Rad17 relationship to MRN is clearly different from that of Mdc1/γ-H2AX, however, in that Rad17 also requires MRN for the formation of Rad17 foci, whereas Mdc1 foci formation at γ-H2AX sites is independent of MRN (Goldberg et al, 2003).
These results are provocative and novel, but raise many more questions than they answer. Several studies have shown that the MRN complex is recruited to γ-H2AX at chromatin adjacent to DSB sites through the interaction between the forkhead-associated (FHA) domain of Nbs1 and phosphorylated SDTD repeats on Mdc1 (Goodarzi & Jeggo, 2013). In their new study, the authors find that the FHA domain of Nbs1 also interacts with phosphorylated Rad17 but the recognition site is completely different from that of Mdc1. It is possible that one domain interacts with multiple targets but a more definitive clarification of the FHA domain residues required for the Rad17 interaction is clearly needed. Interestingly, previous studies have shown that overexpression of part or all of the Mdc1 domain that normally interacts with Nbs1 blocks MRN foci formation at relatively early time points after IR (Goldberg et al, 2003; Xu & Stern, 2003), an effect which, in light of the current data, could be in part due to competitive interactions with Rad17.
Another major question relates to the mechanism by which Rad17 localizes to DSB sites. Rad17 is part of the Rad17-RFC complex, which binds to replication intermediates (nicked, gapped, and primed ssDNA structures) and loads the 9-1-1 complex as part of the replication checkpoint (Navadgi-Patil & Burgers, 2009). Is it possible that this entire complex localizes to DSB sites? Or does recruitment of MRN occur preferentially at sites of active replication? Mammalian Rad9 protein has also been shown to be required for homologous recombination (Pandita et al, 2006) and to localize to DSB sites (Greer et al, 2003), perhaps an additional sign that the checkpoint clamp loader and clamp play important roles in DSB repair. Rad17 is a member of the AAA+ family of ATPase, and its ATPase activity is required for 9-1-1 complex loading onto DNA (Navadgi-Patil & Burgers, 2009). Is the ATPase activity of Rad17 required for recruitment of the MRN complex to DSBs? And is Rad17-dependent recruitment of MRN actually to the sites of DSB ends, or also in this case to sites adjacent to the actual break? Elucidating the mechanism of DSB recognition and accumulation of Rad17 will be essential to solidify this model.
In conclusion, the new study suggests that early recruitment of MRN occurs through a completely unexpected source—the checkpoint clamp loader originally thought to only affect ATR signaling, not ATM. It is likely that our models of the DSB signaling cascade will need to change to incorporate these new data, but the new model provides a rich source of ideas for these future experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. Adv Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- Greer DA, Besley BD, Kennedy KB, Davey S. hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res. 2003;63:4829–4835. [PubMed] [Google Scholar]
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004;23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. A tale of two tails: activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair. 2009;8:996–1003. doi: 10.1016/j.dnarep.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Inoue E, Takada S, Abe T, Akita M, Yoshimura A, Tada S, Kobayashi M, Yamamoto K, Seki M, Enomoto T. A novel role for Rad17 in homologous recombination. Genes Genet Syst. 2008;83:427–431. doi: 10.1266/ggs.83.427. [DOI] [PubMed] [Google Scholar]
- Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, Gupta A, Wellinger RJ, Zhang J, Powell SN, Roti Roti JL, Lieberman HB, Pandita TK. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006;26:1850–1864. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi A, Nakahara M, Sale JE, Kikuchi K, Arakawa H, Buerstedde JM, Yamamoto K, Takeda S, Sonoda E. The 9-1-1 DNA clamp is required for immunoglobulin gene conversion. Mol Cell Biol. 2008;28:6113–6122. doi: 10.1128/MCB.00156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Goldstein M, Alexander P, Wakeman TP, Sun T, Feng J, Lou Z, Kastan MB, Wang XF. Rad17 recruits the MRE11-RAD50-NBS1 complex to regulate the cellular response to DNA double-strand breaks. EMBO J. 2014;33:862–877. doi: 10.1002/embj.201386064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Stern DF. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. FASEB J. 2003;17:1842–1848. doi: 10.1096/fj.03-0310com. [DOI] [PubMed] [Google Scholar]