

Abstract
The most common enzyme defect in humans is glucose-6-phosphate dehydrogenase (G6PD) deficiency, which affects more than 400 million people. G6PD shunts glucose into the pentose phosphate pathway (PPP) to generate nucleotides and reducing potential in the form of NADPH. In this issue, Wang et al (2014) show that G6PD activity is post-translationally regulated by SIRT2, a cytoplasmic NAD+-dependent deacetylase, thereby linking NAD+ levels to DNA repair and oxidative defences, and identifying potential new approaches to treating this common genetic disease.
If you have ever opened a textbook or sat through an undergraduate lecture on glucose metabolism, you could be forgiven for thinking the major questions of the field have been solved. But in the past few years, thanks to advances in mass spectrometry and our ability to detect protein modifications on a proteomic scale, the field has experienced a renaissance. A case in point is G6PD deficiency, the most common enzyme deficiency in the world, primarily in people of Asian and African descent, likely as a genetic defence against malaria. This X-linked recessive genetic disease manifests, to various degrees, as increased oxidative damage and a high risk of haemolytic anaemia due to certain medicines, infections and foods. In some cases, chronic anaemia ensues, and there are reports of a higher incidence of age-related diseases such as type 2 diabetes (Heymann et al 2012).
In the 1930s, Warburg and Christian first isolated G6PD as a yellow protein from yeast they called Zwischenferment, shown shortly thereafter to reduce (i.e. regenerate) the antioxidant glutathione in the presence of glucose (Warburg & Christian, 1933). G6PD activity controls a major fork in the road for glucose utilization: it can be used for energy or it can be shunted to the pentose phosphate pathway (PPP), which takes glucose-6-phosphate (G6P), an early glycolytic intermediate, and metabolises it into the nucleotide precursor ribose-5-phosphate (R5P) via a process that generates two molecules of NADPH for reductive biosynthetic processes such as de novo lipogenesis and for the regeneration of glutathione. Because G6PD catalyses the irreversible conversion of glucose-6-phosphate into 6-phosphogluconate, the rate-limiting step in the PPP, a deficiency in its activity has major implications for DNA repair, lipogenesis and antioxidant defences.
The classic view of G6PD regulation is that it is mediated solely by substrate availability and the ratio of NADPH to NADP+. Recent studies, however, indicate that this model is too simplistic. The G6PD gene is subject to extensive regulation at the transcriptional level, and Src-mediated phosphorylation (Pan et al, 2009) modulates its activity by approximately 20%.
In this issue, Wang et al (2014) show that large changes in G6PD activity are controlled by the acetylation status of just one evolutionarily conserved lysine residue within the NADP+ structural binding domain of G6PD (K403). What makes the study particularly interesting and potentially medically relevant is that the acetylation status of K403 is controlled by SIRT2, a member of the sirtuin family of lysine deacylases that are NAD+-dependent. Other members of the sirtuin family (SIRT1-7) are localized to the nucleus and mitochondria where they control cell responses to energy availability and biological stress. The functions of SIRT2, however, are poorly understood. One known function of SIRT2 is that it maintains faithful chromosome division and replication (Kim et al, 2011).
By potentially connecting the PPP to NAD+ availability, this work could explain how a cell is able to rapidly and exquisitely regulate PPP activity in response to DNA damage, redox status (Ralser et al, 2007) and nutrient intake. The work also raises the possibility of a positive feedback in which the end product of the PPP, the nucleotide precursor ribose-5-phosphate is used to generate NAD+ via 5-phospho-α-D-ribosyl 1-pyrophosphate (PRPP) and the stress- and nutrient-responsive NAD+ biosynthetic enzyme NAMPT (see figure). In this way, SIRT2 and PPP could be mutually reinforcing and activate other sirtuins, amplifying and sustaining a weak DNA damage or metabolic signal.
The work is also significant because it points to a new strategy to alleviate G6PD deficiency. Strategies that increase NAD+ or sirtuin activity can provide health benefits in mammals including improved insulin sensitivity, decreased inflammation and improved mitochondrial function (Yoshino et al, 2011; Canto et al, 2012; Gomes et al, 2013).
These findings may also be relevant to ageing. Genome instability and oxidative stress are hallmarks of old age, and there is abundant evidence that the sirtuins slow the pace of biological ageing. Sirtuin activity and NAD+ levels decline with old age (Gomes et al, 2013), and perhaps not coincidentally, so does the PPP (Niedermuller, 1986). Although a link between these three phenomena has not yet been established, in the light of the new data, one can see how decreased NAD+ and SIRT2 activity could inhibit G6PD and inhibit nucleotide biosynthesis, genome replication and DNA repair, while increasing oxidative damage (Fig1). Thus, maintenance of G6PD activity by keeping NAD+ levels high throughout our lives might be a way to delay key aspects of ageing. If so, the study of glucose utilization is far more interesting and medically relevant than biochemistry textbooks would have us believe.
Figure 1. Potential positive feedback loop involving the pentose phosphate pathway (PPP), NAD+ and SIRT2.
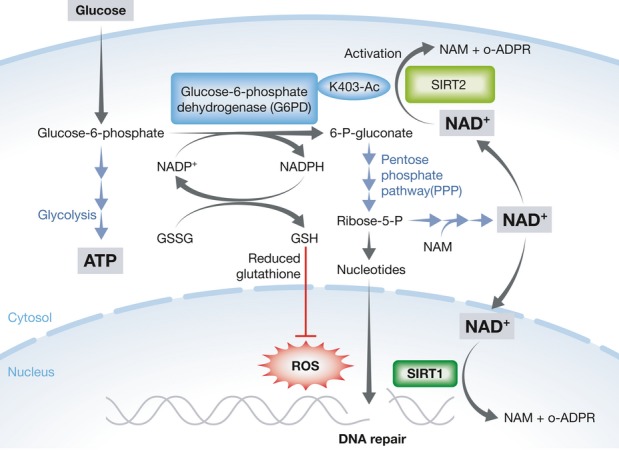
SIRT2 mediates deacetylation and activation of glucose-6-phosphate dehydrogenase (G6PD), which catalyses the first committed step of the PPP. The PPP converts NADP+ to NADPH and regenerates the antioxidant glutathione (GSH). A product of the PPP, ribose-5-phosphate, is used as a substrate for NAD+ synthesis to increase SIRT2 activity and further activate the PPP. In this way, small increases in NAD+ may be amplified and sustained. Patients with a G6PD deficiency may benefit from molecules that raise NAD levels. Conversely, a decline in NAD+ with age could have deleterious effects on antioxidant defences and may underlie aspects of normal aging.
Acknowledgments
LEW is an Early Career Fellow of Cancer Institute NSW, Australia. DS is supported by the NIH, the Juvenile Diabetes Research Foundation, The Schulak family and the Paul F. Glenn Foundation for Medical Research.
Conflict of interest
LEW declares no conflict of interest. DS discloses that he is a consultant to GlaxoSmithKline, OvaScience, Cohbar, Segterra and MetroBiotech.
References
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD(+) induces a Pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann AD, Cohen Y, Chodick G. Glucose-6-phosphate dehydrogenase deficiency and type 2 diabetes. Diabetes Care. 2012;35:e58. doi: 10.2337/dc11-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedermuller H. Effects of aging on the recycling via the pentose cycle and on the kinetics of glycogen and protein metabolism in various organs of the rat. Arch Gerontol Geriatr. 1986;5:305–316. doi: 10.1016/0167-4943(86)90033-6. [DOI] [PubMed] [Google Scholar]
- Pan S, World CJ, Kovacs CJ, Berk BC. Glucose 6-phosphate dehydrogenase is regulated through c-Src-mediated tyrosine phosphorylation in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:895–901. doi: 10.1161/ATVBAHA.109.184812. [DOI] [PubMed] [Google Scholar]
- Ralser M, Wamelink MM, Kowald A, Gerisch B, Heeren G, Struys EA, Klipp E, Jakobs C, Breitenbach M, Lehrach H, Krobitsch S. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J Biol. 2007;6:10. doi: 10.1186/jbiol61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, Ling ZQ, Hu FJ, Sun YP, Zhang JY, Yang C, Yang Y, Xiong Y, Guan KL, Ye D. Regulation of G6PD acetylation by KAT9/SIRT2 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014;33:1304–1320. doi: 10.1002/embj.201387224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Christian W. Über das gelbe Oxydationsferment. Biochemische Zeitschrift. 1933;257:492. [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]