

Abstract
Loss of the coenzyme NAD+, which is required for many energy-dependent cellular processes, has emerged as a potentially unifying mechanism for age-related conditions. A study in this issue of The EMBO Journal identifies a novel link between depletion of NAD+ and age-associated loss of proliferating adult neural stem/progenitor cells in the murine brain (Stein & Imai, 2014). These data have important implications for how brain function might decline with age.
Stem cells are defined by two essential functions: (i) the ability to differentiate into multiple cellular lineages and (ii) the capacity to undergo self-renewing mitosis. Loss of either function can diminish tissue homeostasis and thus cause phenotypes and pathologies associated with aging (Jones & Rando, 2011). Impaired differentiation deprives tissues of essential cell lineages, whereas loss of self-renewal depletes the progenitor pool and ultimately exhausts any lineages that arise from those progenitors. In the case of neural stem/progenitor stem cells (NSPCs), impairment of both renewal and differentiation occurs during aging, but why do these NSPC functions decline with age? Stein and Imai show that this loss is likely due to an age-related decline in the enzyme nicotinamide monophosphoribosyl transferase (Nampt) and hence its product nicotinamide mononucleotide (NMN). NMN is subsequently converted into nicotinamide adenine dinucleotide (NAD+), a crucial co-factor for numerous energy-requiring cellular processes, including energy metabolism, deacetylation, polyadenylation, and calcium signaling (Fig1).
Figure 1. Metabolic pathways link NAD+ to tissue homeostasis.
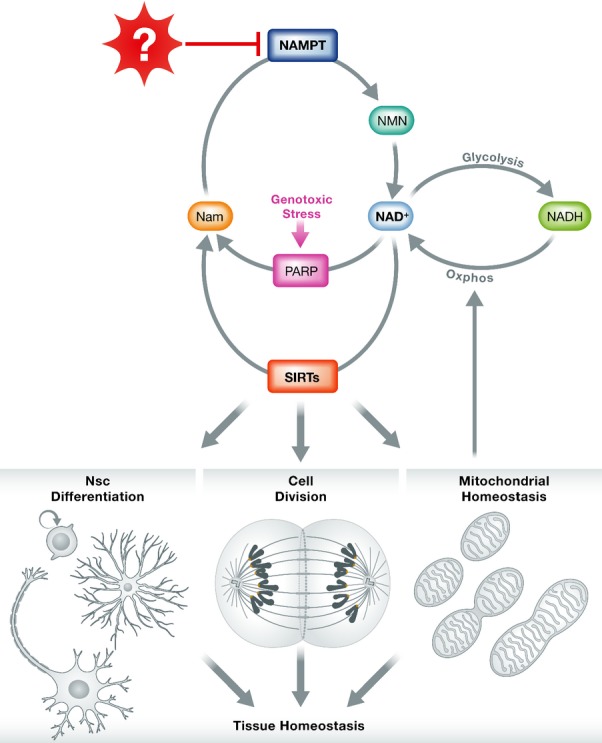
NAD+ levels are regulated by several factors. In response to genotoxic stress, PARPs consume NAD+ and release nicotinamide (Nam). The sirtuins similarly consume NAD+ and release Nam in deacetylation reactions that act in part to maintain homeostasis of various tissues through control of NSPC self-renewal and differentiation (brain), cell division (mitotically active tissues), and mitochondrial function (muscle). NAD cycles between NADH following glycolysis and NAD+ following oxidative phosphorylation in the mitochondria. Nam is then salvaged to NMN by Nampt, which declines with age by an unknown mechanism.
Because Nampt is rate-limiting for the salvage of NAD+ from nicotinamide, Nampt insufficiency can deplete cellular stores of NAD+. NAD+ has been linked to phenotypic changes associated with aging, including type 2 diabetes, cancer, and muscle degeneration (Garten et al, 2009; Gomes et al, 2013). How might NAD+ depletion lead to aging? In the context of aging and longevity, NAD+ is thought to serve as a substrate for the sirtuins, an evolutionarily conserved family of proteins with deacetylase or mono-ribosyltransferase activity. Some sirtuins positively regulate longevity pathways in diverse organisms, ranging from yeast and nematodes to mice and possibly humans (Haigis & Sinclair, 2010). Now, Stein and Imai show that attrition of Nampt—and consequently NAD+—most likely mediates stem cell dysfunction in the brain.
Stein and Imai document loss of both differentiation and self-renewal capacities in NSPCs that have insufficient NAD+. They show that NAD+ levels decline in the hippocampus of mice with age and that this loss of NAD+ coincides with reduced Nampt levels—in agreement with previous studies showing loss of Nampt and/or NAD+ in other (peripheral) aged tissues (Yoshino et al, 2011; Gomes et al, 2013). Nampt levels correlated highly with the level of NSPC function in the aging brain, consistent with a causal relationship between NSPC dysfunction and NAD+ levels. To test this hypothesis, the authors used a transgenic mouse in which it was possible to inducibly eliminate Nampt from NPSCs. Nampt elimination reduced the NPSC cell pool and impaired their capacity for self-renewal. In agreement with these observations, pharmacological or genetic inhibition of Nampt also suppressed the ability of NSPCs to form neurospheres and undergo cell division in culture, with commensurate changes in gene expression indicative of a proliferative arrest in the G1/G0 phase of the cell cycle. The arrest could be rescued by dietary supplementation with NMN, indicating that Nampt enzymatic activity is required for NSPC self-renewal. Most notably, in wild-type mice between the ages of 6 and 18 months, dietary NMN supplements prevented the loss of NPSC markers, suggesting that exogenous NMN could be an efficacious strategy to treat or prevent age-related loss of NSPC function. It is not yet known whether the partial rescue of NSPC self-renewal is due to insufficient rescue of NAD+ levels in the brain, or whether other factors contribute to the loss of NPSC function with age. Nonetheless, the data clearly show that Nampt-mediated synthesis of NAD+ is essential for the self-renewal of NSPCs.
In addition to a loss of self-renewal, NAD+ insufficiency also suppressed the ability of NSPCs to differentiate. In cell culture assays, Nampt-deficient NSPCs were defective in differentiating into oligodendrocytes—cells responsible for the myelination of neurons in the brain. Specifically, Nampt-deficient NSPCs were unable to form oligodendrocyte precursor cells (OPCs). Further, OPC differentiation into oligodendrocytes not only required NAD+, but also required the redundant activities of the sirtuins SIRT1 and SIRT2. Genetic or pharmacological inhibition of both sirtuins—but not each individually—strongly prevented OPC differentiation. This failure of differentiation coincided with increased expression of p21 (CDKN1A), a cell cycle inhibitor that is important for quiescence and senescence.
An important prediction of these findings is that loss of NAD+ with age might prevent regenerative remyelination after brain injury. To test this possibility, Stein and Imai used cuprizone to induce demyelination and subsequent remyelination. After cuprizone feeding, wild-type NSPCs appeared in the corpus callosum, a highly myelinated region of the brain. Genetic ablation of Nampt suppressed this process, resulting in defective remyelination. Thus, NAD+ was required for differentiation of the oligodendrocyte lineage both in culture and in vivo.
This new study raises several intriguing questions. The finding that SIRT1 and SIRT2, which have deacetylase activity, are redundantly required for oligodendrocyte differentiation suggests the existence of an acetylated factor or factors that mediate this differentiation. As the authors point out, the tumor suppressor p53 stands out as a relevant SIRT1/2 target in this context. p53 is activated by acetylation, is deacetylated by both sirtuins, and induces p21 transcription in many cell types. Further, p53 acetylation and activity is antagonized by Olig2, a transcription factor that is needed for oligodendrocyte genesis (Mehta et al, 2011; Sun et al, 2011). Thus, p53 is a strong candidate as a downstream suppressor of oligodendrocyte differentiation, which is reversed by NAD+-dependent SIRT1 and SIRT2 activity. This study also comes on the heels of another recent publication in which NAD+ was shown to be essential for the maintenance of mitochondrial homeostasis in the muscles of aging mice (Gomes et al, 2013).
Given that loss of NAD+ has now been linked to multiple age-associated conditions (Yoshino et al, 2011; Gomes et al, 2013), an important unanswered question is whether the age-related depletion of NAD+ results from a common source, such as loss of NAMPT, or whether there are other causes of NAD+ depletion (Fig1). Two potential alternative sources of NAD+ depletion are genotoxic stress and mitochondrial dysfunction. Genotoxic stress activates poly-ADP-ribose polymerases (PARPs), which consume NAD+ as a cofactor. Mitochondrial dysfunction, by comparison, can result in an accumulation of NADH at the expense of NAD+. Thus, while loss of NAD+ is a potential common mediator of aging phenotypes, it remains unclear whether this deficit is the result of a single underlying mechanism. Since depletion of Nampt with age is responsible for the loss of NAD+ in NSPCs (and potentially other stem or progenitor cells), it is important to uncover what drives Nampt loss. Surprisingly, little is known about the regulation of Nampt. Previous work implicated a host of candidate regulators, but none that would be considered an obvious candidate for mediating the age-related decline in Nampt levels. For example, NAMPT is regulated by the circadian transcription factor CLOCK (Ramsey et al, 2009) and AMP-activated protein kinase (Fulco et al, 2008), but it is unclear how either of these mechanisms might be linked to neural aging. Finally and perhaps most importantly, the Stein and Imai study suggests a simple potential intervention in the aging brain. If dietary NMN can partially prevent the effects of Nampt loss, might this metabolite be used therapeutically to prevent the loss of NSPC function during aging or following injury? If so, can any of the neurodegenerative disorders associated with aging be rescued by this approach? Notably, these data suggest that treatable deregulation of NAD+, rather than irreversible macromolecular damage, might mediate part of the aging process in the brain, providing a promising foundation for future intervention.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Suave AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nature Cell Biol. 2011;13:506–512. doi: 10.1038/ncb0511-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, Alberta JA, Kane MF, Theisen M, Ligon KL, Rowitch DH, Stiles CD. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell. 2011;19:359–371. doi: 10.1016/j.ccr.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, Takahashi JS, Imai S, Bass J. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Albert JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, Powers JF, Runquist IS, Rowitch DH, Stiles CD. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011;69:906–917. doi: 10.1016/j.neuron.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]